

Abstract
In bacteria, ribosomal hibernation shuts down translation as a response to stress, through reversible binding of stress‐induced proteins to ribosomes. This process typically involves the formation of 100S ribosome dimers. Here, we present the structures of hibernating ribosomes from human pathogen Staphylococcus aureus containing a long variant of the hibernation‐promoting factor (SaHPF) that we solved using cryo‐electron microscopy. Our reconstructions reveal that the N‐terminal domain (NTD) of SaHPF binds to the 30S subunit as observed for shorter variants of HPF in other species. The C‐terminal domain (CTD) of SaHPF protrudes out of each ribosome in order to mediate dimerization. Using NMR, we characterized the interactions at the CTD‐dimer interface. Secondary interactions are provided by helix 26 of the 16S ribosomal RNA. We also show that ribosomes in the 100S particle adopt both rotated and unrotated conformations. Overall, our work illustrates a specific mode of ribosome dimerization by long HPF, a finding that may help improve the selectivity of antimicrobials.
Keywords: cryo‐electron microscopy, hibernation, pathogen, ribosome
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Protein Biosynthesis & Quality Control; Structural Biology
Introduction
Survival under unfavorable conditions is one of the key factors of a successful bacterial infection. Considering the variety of stresses bacteria may be exposed to during their life cycle, bacterial responsiveness should be timely and effective and require little energy. In that sense, hibernation of the translation machinery appears as beneficial, since protein synthesis is the most energy‐consuming cellular process (Russell & Cook, 1995; Szaflarski & Nierhaus, 2007).
The hibernation of ribosomes is prompted by the binding of small stress‐induced proteins to 70S ribosomes, which frequently triggers their association into 100S ribosome dimers (“disomes”; see Yoshida & Wada, 2014 for a recent review). These hibernation agents vary according to species or bacterial strain (Ueta et al, 2013). For example, Escherichia coli possesses a hibernation‐promoting factor (EcHPF) and a ribosome modulation factor (EcRMF) that act in concert (Ueta et al, 2005, 2008), while S. aureus comprises instead a single but longer variant of the HPF protein (SaHPF; Ueta et al, 2010, 2013; Tagami et al, 2012). The C‐terminal end in SaHPF (CTD) is only marginally similar to RMF (Ueta et al, 2010), but nonetheless conserved across bacteria (Ueta et al, 2013). The expression patterns of these factors differ between bacteria, as for example, EcHPF and EcRMF are expressed during the transition to the stationary phase, while SaHPF is expressed during every growth phase (Ueta et al, 2010). These species‐specific characteristics account for differential responses to stress conditions. For instance, dimerization increases long‐term viability and stress tolerance during the stationary phase in E. coli (Yoshida & Wada, 2014), while in Bacillus subtilis, it facilitates rapid regrowth of cells upon stress removal (Akanuma et al, 2016). In S. aureus, SaHPF prevents ribosome degradation and interferes with translation initiation (Basu & Yap, 2016).
As their variations in structure, expression pattern, and functional role suggest, hibernation factors promote dimerization in different ways. In E. coli, dimerization occurs in two steps: 90S dimers are formed upon binding of EcRMF to the anti‐Shine‐Dalgarno (SD) region of an mRNA‐free ribosome, before they are converted to 100S dimers upon binding of EcHPF (Ueta et al, 2005, 2008) to tRNA binding sites (Polikanov et al, 2012). In S. aureus, dimerization mediated by the longer SaHPF does not go through a 90S stage (Ueta et al, 2010). In addition, the stability of 100S dimers is proportional to the length of HPF (Ueta et al, 2013), which further emphasizes the importance of comparatively analyzing dimerization in bacteria (Khusainov et al, 2016a).
At the molecular level, dimers of E. coli ribosomes analyzed by cryo‐electron microscopy (cryo‐EM) and tomography at resolutions > 30 Å were shown to be mediated by ribosome–ribosome contacts, which involve ribosomal proteins uS2, uS9, uS10, and helix 39 of the 16S rRNA (Kato et al, 2010; Ortiz et al, 2010). As no intermolecular interactions were observed between hibernation factors, the role in particular of the C‐terminal extension in longer HPF (as in SaHPF) remained unclear. Structural information available regarding hibernation factors consisted of NMR and X‐ray crystal structures that revealed similar folds for the N‐terminal domain (NTD), and a similar binding mode to the 30S subunit (Vila‐Sanjurjo et al, 2004; Sato et al, 2009; Polikanov et al, 2012). One structure for the CTD assigned as ribosome‐associated protein Y from Clostridium acetobutylicum was found in the Protein Data Bank (PDB ID 3KA5).
Here, we obtained the structure at near‐atomic resolution (3.7 Å) of hibernating SaHPF‐bound ribosomes from S. aureus by cryo‐electron microscopy. The structures show that the NTD is bound to the decoding center, while the CTD protrudes out of each ribosome through the E site. The structures of full dimers of hibernating ribosomes solved at 9–11 Å revealed the direct involvement of the SaHPF‐CTD in dimerization, as we also observed in our NMR structure of an isolated SaHPF‐CTD. Moreover, h26 makes additional contacts between ribosomes, thereby supporting its implication in translation regulation. Furthermore, dimers accommodate any combination of unrotated or rotated states of the ribosome, suggesting an efficient mechanism for stalling and resuming translation in a timely manner. Overall, our work paves the way for deciphering the diverse nature of stress response across bacteria, which opens up new avenues for developing drugs with increased selectivity and potency.
Results
In vitro reconstitution of 100S dimers
Ribosome dimers from S. aureus were reconstituted in vitro from purified 70S ribosomes. First, high‐salt washed vacant 70S ribosomes were purified by sucrose‐density gradient centrifugation (Fig 1A). We then purified His‐tagged SaHPF expressed in E. coli by affinity chromatography (using Ni sepharose) followed by size‐exclusion chromatography. Two peaks were eluted that both contained SaHPF in dimeric form as revealed by native polyacrylamide gel electrophoresis (native PAGE; Fig EV1A–C). SaHPF from the second peak contained a higher absorbance at 260 nm and was more efficient at promoting ribosome dimerization (Fig EV1A–E). The best production of 100S ribosomes was achieved using a pool of fractions containing the highest absorbance at 260 nm (with a 2:1 ratio for A260/A280; Fig EV1A, F and G).
Figure 1. Purification and characterization of 100S ribosome dimers.
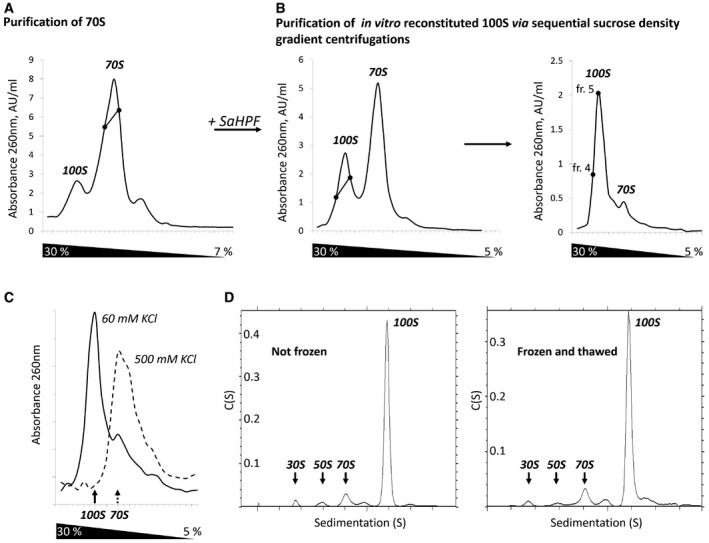
- Sedimentation profile performed to separate 70S ribosomes from 100S particles present in the cell.
- Sedimentation profiles were carried out sequentially in order to separate 100S from 70S particles. SaHPF was added to fractions containing 70S particles (intercept on panel A). Fractions containing 100S dimers (intercept on left panel) were further purified on a similar gradient (right panel). Fractions 4 and 5 were used to assess stability and to perform cryo‐EM analysis.
- 100S dimers are stable at low ionic strength. Sucrose gradient performed under similar conditions to that shown in panel (B).
- 100S dimers do not suffer from freezing. The sedimentation profiles from analytical ultracentrifugation experiments are shown for a sample that was either kept at 4°C prior to analysis (left), or frozen at −80°C and thawed at 4°C (right).
Figure EV1. Purification and characterization of active His‐tagged SaHPF protein.
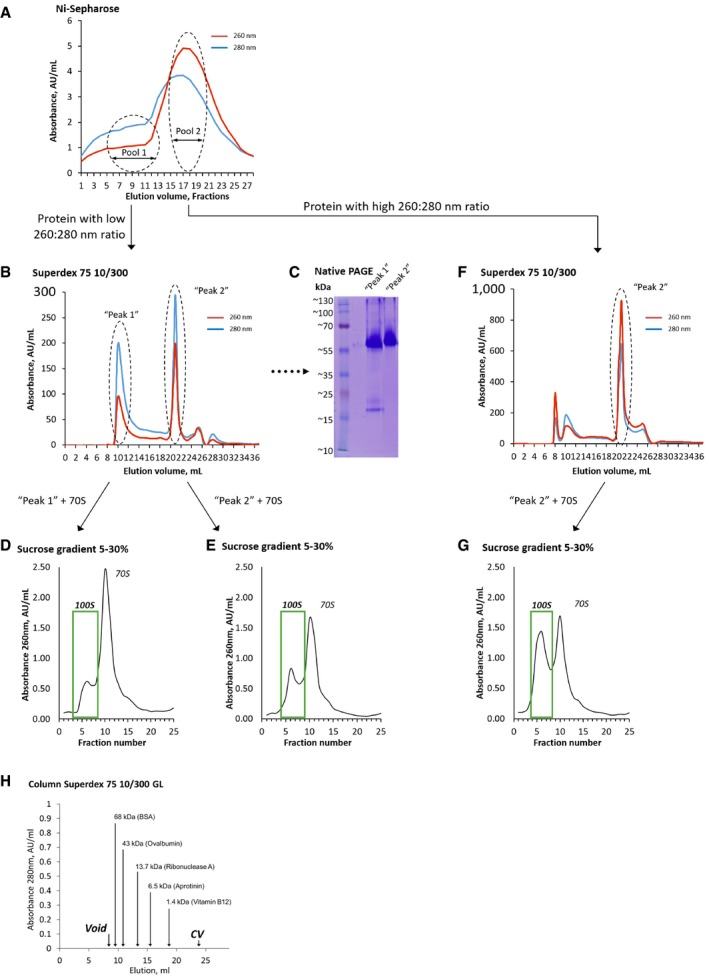
-
AElution profile of SaHPF after Ni sepharose affinity chromatography. Fractions containing mostly absorbance at 280 nm were pooled (pool 1) separately from those containing mostly absorbance at 260 nm (pool 2). Each pool was subjected to size‐exclusion chromatography.
