

Abstract
Aims
A population pharmacokinetic (PK) model was developed for cediranib to simulate cediranib exposure for different doses, including comedication with strong uridine glucuronosyl transferase/P‐glycoprotein inducers such as rifampicin, in cancer patients.
Methods
Plasma concentrations and covariates from 625 cancer patients after single or multiple oral cediranib administrations ranging from 0.5 to 90 mg in 19 Phase I and II studies were included in the analysis. Stepwise covariate modelling was used to develop the population PK model. The final model was used to simulate cediranib exposure in cancer patients to evaluate cediranib target coverage and the need for dose adjustment for covariates or coadministration with rifampicin.
Results
A two‐compartment model with sequential zero‐ and first‐order absorption and first‐order elimination adequately described the cediranib concentration–time courses. Body weight and age were identified as having statistically significant impact on cediranib PK, but only <21% impact on AUC and maximum concentrations. Simulated lower bounds of 90% prediction interval or median of unbound cediranib concentrations after cediranib 15 or 20 mg exceeded the IC50 for vascular endothelial growth factor receptors‐1, ‐2 and ‐3. Exposures of cediranib 20 or 30 mg with coadministration of rifampicin were comparable to those of 15 or 20 mg, respectively, without coadministration.
Conclusions
No covariate was identified to require dose adjustment for cediranib. Cediranib exposure following 15 or 20 mg daily dose administration is adequate overall for inhibition of in vitro estimated vascular endothelial growth factor receptor‐1, ‐2 and ‐3 activities. An increase in cediranib dose may be needed for cediranib coadministered with strong uridine glucuronosyl transferase/P‐glycoprotein inducers such as rifampicin.
Keywords: AZD2171, cediranib, exposure prediction, pharmacometrics, population pharmacokinetics, vascular endothelial growth factor receptor inhibitors
What is Already Known about this Subject
Cediranib is a potent small molecule targeting vascular endothelial growth factor receptors (‐1, ‐2, ‐3) and has linear pharmacokinetics suitable for once‐daily (qd) oral dose administration.
Cediranib 20 mg qd tablet coadministered with platinum‐based chemotherapy followed by cediranib 20 mg qd in patients with ovarian cancer significantly increased progression‐free survival compared with placebo plus platinum‐based chemotherapy.
What this Study Adds
A population pharmacokinetic model for cediranib was developed to characterize cediranib pharmacokinetics in cancer patients.
Model‐predicted unbound cediranib exposure after 15 or 20 mg qd dosing was above the in vitro IC50 for vascular endothelial growth factor receptors‐1, ‐2 and ‐3 overall, supporting these doses in cancer patients.
Table of Links
| TARGETS |
|---|
| Enzymes 2 |
| VEGFR‐1 http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2534 |
| VEGFR‐2 |
| VEGFR‐3 |
This Table lists key protein targets in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Ovarian cancer is the fifth most common cause of cancer death in women in the USA and the leading cause of death from gynaecological cancers in the UK 3, 4. The current standard of care for treating platinum‐sensitive relapsed (PSR) ovarian cancer is platinum‐based doublet chemotherapy. Recently, angiogenesis has been validated as a therapeutic target in advanced ovarian cancer patients through the use of the monoclonal anti‐vascular endothelial growth factor (VEGF) antibody, bevacizumab 5. However, although bevacizumab has shown activity when combined with chemotherapy, the current indication does not support use in patients who have previously received this agent, or another VEGF inhibitor, in a first‐line setting. Moreover, an increase in the use of alternative VEGF ligands has been reported in patients progressing on chemotherapy plus bevacizumab, suggesting that more comprehensive inhibition of VEGF signalling may be appropriate at that point 6. Therefore, there remains a need for effective treatment options with an alternative anti‐angiogenic treatment for patients at their first platinum‐sensitive relapse.
Cediranib is a potent, orally administered, once‐daily (qd) small‐molecule tyrosine kinase inhibitor of VEGF signalling and angiogenesis that targets all three VEGF tyrosine kinase receptors (VEGFR‐1, ‐2, ‐3); it also exhibits additional activity against stem cell factor receptor (c‐kit)‐dependent tumour growth 7, 8, 9, 10, 11, 12, 13.