-
BElution profile of pool 1 after size‐exclusion chromatography. Two peaks contained SaHPF: “peak 1” corresponds to pure protein (size ˜50 kDa, suggesting the presence of SaHPF as a dimer); “peak 2” contains an extra absorbance at 260 nm and eluted at a position near 1 column volume, possibly due to interaction of this pool with the resin.
-
CNative PAGE of samples shown in (B), indicating that both peaks contain SaHPF dimers (˜55 kDa).
-
D, ESucrose‐density gradient centrifugation profiles demonstrate the higher efficiency of “peak 2” in ribosome dimerization.
-
FSame as (B) for pool 2 after size‐exclusion chromatography. Here, peak 1 is smaller and peak 2 contains an extra absorbance at 260 nm. Peak 2 was collected and used for dimerization of ribosomes.
-
GSucrose‐density gradient centrifugation profile obtained with peak 2 highlighted in (F).
-
HCalibration graph of Superdex 75 10/300 GL column according to the manufacturer.
These dimers were then subjected to two sequential sucrose‐density gradient centrifugations, leading to a homogeneous population of disomes (Fig 1B). By dialyzing this dimer solution from 50 to 500 mM KCl, and back to 60 mM, followed by ultracentrifugation analysis, we showed that disome association is reversible (Fig 1C). We also tested disome stability by subjecting an aliquot of pure 100S to one freeze–thaw cycle (from −80°C to 4°C), followed by analytical ultracentrifugation. The resulting profiles indicated that > 90% of 100S dimers remained formed after one freeze–thaw cycle, although we observed minor aggregated contaminants (Fig 1D). Together, these findings indicate that we could reconstitute stable 100S dimers of S. aureus ribosomes in vitro from independently expressed ribosomes and SaHPF.
Strategy for visualizing hibernating ribosomes at different resolutions
Cryo‐EM images revealed a mixture of monomers and dimers (Fig 2A), with some dimers less rigid than others. In order to simultaneously get a near‐atomic resolution of hibernating ribosomes and an overall view of their dimeric architecture, we processed the same set of cryo‐EM images in two different ways. First, we chose a large box in order to only pick and classify ribosome dimers (Fig 2A, red ovals). At that stage, we generated 2D reconstructions for the flexible disomes that comprised one well‐resolved and one blurred ribosome (Figs 2B and EV2). After processing, we retrieved two categories of dimers, “tight” and “loose” (Fig EV2). Within the tight dimer, both 70S were better resolved than in the loose dimer (Figs EV2 and EV3B). Within each tight or loose category, we counted three types of dimers, comprising either only unrotated ribosomes, only rotated ribosomes, or a combination of both (Figs EV2 and EV3, and Table 1). Cryo‐EM density maps for the tight and loose dimers were further refined to resolutions of 11 and 9 Å, respectively (at FSC = 0.143; Figs 2C, EV2 and EV3A–C). In each dimer, the contact region was observed as a continuous density coming from around the platform and the body of the 30S particle. While we identified only one contact region in the loose dimer, two were found in the tight dimer.
Figure 2. Two strategies for processing cryo‐EM density maps of Staphylococcus aureus ribosome dimers.
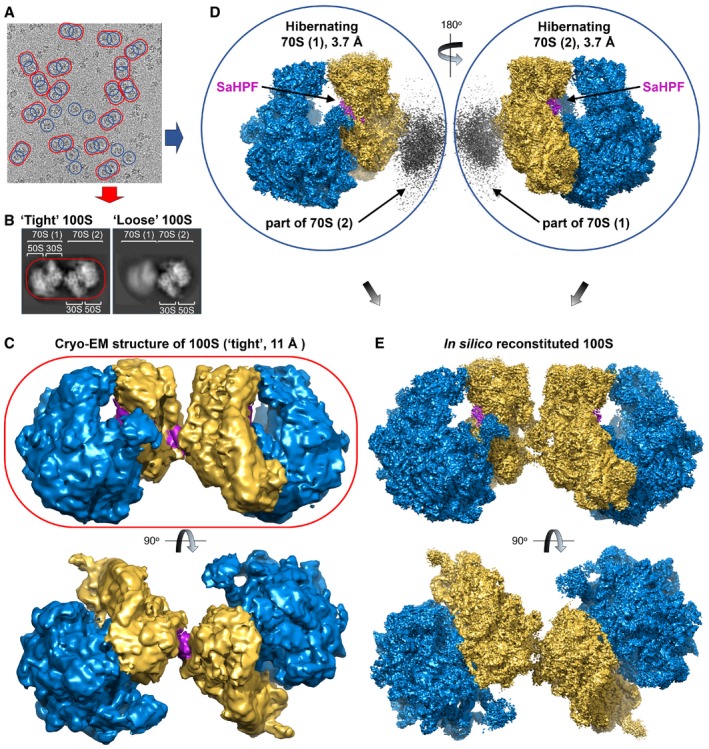
- Representative electron micrograph showing the distribution of 100S particles. Map refinement was carried out at either low resolution using a dimer as a reference (red oval), or at high resolution using a monomer as the search model (blue circles).
- Representative close‐ups of “tight” and “loose” dimers.
- Structure of the 100S dimer at 11 Å resolution (yellow, SSU; blue, LSU).
- Map processing using a monomer as the reference structure leads to a resolution comparable to that of the vacant 70S (Khusainov et al, 2016b). The gray density off from the SSU indicates the site of interaction between two monomers.
- Duplication followed by rotation of the SaHPF‐bound monomer solved at high resolution leads to an in silico reconstituted dimer.
Figure EV2. Sorting out various classes of ribosomes within a dimer.
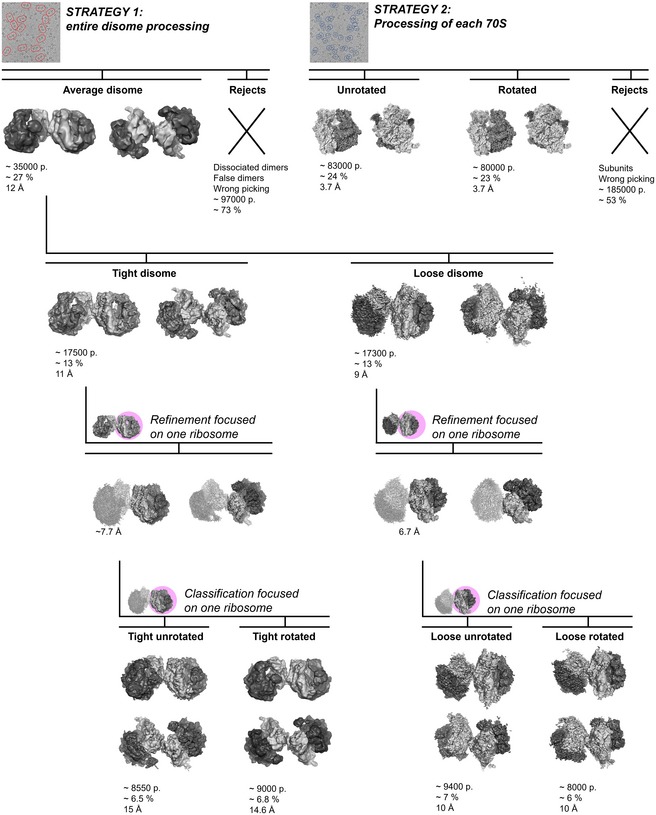
Strategy 1: Cryo‐EM 3D particle sorting reveals two types of disome reconstructions after several runs of classification. Strategy 2: 3D reconstruction of each 70S revealed heterogeneity of the disomes. The total number of particles (p) used for the 3D reconstructions is indicated below each structure, along with the percentage calculated over the total number of particles, and the final average resolution of each class after refinement.
Figure EV3. Local resolution estimation of cryo‐EM density maps.
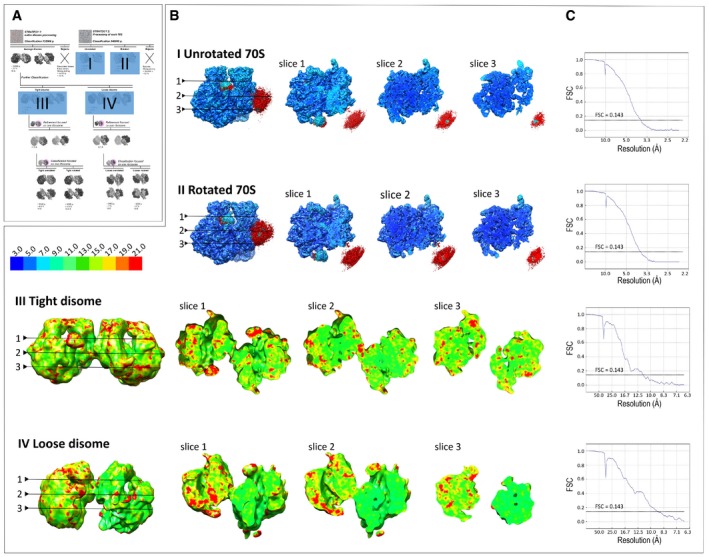
- Miniaturized scheme of cryo‐EM 3D particle sorting (shown in Fig EV2) highlighting 3D reconstructions (I–IV) used for local resolution estimation.
- Local resolution (rainbow scale) displayed on representative slices of the density maps I–IV.
- Fourier shell correlation (FSC) curve indicating the overall resolution of the cryo‐EM maps I–IV.
Table 1.
Data collection, refinement, and validation statistics
| Data collection | |
| Microscope | Titan Krios S‐FEG |
| Camera | CMOS (Falcon II) |
| Voltage (kV) | 300 |
| Magnification | 59,000× |
| Pixel size (Å.px−1) | 1.1 |
| Defocus range (μm) | (−4.5) – (−0.6) |
| Total dose (ē/Å2) | 60 |
| Dose per frame | ~3.5 |
| Refinement | (tight; loose) dimer/(unrotated; rotated) monomer |
| Number of particles (total) | 132,000/348,000 |
| Number of particles (used for 3D reconstruction) | 17,500; 17,300/83,000; 80,000 |
| Resolution (Å; at FSC = 0.143) | 11.0; 9.0/3.7; 3.7 |
| CC (model to map fita) | n.d.; n.d./0.73; 0.74 |
| Model composition | (unrotated; rotated) monomer |
| Non‐hydrogen atoms | 141,613; 141,636 |
| Residues | 10,090; 10,093 |
| RMS deviations | |
| Bonds (Å) | 0.01; 0.01 |
| Angles (°) | 1.08; 1.2 |
| Chirality (°) | 0.05; 0.08 |
| Planarity (°) | 0.005; 0.007 |
| Validation b | |
| Clashscorec | 6.48 (89th p.); 7.82 (82nd p.) |
| Proteins | |
| MolProbity score | 2.06 (72nd p.); 2.16 (67th p.) |
| Favored rotamers | 4,502 (96.22%); 4,400 (94.00%) |
| Ramachandran favored | 4,449 (82.28%); 4,343 (80.28%) |
| Ramachandran allowed | 923 (17.26%); 1,040 (19.22%) |
| Ramachandran outliers | 25 (0.46%); 25 (0.5%) |
| RNA | |
| Correct sugar puckers | 99.78%; 99.42% |
| Correct backbone conformations | 79.23%; 75.41% |
| Bad bonds | 0/109,772; 0/109,772 |
| Bad angles | 5/171,185; 8/171,185 |
FSC, Fourier shell correlation; CC, correlation coefficient; RMS, root‐mean‐square.
Only across atoms in the model; compiled using Phenix (Afonine et al, 2013).
Compiled using MolProbity (Chen et al, 2010); p, percentile.
Clashscore is the number of serious steric overlaps (> 0.4 Å) per 1,000 atoms.
In the second processing strategy, we treated each 70S ribosome on our grid equally, without considering whether it was within a dimer or present as a monomer (Fig 2A, blue circles). We thereby left the box dimensions large enough to include a part of the neighboring ribosome, in order to locate the contact regions within dimers. Three‐dimensional particle classification led to an equal partition of unrotated and rotated ribosomes, which all had SaHPF bound (Fig EV2). Following this procedure, we obtained a high‐resolution structure of the hibernating 70S ribosome in the context of 100S disomes (3.7 Å at FSC = 0.143; Figs EV2 and EV3, Table 1).
Further, in order to interpret the 11 Å resolution structure of 100S in greater details, we fitted each 3.7 Å hibernating 70S ribosome into the structure of the tight disome, using the densities of the body of the small subunit (SSU) and of the neighboring 70S (Fig 2D, gray) as a reference for the alignment. This structure is the one we analyze and discuss in the following sections, as its level of detail enables a thorough investigation of the ribosome dimerization mode.
Structure of SaHPF bound to the ribosome
SaHPF comprises an N‐terminal domain (NTD; residues 1–95) and a C‐terminal domain (CTD; residues 130–190), connected by a linker of 35 amino acids. SaHPF‐NTD was modeled by homology with the E. coli hibernation factors HPF and YfiA (Rak et al, 2002; Polikanov et al, 2015). SaHPF‐NTD therefore adopts a similar β1‐α1‐β2‐β3‐β4‐α2 arrangement, as it is wedged between the head and the body of the SSU (Figs 3A and B, and EV4A–D). Its β sheet interacts with 16S rRNA on the head (stacking interactions between Glu5/His7 and A975, and between Arg66 and G976), while conserved positively charged residues belonging to the α helices (Lys27, Arg30, Lys84, Arg90, Arg95) stabilize the interaction with 16S rRNA from the body (Fig EV4A and B). This observation rationalizes previous biochemical analysis that highlighted the essential role of these amino acids in binding to the ribosome in order to promote dimerization (Basu & Yap, 2016). Noteworthy, the interactions between SaHPF‐NTD and the body are lost upon subunit rotation, suggesting that the most stable contacts to the ribosome are those made with the head (Fig EV4A and B).
Figure 3. Ribosome dimerization occurs via interaction of the C‐terminal domain of SaHPF, and via interactions involving h26.
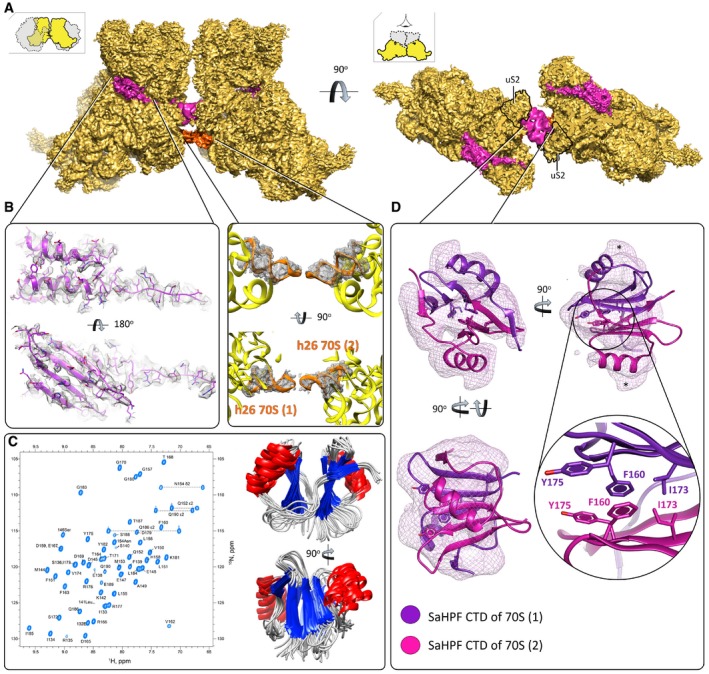
- View of the density maps for unrotated ribosome‐bound SaHPF, emphasizing the dimer interface (magenta, SaHPF; orange, h26; LSU omitted for clarity; orientations as in Fig 2). The density for CTD (interface) derives from the same but Gaussian‐filtered (2.0 SD) high‐resolution map.
- Side chains of amino acids from the N‐terminal domain of SaHPF are visible in the density map (left; unfiltered map). Helix 26 stabilizes dimer formation in the “tight” dimer (center; unfiltered map).
- SaHPF‐CTD forms a dimer in solution. 15N,1H‐HSQC spectrum of the C‐terminal domain of SaHPF showing backbone amide resonances (left). The spectrum was recorded at a proton resonance frequency of 700 MHz at 35°C, in PBS buffer (90% H2O + 10% D2O) at pH 7.6 with 200 mM NH4Cl concentration. Backbone superimposition of the 10 final simulated annealing structures in two orientations (right).
- Dimerization of the C‐terminal domain of SaHPF from two ribosomes within a dimer (map filtered at 2.0 SD using a Gaussian filter). Residues involved in hydrophobic interactions at the interface of the two CTDs are shown as sticks. The region of potential interaction of CTD with bS2 is marked by *.
Figure EV4. Comparison of SaHPF‐NTD‐ribosome contacts between unrotated and rotated ribosomes, as well as with EcYfiA and EcHPF .
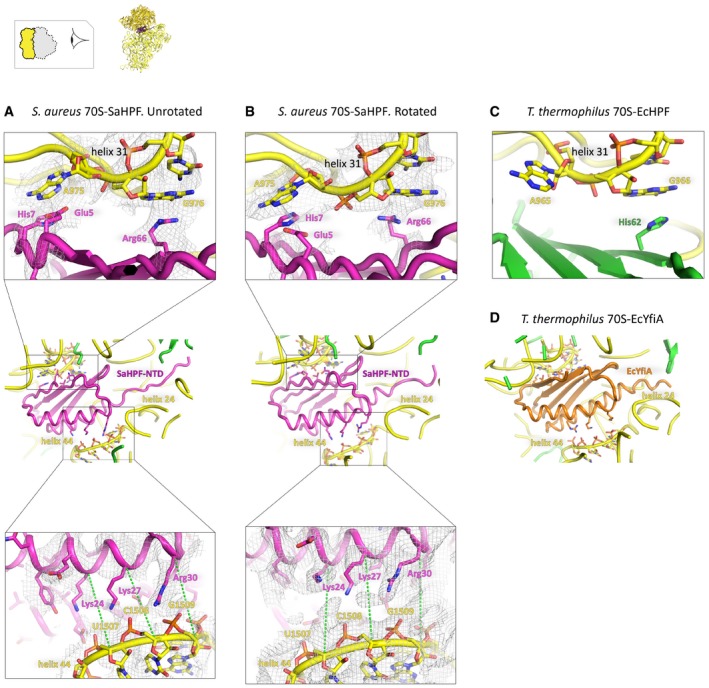
- Binding site of SaHPFNTD in the unrotated state. Amino acids of the NTD and nucleotides within 5 Å of one another in the head and body of the 16S rRNA are shown as sticks. The insets show a close‐up of the interface between the β sheet and helix 31 (top; unfiltered map), and between positively charged amino acids in the α helical domain and helix 44 (bottom; unfiltered map). Green dashed lines are drawn from the same protein and backbone atoms in the two structures, in order to help visualize conformational dynamics that occur upon rotation.
- Same as panel (A), but for the rotated state. Green dashed lines are drawn from the same protein and backbone atoms in the two structures, in order to help visualize conformational dynamics that occur upon rotation.
- Same as the top view in panels (A and B), but for EcHPF in the T. thermophilus ribosome (PDB ID 4V8H; Polikanov et al, 2012).
- Same as the middle view in panels (A and B), but for EcYfiA in the T. thermophilus ribosome (PDB ID 4V8I; Polikanov et al, 2012).
The last 60 amino acids that make the CTD were first modeled as adopting a β1‐α‐β2‐β3‐β4 structure, based on sequence similarity with the Lmo2511/Y protein (see Materials and Methods). In order to provide experimental support to this model, we analyzed the isolated CTD in solution using NMR. First, interproton distances were derived from 2D 1H‐1H NOESY for protein in 100%D2O, 3D 15N edited NOESY‐HSQC and 3D 13C edited NOESY‐HSQC. The intramonomer NOEs were obtained from isotope filtering 13C/15N – 12C/14N edited NOESY‐HSQC. Data processing led to 100 structures, of which we compared the 10 with the lowest energy (Fig 3C). The average root‐mean‐square deviations calculated from these structures were 2.0 Å for backbone atoms, and 2.6 Å for heavy atoms (see Table EV1 for complete statistics). In these structures, SaHPF‐CTD adopts a β1‐α‐β2‐β3‐β4 topology, so that the α‐helix comprises residues 146–156, and the β‐sheet is composed of β2 (residues 160–164), β3 (171–175), and β4 (182–187) from one molecule, and β1 (133–135) from the other one. Hydrophobic residues Phe160, Val162, Thr164, Thr171, Ile173, Tyr175 are likely to play a key role in forming the dimer structure. In the end, the three‐dimensional structure of SaHPF‐CTD solved by NMR was used for the interpretation of the density maps of the ribosomes.