Cediranib is under development as a starting tablet dose of 20 mg in combination with platinum‐based chemotherapy, followed by cediranib maintenance monotherapy (20 mg qd), for the treatment of PSR ovarian cancer. A 15‐mg tablet is also used for patients who cannot tolerate the 20‐mg qd dose 14, 15, 16. In a randomized, double‐blind, placebo‐controlled, Phase III trial (ICON 6), cediranib was compared with placebo in combination with carboplatin and paclitaxel in PSR ovarian cancer patients. The results revealed superiority of the combination of carboplatin and paclitaxel with cediranib, followed by cediranib as maintenance therapy, with improved progression‐free survival (11.4 vs. 8.7 months; hazard ratio [HR] 0.56; P < 0.0001) compared with placebo control 16.
Intensive or sparse plasma concentrations were collected in 19 previous Phase I or II studies in patients with various types of cancer. Noncompartmental analyses to assess the pharmacokinetics (PK) of cediranib were conducted in individual studies with intensive plasma sampling 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. Cediranib is administered in tablets as cediranib maleate and can be classified as a Biopharmaceutical Classification System 3 compound exhibiting high solubility and low permeability. Cediranib demonstrated linear PK with doses ranging from 0.5 to 60 mg. Cediranib was absorbed moderately slowly, with peak plasma concentrations observed typically within 1–5 h postdose. Plasma protein binding in human plasma for cediranib is ~95%, with mean apparent volume of distribution at steady state ranging from 429 to 1290 l. Cediranib is metabolized via flavin‐containing monooxygenase 1 and 3 (FMO1, FMO3) and uridine glucuronosyl transferase (UGT)1A4. Cediranib and its metabolites are mainly excreted in faeces (59%), with <1% of unchanged drug excreted in the urine. The mean apparent oral clearance of cediranib and its terminal half‐life were 28.2 l h–1 and 22 h, respectively. Cediranib is a substrate of P‐glycoprotein (Pgp). Coadministration with ketoconazole, a potent Pgp inhibitor, increased the cediranib area under the concentration–time curve at steady state (AUCss) in patients by 21%, while coadministration with rifampicin, a potent inducer of Pgp, decreased cediranib AUCss by 39%. However, a systemic assessment of cediranib PK in cancer patients, including ovarian cancer patients, and the potential covariate effect on cediranib PK is lacking. Hence, the objectives of the present analysis are to develop a population PK (PPK) model that could describe the cediranib concentration–time courses in cancer patients, to explore the impact of covariates on relevant PK parameters, to compare the PK/exposure in patients with ovarian cancer and other cancers, to evaluate the influence of coadministration of antihypertensive drugs on PK/exposure of cediranib, to evaluate the target engagement/coverage, and to investigate the dose adjustment when coadministered with strong UGT/Pgp inducers such as rifampicin.
Methods
Data
Clinical study and plasma sampling
Cediranib plasma concentrations measured in patients in 19 Phase I and II studies were included in the analysis, with cediranib doses ranging from 0.5 to 90 mg and a majority of patients treated with 20‐, 30‐ or 45‐mg doses. Of the 19 studies, 14 involved cediranib as monotherapy and five involved cediranib in combination with other chemotherapies. Most studies had a large (e.g., predose, 1, 2, 3, 4, 6, 8, 12, 24 h postdose) or moderate (e.g., predose, 1, 2, 4, 6, 8, 24 h postdose) number of cediranib plasma concentration samples 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. All patients provided written informed consent, and all trials were approved by the respective independent ethics committees and were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32.
Covariate data
Covariates of sex (male or female), age, race (Caucasian, black, Asian or other), body weight (WT), alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, total bilirubin, creatinine clearance, albumin, and platinum‐containing chemotherapy were evaluated for their potential impact on cediranib PK parameters.
The final data for the analysis included a total of 7011 observations of cediranib concentrations from 625 patients with various types of cancer.
Model development
A base model was first developed, including random effects, interindividual variability (IIV), interoccasion variability (IOV) and residual error, and was used for subsequent covariate model development.
Base model
A one‐compartment model with first‐order absorption and first‐order elimination was considered as an initial structural model to describe cediranib concentration–time courses. Subsequently, more complex structural models were examined, including a two‐compartment model with sequential zero‐ and first‐order absorptions. The IIV of the typical PK parameters was modelled to be log‐normally distributed. IOV was also evaluated for occasions with intensive cediranib plasma concentrations. Owing to a wide cediranib concentration range for the 5–90 mg doses, concentration data were log‐transformed, and the residual variability was primarily modelled using an additive error on the log scale (proportional on untransformed scale; referred to as ‘proportional error’ hereafter).