100S formation is mediated by CTD interactions
Within the globular and symmetric dimer observed by NMR (Fig 3C), we observed that the average T1/T2 relaxation ratio for 15N‐labeled protein from 1H‐15N HSQC spectra was ~2‐fold larger than that expected for a monomeric form (Fig EV5A and B). We further carried out isotope filtering NMR experiments (13C/15N – 12C/14N edited NOESY‐HSQC) with a 1:1 mixture of labeled and non‐labeled proteins that enabled us to observe the following 36 intermonomer NOEs (total number of distance constraints = 1,326; Figs 3C and EV5B): (i) between the γ‐methyl and δ‐methyl groups of Ile173; (ii) between the aromatic ring of Phe160 and the δ‐methyl group of Ile173; (iii) between the aromatic ring of Phe160 and the β‐methylene group of Tyr175; and (iv) between the aromatic rings of Phe160 that interacts via π‐stacking (Fig 3D). Together, these observations indicated that SaHPF‐CTD exists as a homodimer in solution, even in the absence of the rest of the SaHPF protein and of the ribosomes. Overall, this NMR analysis supported the interpretation that the interaction between the CTDs induces the formation of 100S dimers.
Figure EV5. SaHPF‐CTD forms a dimer in solution.
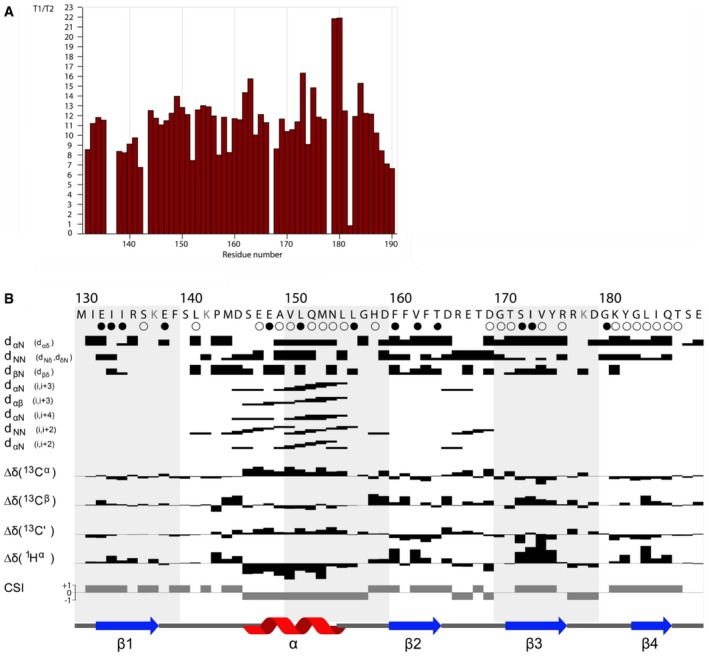
- T1/T2 relaxation time ratio for SaHPF‐CD residues (130–190).
- Summary of the sequential and medium‐range NOEs, chemical shift index (CSI; Berjanskii & Wishart, 2005), and TALOS secondary structure. Residues shown in gray have not been detected in 15N–1H heteronuclear NMR spectra. The cartoons represent the portions of the SaHPF‐CD predicted by TALOS to be α‐helical (red) or β‐strand (blue). Slowly and intermediate exchanging protons are identified by filled and empty circles, respectively.
We fitted the NMR structure of the CTD dimer into the extra density we observed close to the tRNA exit in all disome structures that we obtained after processing (Figs 2 and 3). The SaHPF‐CTD does not directly interact with the ribosome, as it protrudes instead next to ribosomal protein uS2. The path of the ~35 amino acids that connect the NTD to the CTD is not visible, indicating these residues are flexible even in the dimeric state. Additional contacts are observed between helix 26 of the two ribosomes at the interface of the “tight” dimer. However, since the h26‐h26 contacts are absent in the “loose” dimers, they are probably secondary in providing dimer stability. Therefore, we conclude that the interactions involving the CTDs are the primary driver for dimerization of ribosomes during hibernation.
100S dimers accommodate both unrotated and rotated conformations of 70S ribosomes
As mentioned above, unrotated and rotated 70S ribosomes were evenly distributed among our reconstructions at the highest resolution (Fig EV2). In this particular case of dimer formation occurring via the small subunit (SSU)—and not involving the large subunit (LSU)—we consider the rotation of the LSU with respect to the SSU, and not vice versa as more conventionally accepted (Fig 4A). The rotated state within a dimer provokes a shift of atom coordinates by as much as ~35 Å at the level of the central protuberance (the smaller shifts were found near the peptide tunnel exit; Fig 4B). The comparison between the unrotated 70S in its vacant state (Khusainov et al, 2016b) and SaHPF‐bound states (this work) revealed that the binding of SaHPF principally induced conformational changes in flexible ribosomal elements in the LSU, such as the L1 stalk, the L7/L12 stalk, and helix H69 (Appendix Fig S1).
Figure 4. Both unrotated and rotated ribosomes are found within dimers.
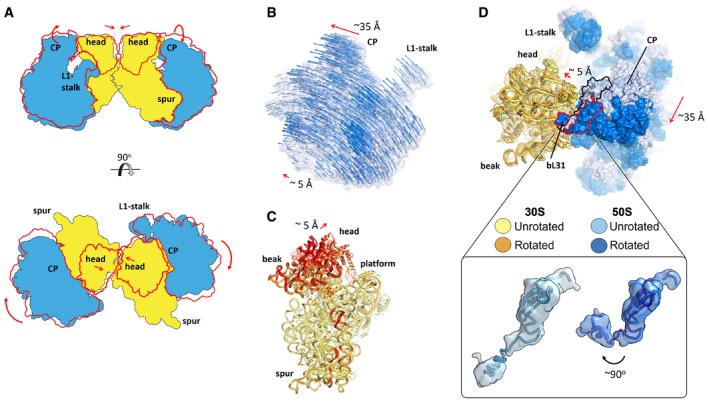
- Schematic representation of subunits movements within a disome (red contour, rotated state of the LSU, and the head of the SSU).
- Movement of the LSU from the unrotated to the rotated conformation. Vectors overlayed on the density map of the unrotated structure help visualize the direction of the rotation, as well as its amplitude. Vectors were calculated by measuring the distance between phosphate atoms (for RNA) and between C‐α atoms (for proteins). For clarity, only one in three vectors is represented.
- The head of the SSU buckles away upon subunit rotation. Superimposition of the unrotated and rotated SSU, based on structural elements from the body only (red, root‐mean‐square deviations > 3 Å between the two sets of coordinates).
- Superimposition as in (B) but from looking toward the head of the SSU, and displaying the LSU as a surface. Inset, close‐up on the relative orientation of bL31 in the unrotated and rotated ribosomes (map filtered at 1 SD using a Gaussian filter).
The average amplitude of the movements of the SSU during rotation is smaller (~10 Å), with the largest rearrangements occurring in the head and at h44 in close proximity to the decoding center (Fig 4C). In response to the central protuberance movements of the LSU, the head of the SSU buckles away toward the solvent‐exposed side (Fig 4C). These concerted conformational changes between the two subunits are controlled by a variant of ribosomal protein bL31 that comprises a 15‐amino acid extension (“B” type) in place of Zn2+‐coordinating cysteines (“A” type; Fig 4D; Khusainov et al, 2016b). As observed previously for the A type (Jenner et al, 2010; Fischer et al, 2015), and as we recently described for the B type in the vacant S. aureus ribosome (Khusainov et al, 2016b), protein bL31 interacts with the 5S rRNA through its N‐terminal domain, and with h42 on the SSU through its C‐terminal domain. Because we obtained both unrotated and rotated conformations in the S. aureus dimer, we can observe that the B‐type bL31 accommodates subunit dynamics in a similar manner to the A type, via its flexible linker between its N‐ and C‐terminal domains. Our structure suggests that SaHPF could bind to ribosomes independently from their unrotated or rotated state.
Discussion
The 60‐aminoacid CTD found in long variants of HPF is responsible for ribosome dimerization in S. aureus. In both the “tight” and “loose” dimers we observed by cryo‐EM, the CTDs of ribosome‐bound SaHPF protrude out of the platform of the small subunit next to uS2. The CTDs interact via electrostatic and hydrophobic interactions, as we characterized in more detail using NMR. In the tight disomes, additional interactions involve helix 26 (Figs 3A and B, and 5A and B). Hence, the interface is markedly different between 100S from S. aureus and E. coli (Fig 6A and B). In the E. coli dimer, ribosomes are closer to one another in space, and they interact principally through the heads and platforms of the small subunits (Kato et al, 2010; Ortiz et al, 2010). Prior to this work, no hibernation factors had been described that directly mediated dimerization.
Figure 5. Structural variation between tight and loose disomes.
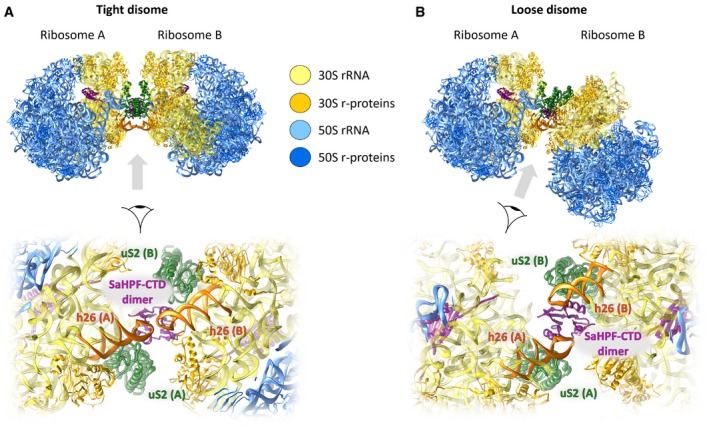
- Molecular model of a tight disome, oriented as in Fig 2 (top). Close‐up of the dimer interface, viewed from the bottom of panel (A) (bottom).
- Molecular model of a loose disome, with ribosome A oriented as in panel (A). Both disomes were reconstituted by placing the structure of the monomer into the density map of the disome, using the body of the SSU as a reference. Only unrotated ribosomes are shown.
Figure 6. Overview of hibernation modes across bacteria.
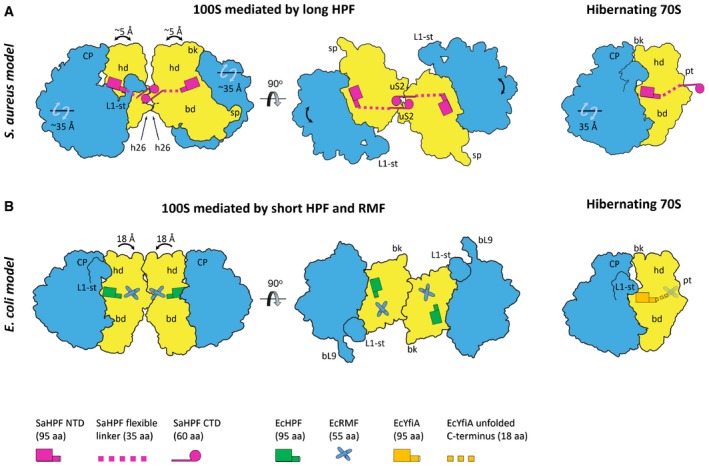
- Ribosome hibernation in Staphylococcus aureus occurs upon binding of SaHPF‐NTD to the SSU, while dimerization is mediated by interactions of SaHPF‐CTDs. Secondary contacts involve helix 26. Relative rotations of the subunits and head movements are indicated in this panel and panel (B).
- Ribosome hibernation in Escherichia coli involves three proteins. Simultaneous binding of EcRMF and EcHPF leads to the conformational change in the head of the SSU, which in turn allows two ribosomes to interact via contacts between ribosomal proteins and rRNA (Kato et al, 2010; Ortiz et al, 2010). Hibernation can also occur upon binding to the YfiA protein, which prevents binding of EcRMF due to its extended C‐terminal tail, thereby not promoting dimer formation (Polikanov et al, 2012).
Structural differences at the interface likely account for reported variations in dimer stability. The extensive network of non‐covalent interactions in the S. aureus 100S seems to be responsible for the enhanced stability of dimers that are mediated by a long HPF rather than a short HPF and RMF (Ueta et al, 2013). Whether the interactions involving h26 in the “tight” dimer (Figs 3A and B, and 5A) contribute to this higher stability is unclear. Other bacteria like Thermus thermophilus possess a long HPF, although h26 in these species is shorter (Sohmen et al, 2015; Khusainov et al, 2016b). Because h26 is shorter by three base pairs in T. thermophilus, it could not form the interactions we see in the S. aureus 100S, unless the two ribosomes within the 100S from T. thermophilus were to tilt in order to bring their respective h26 within interaction distance. Here, the role of the flexible linker between the NTD and the CTD could be to absorb the structural constraints induced on the interacting CTDs by subunit rotation or tilting. In any case, it is possible that the interface between ribosomes within a dimer would display distinctive features across species containing a long HPF, according to the length of h26. Due to their proximity to HPF, we cannot exclude that ribosomal proteins such as uS2 would also participate in ribosome–ribosome contacts within certain dimers. Overall, although dimer formation is a way of responding to stress that is shared among bacteria, this analysis suggests that the mechanisms of dimer formation and the dimer architectures significantly vary across bacterial species.
Shutting down translation by occupying functional binding sites for tRNAs and initiation factors seems to be a conserved mode of action among hibernation factors from various bacteria. Our structure shows that the NTD of SaHPF binds to the small subunit similarly to its homologs EcHPF, EcYfiA, and a plastid‐specific YfiA (Vila‐Sanjurjo et al, 2004; Sharma et al, 2010; Polikanov et al, 2012; Bieri et al, 2017; Figs 7A and EV4). Requiring a single (as in S. aureus) versus a combination of hibernating factors (as in E. coli) could enhance bacteria's timeliness in blocking translation and enabling dimerization. In order to further assess this possibility, it would be interesting to study binding kinetics and cooperativity of the NTD and the CTD of long HPF versus those of short HPF and RMF. On the other hand, a decrease in the number of factors required for hibernation could indicate a more limited variability of regulatory pathways.
Figure 7. SaHPF promotes hibernation by preventing the binding of functional partners and antibiotics.
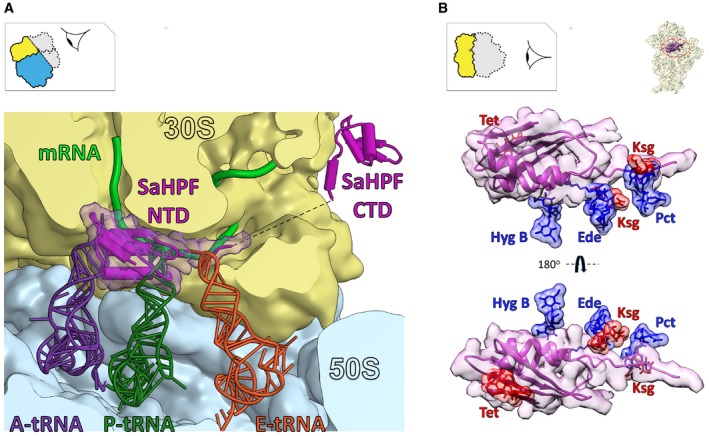
- The binding site of SaHPF overlaps with that of tRNAs and mRNA (magenta, surface rendering). tRNAs are superimposed from the structure of a 70S ribosome from T. thermophilus (PDB ID 5E81; Rozov et al, 2016).
- Superimposition of ribosome bound to SaHPF and bacterial ribosomes bound to various antibiotics. Hyg B, hygromycin B (PDB ID 3DF1–4; Borovinskaya et al, 2008); Tet, tetracycline (PDB ID 4G5K–N; Jenner et al, 2013); Ede, edeine (PDB ID 1I95; Pioletti et al, 2001); Ksg, kasugamycin (PDB ID 2HHH; Schluenzen et al, 2006); Pct, pactamycin (PDB ID 1HNX; Brodersen et al, 2000).
Noteworthy, rotated and unrotated states of the ribosomes are equally distributed within 100S from S. aureus, which suggests that the intermediate states could be thermodynamically unfavorable in the absence of any translation factors and tRNAs. But the question remains open as to whether the factor influences the transition between these two stages, or binds to the pre‐formed conformations. Nevertheless, this observation suggests that the mechanism of SaHPF binding to the ribosome is permissive enough that it should not be affected by ribosomal movements inherent to translation. In any case, it would be interesting to explore the relationship between translation arrest efficiency and the types of ribosome conformations—or translation stages—trapped within dimers, in particular by comparing the structures of dimers from various species.
Large rearrangements of the subunit interface are coupled with movements of the bridging protein B‐type bL31, which are similar to those described earlier (Jenner et al, 2010; Fischer et al, 2015). This suggests that B‐type bL31 is able to stabilize the two subunits in a similar manner to A‐type bL31, as well as to allow a plasticity for their mutual movements. This interplay could be crucial for the redistribution of ribosomes from hibernating to translationally competent states, upon release of SaHPF and dissociation of 100S ribosomes into free 30S and 50S subunits for canonical translation. Alternatively, vacant 70S particles could, for example, be involved in the translation of leaderless mRNAs, one of the beneficial mechanisms of survival under stress (Moll & Engelberg‐Kulka, 2012), because this process does neither require a dissociation into individual subunits, nor a complete pool of translation factors (Moll et al, 2004). Therefore, we hypothesize that the presence of two extreme states of subunit rotation within hibernating ribosomes is a hallmark of their ability to be rapidly recruited for translation.
In bacteria, ribosome hibernation does not only shut down translation, but it also provides protection against antibiotics. For example, E. coli cells deprived of their rmf gene were shown to become more sensitive to gentamicin (McKay & Portnoy, 2015). Our structure indicates that the NTD of SaHPF is in proximity to the aminoglycoside binding site within helix 44, therefore probably interfering with the flipping of the two adenines used in decoding (Appendix Fig S2). Furthermore, upon binding to the small subunit, the NTD of SaHPF occludes several antibiotic binding sites at the A site (hygromycin B, tetracycline), P site (edeine), and E site (pactamycin, kasugamycin; Fig 7B). Investigating the long‐term survival of S. aureus and other bacteria under antibiotic pressure could lead to advances in antibiotherapy.
Because dimerization happens through contacts between the CTDs, we may wonder about the mechanism for hibernation in S. aureus. Does SaHPF exist in a free form in the cell, which would be able to form dimers prior to binding to the ribosome? If that were the case, would the two SaHPF molecules need to dissociate so that ribosomes would dimerize? In S. aureus, a high expression level of the SaHPF‐coding gene during the exponential growth phase (Ruiz de los Mozos et al, 2013) does not necessarily coincide with a high ratio of 100S/70S ribosomes (Ueta et al, 2010). Hence, we could assume that a large pool of free SaHPF would be present in the exponentially growing cells.
This free SaHPF could be protected from unspecific binding to the ribosome by post‐translational modifications or by binding of additional factors. The high A260/280 ratio we observed in the fraction of SaHPF that was able to promote dimer formation could be in support of binding of nucleic acids to the NTD, such as an oligonucleotide or even a single nucleotide. The absence of nucleic acids in the first fraction could have led to the formation of an unproductive SaHPF.
There are still many open questions on the mechanism of SaHPF binding and dimerization. Recently, SaHPF was found in the 30S fraction during exponential growth of S. aureus (Basu & Yap, 2016). This observation reflects the ability of hibernation factors to trap the 30S particles after recycling and before entering initiation stages in vivo, albeit in that case with artificially elevated expression levels of SaHPF, which would bias the competition with tRNAs and translation factors for binding to the small subunit. Interestingly, upon approaching the stationary phase, the preferential mechanism of 100S formation could be different. Thus, upon its translation, SaHPF could immediately occupy the full 70S, which would be just rescued from stalling, for example, by tmRNA, ArfA, or ArfB (see for review Janssen & Hayes, 2012; Keiler, 2015), but not yet dissociated. Whichever sequence of events lead to hibernation in bacteria remains to be discovered, along with the mechanism for its regulation. Our structural analysis is corroborated by a recently published study from B. subtilis (Beckert et al, 2017) and represents one of the first milestones toward unraveling the mechanism for ribosome hibernation in S. aureus, which should help guide the development of more selective treatments against this pathogen.
Materials and Methods
70S ribosome purification
70S ribosomes from S. aureus were obtained as described previously (Khusainov et al, 2016b). In brief, two liters of S. aureus RN6390 culture was grown at 37°C (180 rpm) in brain–heart infusion broth (BHI) and harvested in early logarithmic phase (A600 = 1.0 AU ml−1). Cells were washed twice with 10 mM Tris–HCl pH 7.5 and pelleted at 4,750 × g, and the cell pellet was frozen at −80°C. A typical yield was 4.5–5.0 g of cells from 2 l of cell culture.
For 5 g of cells, the pellet was re‐suspended in 30 ml of buffer A (20 mM HEPES‐KOH pH 7.5, 100 mM NH4Cl, 21 mM Mg(OAc)2, 1 mM EDTA, 1 mM DTT), supplemented with the addition of protease inhibitor cocktail, DNase I (Roche), and of 3.5 mg lysostaphin (Sigma‐Aldrich), before being lysed at 37°C for 45 min. Cell debris was removed by centrifugation at 30,000 × g for 90 min.
The supernatant was supplemented with 2.8% w/v PEG 20,000 (Hampton Research) for the first fractionation. The supernatant was then recovered, and PEG 20,000 was increased to 4.2% w/v for the second fractionation. The solution was then centrifuged at 20,000 × g for 10 min, and the ribosome pellet was resuspended in 35 ml buffer A and layered on 25 ml of a sucrose cushion (10 mM HEPES‐KOH pH 7.5, 500 mM KCl, 25 mM Mg(OAc)2, 1.1 M sucrose, 0.5 mM EDTA, 1 mM DTT). Centrifugation was subsequently carried out at 158,420 × g for 15 h using a Beckman Type 45 Ti rotor.
The pellet containing ribosomes was resuspended in buffer E (10 mM HEPES‐KOH pH 7.5, 100 mM KCl, 10 mM Mg(OAc)2, 0.5 mM EDTA, 1 mM DTT) up to a concentration of 7 mg ml−1. 0.5 ml was loaded onto 7–30% sucrose‐density gradients and centrifuged at 38,694 × g for 15.5 h using a Beckman SW28 rotor. The fractions corresponding to 70S particles were pooled, the concentration of Mg(OAc)2 was adjusted to 25 mM, and PEG 20,000 was added to a final concentration of 4.5% w/v. Ribosomes were pelleted by centrifugation at 20,000 × g for 12 min, the pellet was gently dissolved in buffer G (10 mM HEPES‐KOH pH 7.5, 50 mM KCl, 10 mM NH4Cl, 10 mM Mg(OAc)2, 1 mM DTT) to a final concentration of 25–30 mg ml−1. Aliquots of 30 μl were flash‐frozen in liquid nitrogen and stored at −80°C. A typical yield was 10–12 mg of ribosomes from 5 g of cells.
Isolation and purification of full‐length SaHPF
Escherichia coli BL21 Star (DE3) cells (Invitrogen) were transformed using a modified pGS21A vector (GenScript) containing SaHPF (UniProt accession number D2Z097) followed by a His‐tag (comprising six histidines fused to the C‐terminus). Cells were grown in 6 l of Luria‐Bertani (LB) broth in the presence of 100 mg/l ampicillin to a cell density (A600) of 0.3. For large‐scale protein production, IPTG was added up to 0.5–1 mM, and induction proceeded for 4 h at 37°C. Cells were harvested and stored at −80°C until use.
The SaHPF protein was purified according to a protocol modified from Ayupov et al (2016) and Polikanov et al (2012). 1 g of cells was resuspended in 1.5 ml of buffer I (20 mM Tris–HCl pH 7.5 at 25°C, 100 mM NH4Cl, 10 mM MgCl2); this suspension was then lysed using a Constant Cell Disruption System (Constant Systems Limited). Cell lysate was then centrifuged at 25,000 × g for 30 min at 4°C following by high‐speed centrifugation 150,000 × g for 30 min at 4°C. The supernatant was applied to a pre‐packed nickel‐sepharose chromatography column (GE Healthcare) equilibrated in buffer I and containing 5 mM imidazole. The column was extensively washed with equilibration buffer, and the SaHPF protein was eluted using buffer I in the presence of 300 mM imidazole, pH 7.5. Next, size‐exclusion chromatography was performed using a Superdex 75 10/300 column (GE Healthcare) equilibrated in buffer II (10 mM HEPES‐KOH (pH 7.5), 50 mM KCl, 10 mM NH4Cl, and 10 mM MgCl2). Two peaks containing SaHPF were eluted, the first one corresponding to pure SaHPF eluted at a position of ~50 kDa according to the calibration graph (Fig EV1B and H), while the second contained extra absorbance at 260 nm and eluted near one column volume (Fig EV1B, F, and H), even though it also contains SaHPF in a dimeric form (Fig EV1C). The second peak, which was conducive to dimer formation, was frozen in liquid nitrogen and stored at −80°C until subsequent analysis.
Native PAGE of the fractions eluted from the gel filtration was performed using a modified Laemmli protocol (Laemmli, 1970). Running buffer (25 mM Tris, 0.192 M glycine), stacking buffer (4.5% acrylamide:bis‐acrylamide 29:1, Tris–HCl pH 6.8), and resolving buffer (15% acrylamide:bis‐acrylamide 29:1, 375 mM Tris–HCl pH 8.8) were SDS‐free. For sample preparation, each fraction was mixed with an equal volume of 2× loading buffer (120 mM Tris–HCl pH 7.5, 40% glycerol, 0.09% bromophenol blue, 20 mM DTT) at room temperature (~20°C). This sample was then loaded onto the gel, which ran for 60 min at 180 V. Proteins were stained overnight in 0.11% Coomassie brilliant blue made up in 25% ethanol and 10% acetic acid. Gels were diffusion‐destained by repeated washing in a solution containing 25% ethanol and 10% acetic acid.
Isolation and purification of CTD of SaHPF
The SaHPF‐CTD construct (containing a methionine, residues 130–190 of SaHPF, and a six‐histidine tag) was cloned into a modified pGS21A vector and expressed using E. coli Star (DE3) cells (Invitrogen) similarly to the full‐length SaHPF. Uniform labeling with 15N and 13C,15N was expressed in M9 minimal medium containing 15NH4Cl and [13C6]‐glucose as the sole sources of nitrogen and carbon, respectively. Cells were dissolved in the lysis buffer (20 mM Tris–HCl, 500 mM NH4Cl, pH 7.6) supplemented with the addition of 1× of protease inhibitor cocktail (according to the recommendations of the manufacturer; Roche), and of 60 U DNase I (Roche) for each gram of cells. Cells were lysed using a Constant Cell Disruption System (Constant Systems Limited) followed by affinity chromatography using Ni‐NTA agarose beads (Qiagen). The final step of size‐exclusion chromatography was performed using a Superdex 75 10/300 column (GE Healthcare), equilibrated in different aqueous buffers [phosphate‐buffered saline (PBS) containing 90% H2O + 10% D2O; Tris–HCl with various concentration of NH4Cl and pH range 6.0–7.6]. The protein eluted as one pure peak, and no additional absorbance at 260 nm was observed. Peak fractions were pooled, concentrated up to 2 mM using centrifugal Amicon Ultra 3,000 MWCO filters (Millipore), and used for NMR experiments without freezing.
NMR characterization of CTD
The NMR investigation was done using 2 mM samples of non‐labeled, 13C,15N‐labeled, and 15N‐labeled SaHPF‐CD in PBS buffer (pH 7.6) with 200 mM NH4Cl. Isotope‐edited (isotope‐separated) experiments allowed to detect 1H signals attached to 13C/15N nuclei and remove 12C/14N‐attached 1H signals for selective observation of interactions between 13C/15N isotope‐labeled and unlabeled molecules. NMR spectra were acquired at 35°C on a Bruker Avance 700 NMR spectrometer equipped with a cryoprobe. For direct observation of intermonomer NOEs from isotope filtering NMR experiments, a mixture of non‐labeled and 13C,15N‐labeled SaHPF‐CTD in PBS buffer was used at a 1:1 molar ratio.
NMR spectra were analyzed using CCPNMR (Vranken et al, 2005). The 1H, 13C, and 15N assignments were obtained from standard multi‐dimensional NMR methods (Sattler et al, 1999), CBCANH, CBCA(CO)NH, HNCA, HN(CO)CA, HN(CA)CO, HNCO, for main‐chain assignments, and C(CO)NH, H(CCO)NH (Clowes et al, 1993) for side‐chain assignments. Interproton distances were derived from 2D 1H‐1H NOESY for protein in 100% D2O, 3D 15N edited NOESY‐HSQC and 3D 13C edited NOESY‐HSQC. Backbone dihedral φ and ψ angles were derived from TALOS (Cornilescu et al, 1999). The intramonomer NOEs were obtained from isotope filtering 13C/15N – 12C/14N edited NOESY‐HSQC.
The program Xplor‐NIH was used for structural restraint collection (Schwieters et al, 2003). Individual structures were minimized, heated to 1,000 K for 6,000 steps, cooled in 100 K increments to 50 K, each with 3,000 steps, and finally minimized with 1,000 steps of the steepest descent, followed by 1,000 steps of conjugate gradient minimization. A total of 100 structures were calculated and 10 with minimal energy were chosen. None of the 10 structures had any violated nuclear Overhauser effect (NOE) distances. The lowest energy structure among the ensemble was used as representative in order to build a model for the CTD in the context of SaHPF bound to the 70S.
Formation and purification of S. aureus 100S dimers
For the in vitro reconstitution of S. aureus 100S ribosomes, 1.3 nmole of purified 70S ribosomal particles was mixed with 13 nmoles of purified SaHPF, and the ionic conditions were adjusted to those of buffer II, to a final volume of 1 ml. After 30 min of incubation at 37°C, the complex was layered onto 35 ml of 5–30% sucrose gradient with the same ionic condition as the reaction (10 mM HEPES‐KOH pH 7.5, 50 mM KCl, 10 mM NH4Cl, and 10 mM MgCl2). After 15.5 h of centrifugation at 17,100 rpm (38,694 × g) at 4°C using a Beckman SW28 rotor, the gradients were fractionated, while their profiles were recorded at 260 nm (A260). Fractions corresponding to 100S ribosomes were pooled, dialyzed against buffer G (10 mM HEPES‐KOH pH 7.5, 50 mM KCl, 10 mM NH4Cl, 10 mM Mg(OAc)2, 1 mM DTT) and concentrated using centrifugal Amicon Ultra 50,000 MWCO filters (Millipore). Next, the ribosome solution was layered onto 11 ml 5–30% sucrose gradient in buffer G. After 15 h of centrifugation at 27,783 × g at 4°C using a Beckman SW41 rotor, the gradients were fractionated while their A260 profiles were recorded. Fractions number 4 and 5 that correspond to the pure 100S peak (Fig 1B) were separately flash‐frozen in liquid nitrogen and stored at −80°C.
Stability analysis of 100S ribosome dimers from S. aureus
To analyze the stability of 100S ribosome dimers under high‐salt conditions, the sample was dialyzed in buffer II containing 500 mM KCl. Thereafter, it was layered onto the two distinct 5–30% sucrose gradients prepared in buffer II with either 50 mM KCl or 500 mM KCl. After 15 h of centrifugation at 15,000 rpm (27,783 × g) at 4°C using a Beckman SW41 rotor, the gradients were fractionated while their A260 profiles were recorded.
Analytical ultracentrifugation
Experiments were conducted at 4°C using a Beckman Coulter Proteome Lab XL‐I analytical ultracentrifuge, with the eight‐hole Beckman An‐50Ti rotor. The sample of 100S dimers was thawed, diluted 10 times with buffer II to decrease the concentration of sucrose, and concentrated to 1.0 A260U ml−1 using centrifugal Amicon Ultra 50,000 MWCO filters. The sample (400 μl) was loaded into one of the two quartz cuvettes of the centrifuge tube. The reference cuvette was filled with 410 μl of buffer G. Sedimentation at 20,644 × g was monitored by measuring the A260 and A280 values, with scans taken every 4 min. The density and viscosity of buffer II were calculated using the Sednterp software (Laue et al, 1992), taking into account the presence of ~3% of sucrose in solution. Data were analyzed using a c(s) model in SEDFIT (Schuck, 2000).
Electron microscopy of disomes
Grid preparation
4 μl of pure 100S [~0.075 mg ml−1 concentration (20 nM)] was applied to 400‐mesh carbon‐coated holey carbon Quantifoil 2/2 grids (Quantifoil Micro Tools; glow discharge time = 20 s), blotted with filter paper from both sides for 1.5 s in a temperature‐ and humidity‐controlled Vitrobot apparatus Mark IV (FEI, Eindhoven, the Netherlands, T = 4°C, humidity 100%, blot force 5, blot waiting time 30 s), and vitrified in liquid ethane pre‐cooled by liquid nitrogen.
Image acquisition
Data were collected on the spherical aberration (Cs)‐corrected Titan Krios S‐FEG instrument (FEI, Eindhoven, the Netherlands) operating at 300 kV acceleration voltage, at a nominal underfocus of Δz = −0.6 to −4.5 μm using the second‐generation back‐thinned direct electron detector CMOS (Falcon II) 4,096 × 4,096 camera and automated data collection with EPU software (FEI, Eindhoven, the Netherlands). The Falcon II camera was calibrated at a nominal magnification of 59,000×. The calibrated magnification on the 14 μm pixel camera was 127,272×, resulting in 1.1 Å pixel size at the specimen level. Frames 2–8 out of 17 possible were collected and used for image processing. Total exposure was 1 s with a dose of 60 ē/Å2 (or 3.5 ē/Å2 per frame).
Image processing
A framework for image processing employing several software packages (de la Rosa‐Trevin et al, 2016) was used to obtain the 3D reconstruction of the S. aureus 100S. Before particle picking, seven frames in the stack were aligned using the Optical Flow algorithm integrated in Xmipp3 (de la Rosa‐Trevin et al, 2013). Then, an average image of the whole stack was used to determine the contrast transfer function by CTFFIND4 (Rohou & Grigorieff, 2015) and to select particles semi‐automatically in SCIPION (Abrishami et al, 2013). Particle picking and extraction were performed using Xmipp3, focusing either on the full 100S particles or on each 70S ribosome of a dimer. Particle sorting was done first by 2D classification using RELION, followed by extensive 3D classification using RELION (Scheres, 2012). The resulting two major classes were as follows: “tight” or “loose” disomes for particles extracted with large box (640 × 640 × 640 Å), and unrotated or rotated monosomes for particles picked with small box size (340 × 340 × 340 Å). To clarify whether disomes were composed of two unrotated or two rotated ribosomes, or a combination of both, we performed the refinement by focusing on one ribosome within the dimer, followed by additional 3D classification with large shift increments focused on the same ribosome. This resulted in equal distribution of unrotated and rotated ribosomes within the disomes (Fig EV2). All classes were refined using RELION's 3D autorefine function, and the final refined classes were then post‐processed using the procedure implemented in RELION, which applies appropriate masking, B‐factor sharpening, and resolution validation to avoid over‐fitting (Scheres, 2012). The average resolution was ~3.7 Å for both rotated and unrotated monosomes and ~9–11 Å for “loose” and “tight” disomes, respectively (Fig EV2). The resolution of the other classes ranged from 10 to 15 Å (Fig EV2). Determination of the local resolution of the final density maps for tight and loose disomes, as well as unrotated and rotated ribosomes, was performed using ResMap (Kucukelbir et al, 2014).
Map fitting and model refinement
SaHPF was initially modeled by the Swiss‐Model webserver (Biasini et al, 2014), using the following structures as templates: E. coli YfiA (PDB ID 1N3G, 4Y4O) for the N‐terminal domain (Rak et al, 2002; Polikanov et al, 2015), and Clostridium acetobutylicum ribosome‐associated protein Y (PDB ID 3KA5; Northeast Structural Genomics Consortium target ID: CaR123A, http://sbkb.org/pdbid/3ka5), as well as Listeria monocytogenes Lmo2511 protein (PBD ID 3K2T; Northeast Structural Genomics Consortium target ID: LkR84A, http://sbkb.org/pdbid/3k2t) for the C‐terminal domain. The atomic model of SaHPF‐NTD was traced from Phe4 until Arg95 and fitted it its density in the 70S‐SaHPF structure.
Real‐space refinement in Phenix (Adams et al, 2010; Afonine et al, 2013) was performed using our vacant 70S structure (Khusainov et al, 2016b) as a template. Mobile ribosomal elements, such as the L1‐stalk, the L7/L12‐stalk, and 23S rRNA helix 69, as well as SaHPF‐NTD, were set up as rigid bodies for the first cycle of refinement. Following eight steps of real‐space refinement included simulated annealing (starting temperature = 800 K) and global minimization taking into account RNA and protein secondary structure restraints (search_method = from_ca). RNA geometry and fit in density were improved by running Erraser within Phenix (Chou et al, 2013; Jain et al, 2015), for rRNA fragments of ~990 nt that overlapped by 2–4 nt. Model and map were inspected and adjusted in Coot (Emsley & Cowtan, 2004), using tools for real‐space refinement, geometry regularization, and morphing. Final minimization of coordinates was carried out in Phenix (global minimization with hydrogen atoms), with secondary structure restraints (search_method = cablam). For model validation, we used the MolProbity webserver (Chen et al, 2010) and model‐to‐map correlation statistics from Phenix.
Structural analysis
Vectors between the backbone atoms of the unrotated and rotated LSU were calculated and rendered in Pymol using the modevectors.py script (author: Sean M. Law; available from https://pymolwiki.org/index.php/Modevectors). Root‐mean‐square deviations (RMSD, in Å) between the unrotated and rotated SSU were calculated in Pymol v. 1.8.0.5 (Schrödinger) using the colorbyrmsd.py script (authors: Shivender Shandilya, Jason Vertrees, Thomas Holder; available from https://pymolwiki.org/index.php/ColorByRMSD). Superimpositions of SaHPF‐bound ribosomes and ribosomes bound to tRNAs or antibiotics were performed in Chimera (Pettersen et al, 2004).
Accession numbers
Coordinates of the three‐dimensional structure and cryo‐EM map were deposited to the EMDB and PDB with accession numbers: EMDB ID: EMD‐3638 (tight 100S); EMD‐3639 (loose 100S); EMD‐3624, PDB ID 5ND8 (hibernating ribosome in unrotated state); EMD‐3625, PDB ID 5ND9 (hibernating ribosome in rotated state); PDB ID 5NKO (CTD‐SaHPF), BMRB ID 34120 (CTD‐SaHPF). Additionally, PDB files of in silico reconstituted tight 100S disomes in rotated and unrotated states are provided as Datasets EV1 and EV2, respectively.
Supplementary datasets
Atomic coordinates of reconstructed 100S disomes are deposited as four pdb files of 70S ribosomes. They are combined into two datasets representing the 100S tight disome in its rotated state (Dataset EV1A and B), and the 100S tight disome in its unrotated state (Dataset EV2A and B).
Author contributions
IK and RA expressed the SaHPF and the CTD cloned by SV, purified 70S ribosomes, and purified and characterized reconstituted 100S dimers under the supervision of GY and MY; IK refined the structures under the supervision of QV and YH; AM and AS operated the microscope and collected the data; YH processed the data; KU performed the NMR analysis under the supervision of BK; IK, QV, and YH wrote the manuscript with input from MY; all authors revised the manuscript; research was supervised by MY and YH.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
We wish to thank the High Performance Computing Center of the University of Strasbourg (funded by the Equipex Equip@Meso project) for information technology support, the IGBMC electron microscope facility (supported by the Alsace Region, FRM, IBiSA Program, INSERM, CNRS, ARC, and the FRISBI ANR‐10‐INSB‐05‐01); Stefano Marzi and Pascale Romby for useful discussions and for careful reading of the manuscript; Yury Polikanov for kindly sharing the plasmid and cells for expression of full‐length SaHPF; and Eric Westhof for ongoing support and helpful discussions. This work was supported by the “Centre National de la Recherche Scientifique” (CNRS) and the “Agence Nationale de la Recherche” as part of the “Investissements d'Avenir” program (LabEx: ANR‐10‐LABX‐0036_NETRNA and ANR‐14‐ACHN‐0024 CryoEM80S to Y.H.; ANR‐15‐CE11‐0021‐01 to G.Y.); the “Fondation pour la Recherche Médicale en France” (FDT20140930867 to I.K.); the European Research Council advanced grant (294312 to M.Y.); and the Russian Science Foundation (Project No. 16‐14‐10014 to I.K., K.U., S.V., R.A., M.Y.).
The EMBO Journal (2017) 36: 2073–2087
See also: B Beckert et al (July 2017) and RL Gonzalez Jr (July 2017)
Contributor Information
Marat Yusupov, Email: marat@igbmc.fr.
Yaser Hashem, Email: y.hashem@ibmc-cnrs.unistra.fr.
References
- Abrishami V, Zaldivar‐Peraza A, de la Rosa‐Trevin JM, Vargas J, Oton J, Marabini R, Shkolnisky Y, Carazo JM, Sorzano CO (2013) A pattern matching approach to the automatic selection of particles from low‐contrast electron micrographs. Bioinformatics 29: 2460–2468 [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P, Headd J, Terwilliger T, Adams P (2013) New tool: phenix. real_space_refine. Comput Crystallogr Newsl 4: 43–44 [Google Scholar]
- Akanuma G, Kazo Y, Tagami K, Hiraoka H, Yano K, Suzuki S, Hanai R, Nanamiya H, Kato‐Yamada Y, Kawamura F (2016) Ribosome dimerization is essential for the efficient regrowth of Bacillus subtilis . Microbiology 162: 448–458 [DOI] [PubMed] [Google Scholar]
- Ayupov RK, Khusainov I, Validov SZ, Yusupova GZ, Yusupov MM (2016) Isolation and purification of Staphylococcus aureus hibernation promoting factor inactivating of the ribosome. Int J Pharm Technol 8: 14392–14398 [Google Scholar]
- Basu A, Yap MN (2016) Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic Acids Res 44: 4881–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckert B, Abdelshahid M, Schäfer H, Steinchen W, Arenz S, Berninghausen O, Beckmann R, Bange G, Turgay K, Wilson DN (2017) Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J 36: 2061–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berjanskii MV, Wishart DS (2005) A simple method to predict protein flexibility using secondary chemical shifts. J Am Chem Soc 127: 14970–14971 [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T (2014) SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42: W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri P, Leibundgut M, Saurer M, Boehringer D, Ban N (2017) The complete structure of the chloroplast 70S ribosome in complex with translation factor pY. EMBO J 36: 475–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovinskaya MA, Shoji S, Fredrick K, Cate JH (2008) Structural basis for hygromycin B inhibition of protein biosynthesis. RNA 14: 1590–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen DE, Clemons WM Jr, Carter AP, Morgan‐Warren RJ, Wimberly BT, Ramakrishnan V (2000) The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103: 1143–1154 [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R (2013) Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10: 74–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowes RT, Boucher W, Hardman CH, Domaille PJ, Laue ED (1993) A 4dDHCC(CO)NNH experiment for the correlation of aliphatic side‐chain and backbone resonances in 13C/15N labeled proteins. J Biomol NMR 3: 349–354 [Google Scholar]
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H (2015) Structure of the E. coli ribosome‐EF‐Tu complex at <3 A resolution by Cs‐corrected cryo‐EM. Nature 520: 567–570 [DOI] [PubMed] [Google Scholar]
- Jain S, Richardson DC, Richardson JS (2015) Computational methods for RNA structure validation and improvement. Methods Enzymol 558: 181–212 [DOI] [PubMed] [Google Scholar]
- Janssen BD, Hayes CS (2012) The tmRNA ribosome‐rescue system. Adv Protein Chem Struct Biol 86: 151–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Demeshkina N, Yusupova G, Yusupov M (2010) Structural rearrangements of the ribosome at the tRNA proofreading step. Nat Struct Mol Biol 17: 1072–1078 [DOI] [PubMed] [Google Scholar]
- Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, Yusupova G (2013) Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Natl Acad Sci USA 110: 3812–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Yoshida H, Miyata T, Maki Y, Wada A, Namba K (2010) Structure of the 100S ribosome in the hibernation stage revealed by electron cryomicroscopy. Structure 18: 719–724 [DOI] [PubMed] [Google Scholar]
- Keiler KC (2015) Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol 13: 285–297 [DOI] [PubMed] [Google Scholar]
- Khusainov I, Marenna A, Cerciat M, Fechter P, Hashem Y, Marzi S, Romby P, Yusupova G, Yusupov M (2016a) A glimpse on Staphylococcus aureus translation machinery and its control. Mol Biol (Mosk) 50: 549–557 [DOI] [PubMed] [Google Scholar]
- Khusainov I, Vicens Q, Bochler A, Grosse F, Myasnikov A, Menetret JF, Chicher J, Marzi S, Romby P, Yusupova G, Yusupov M, Hashem Y (2016b) Structure of the 70S ribosome from human pathogen Staphylococcus aureus . Nucleic Acids Res 44: 10491–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD (2014) Quantifying the local resolution of cryo‐EM density maps. Nat Methods 11: 63–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL (1992) Computer‐aided interpretation of sedimentation data for proteins In Analytical ultracentrifugation in biochemistry and polymer science, Harding SE, Rowe AJ, Horton JC. (eds), pp 90–125. Cambridge: Royal Society of Chemistry; [Google Scholar]
- McKay SL, Portnoy DA (2015) Ribosome hibernation facilitates tolerance of stationary‐phase bacteria to aminoglycosides. Antimicrob Agents Chemother 59: 6992–6999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll I, Hirokawa G, Kiel MC, Kaji A, Blasi U (2004) Translation initiation with 70S ribosomes: an alternative pathway for leaderless mRNAs. Nucleic Acids Res 32: 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll I, Engelberg‐Kulka H (2012) Selective translation during stress in Escherichia coli . Trends Biochem Sci 37: 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz JO, Brandt F, Matias VR, Sennels L, Rappsilber J, Scheres SH, Eibauer M, Hartl FU, Baumeister W (2010) Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ . J Cell Biol 190: 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F (2001) Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J 20: 1829–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Blaha GM, Steitz TA (2012) How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336: 915–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Melnikov SV, Söll D, Steitz TA (2015) Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat Struct Mol Biol 22: 342–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak A, Kalinin A, Shcherbakov D, Bayer P (2002) Solution structure of the ribosome‐associated cold shock response protein Yfia of Escherichia coli . Biochem Biophys Res Commun 299: 710–714 [DOI] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa‐Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, Sorzano CO (2013) Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol 184: 321–328 [DOI] [PubMed] [Google Scholar]
- de la Rosa‐Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez‐Blanco J, Burguet‐Castell J, Cuenca‐Alba J, Abrishami V, Vargas J, Oton J, Sharov G, Vilas JL, Navas J, Conesa P, Kazemi M, Marabini R, Sorzano CO, Carazo JM (2016) Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195: 93–99 [DOI] [PubMed] [Google Scholar]
- Rozov A, Demeshkina N, Khusainov I, Westhof E, Yusupov M, Yusupova G (2016) Novel base‐pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat Commun 7: 10457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de los Mozos I, Vergara‐Irigaray M, Segura V, Villanueva M, Bitarte N, Saramago M, Domingues S, Arraiano CM, Fechter P, Romby P, Valle J, Solano C, Lasa I, Toledo‐Arana A (2013) Base pairing interaction between 5′‐ and 3′‐UTRs controls icaR mRNA translation in Staphylococcus aureus . PLoS Genet 9: e1004001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JB, Cook GM (1995) Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol Rev 59: 48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Watanabe T, Maki Y, Ueta M, Yoshida H, Ito Y, Wada A, Mishima M (2009) Solution structure of the E. coli ribosome hibernation promoting factor HPF: implications for the relationship between structure and function. Biochem Biophys Res Commun 389: 580–585 [DOI] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc 34: 93–158 [Google Scholar]
- Scheres SH (2012) RELION: implementation of a Bayesian approach to cryo‐EM structure determination. J Struct Biol 180: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluenzen F, Takemoto C, Wilson DN, Kaminishi T, Harms JM, Hanawa‐Suetsugu K, Szaflarski W, Kawazoe M, Shirouzu M, Nierhaus KH, Yokoyama S, Fucini P (2006) The antibiotic kasugamycin mimics mRNA nucleotides to destabilize tRNA binding and inhibit canonical translation initiation. Nat Struct Mol Biol 13: 871–878 [DOI] [PubMed] [Google Scholar]
- Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor‐NIH NMR molecular structure determination package. J Magn Reson 160: 65–73 [DOI] [PubMed] [Google Scholar]
- Sharma MR, Donhofer A, Barat C, Marquez V, Datta PP, Fucini P, Wilson DN, Agrawal RK (2010) PSRP1 is not a ribosomal protein, but a ribosome‐binding factor that is recycled by the ribosome‐recycling factor (RRF) and elongation factor G (EF‐G). J Biol Chem 285: 4006–4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohmen D, Chiba S, Shimokawa‐Chiba N, Innis CA, Berninghausen O, Beckmann R, Ito K, Wilson DN (2015) Structure of the Bacillus subtilis 70S ribosome reveals the basis for species‐specific stalling. Nat Commun 6: 6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaflarski W, Nierhaus KH (2007) Question 7: optimized energy consumption for protein synthesis. Orig Life Evol Biosph 37: 423–428 [DOI] [PubMed] [Google Scholar]
- Tagami K, Nanamiya H, Kazo Y, Maehashi M, Suzuki S, Namba E, Hoshiya M, Hanai R, Tozawa Y, Morimoto T, Ogasawara N, Kageyama Y, Ara K, Ozaki K, Yoshida M, Kuroiwa H, Kuroiwa T, Ohashi Y, Kawamura F (2012) Expression of a small (p)ppGpp synthetase, YwaC, in the (p)ppGpp(0) mutant of Bacillus subtilis triggers YvyD‐dependent dimerization of ribosome. Microbiologyopen 1: 115–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta M, Yoshida H, Wada C, Baba T, Mori H, Wada A (2005) Ribosome binding proteins YhbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli . Genes Cells 10: 1103–1112 [DOI] [PubMed] [Google Scholar]
- Ueta M, Ohniwa RL, Yoshida H, Maki Y, Wada C, Wada A (2008) Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli . J Biochem 143: 425–433 [DOI] [PubMed] [Google Scholar]
- Ueta M, Wada C, Wada A (2010) Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells 15: 43–58 [DOI] [PubMed] [Google Scholar]
- Ueta M, Wada C, Daifuku T, Sako Y, Bessho Y, Kitamura A, Ohniwa RL, Morikawa K, Yoshida H, Kato T, Miyata T, Namba K, Wada A (2013) Conservation of two distinct types of 100S ribosome in bacteria. Genes Cells 18: 554–574 [DOI] [PubMed] [Google Scholar]
- Vila‐Sanjurjo A, Schuwirth BS, Hau CW, Cate JH (2004) Structural basis for the control of translation initiation during stress. Nat Struct Mol Biol 11: 1054–1059 [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Wada A (2014) The 100S ribosome: ribosomal hibernation induced by stress. Wiley Interdiscip Rev RNA 5: 723–732 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Review Process File