Covariate model
Evaluation of the impact of intrinsic and extrinsic subject covariates on cediranib PK parameters were prespecified for the apparent clearance (Cl/F) and apparent volume of distribution of the central compartment (Vc/F), based on the clinical pharmacology profiling of cediranib. Covariates of WT, age, sex, race and platinum‐containing chemotherapy were evaluated for both Cl/F and Vc/F, while covariates of aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, total bilirubin, albumin and creatinine clearance were only evaluated for Cl/F. Prior to covariate modelling, correlation between covariates was evaluated; only one of the covariates was included in the covariate evaluation for those highly correlated covariates. The relationship between continuous covariates and typical values of PK parameters (θTV) were modelled by power models:
where θREF is the typical cediranib PK parameter at the median value (XMedian) of the continuous covariate Xi and θx is the fixed covariate effect of the power relationship to the normalized covariate (Xi/XMedian). The relationships between categorical covariates (Xi) and the typical value of PK parameters (θTV) were modelled as:
where θREF is the typical cediranib PK parameter at the reference value (one of the categories of categorical covariates) and θx is the fixed effect for the proportional covariate model of the categorical covariate Xi.
Covariate search was implemented by a forward‐inclusion and backward‐elimination procedure through the stepwise covariate model building tool in Perl‐speaks‐NONMEM 33. The level of significance was P < 0.01 for the forward inclusion of covariates. For retention of a covariate during backward elimination, P < 0.001 was used. Reductions in IIV and/or residual variability were also considered. The model including covariates after the backward‐elimination step was called the final full covariate model.
A summary table briefly describing the studies included in the analysis, information for bioanalysis, inclusion/exclusion of data, handling of missing data, model evaluation, and software for computation is included in the supplementary material.
Model application
Impact of covariate effect on cediranib exposure
To assess the clinical relevance of the estimated covariate effects on cediranib exposure in terms of maximum concentration (Cmax) and AUC, simulations were performed with the distribution of covariates from the dataset used for the development of the final PPK model, and with uncertainty of parameter estimates from refit to the bootstrapping datasets of the final PPK model dataset. A total of 1000 simulations were performed; for each simulated individual dataset, the ratios at the 5th and 95th percentiles of the covariate distribution to the median value for Cmax and AUC were calculated. Subsequently, the medians, as well as the 5th and 95th percentiles, of the simulated ratios were calculated and plotted as a forest plot to evaluate the covariate effects on cediranib exposure 34.
Exposure comparison among different subject populations
The empirical Bayesian estimates for the 625 subjects in the final NONMEM dataset were used to predict cediranib Cmax at steady state (Cmax,ss), minimum concentration at steady state (Cmin,ss), and AUCss following repeated daily dose administration of the same 20 mg dose of cediranib. The exposures for Cmax,ss, Cmin,ss and AUCss were compared between patients with ovarian cancer and those with other cancers, and between patients with and without antihypertensive medication during cediranib treatment, all using box‐and‐whisker plots.
Simulations of cediranib plasma profiles and target coverage
Simulation was performed to describe plasma concentration–time profiles during 8 days of cediranib 15 and 20 mg dose administration with dense sampling. Two thousand individuals with covariate combinations were re‐sampled from the dataset that was used for the PPK modelling. The simulated median and 90% confidence interval (CI) for free cediranib concentrations with a free fraction of 5% every 30 min was constructed and compared with the in vitro target cediranib plasma concentrations required to obtain 50% inhibition (IC50) of VEGFR‐1, ‐2 and ‐3 in a range of cell assays 7.
Simulations of cediranib exposure in the presence of a strong CYP3A4 inducer
Simulations were also conducted to evaluate whether an increased dose would be needed to compensate for the average effect of a strong inducer of cytochrome P450 3A4 (CYP3A4), UGT and Pgp transporter, rifampicin, on cediranib exposure. Two tentative cediranib doses of 20 and 30 mg with coadministration of rifampicin 600 mg qd were selected for the evaluation. A total of 2000 individuals were simulated from the dataset used for the PPK modelling. In a previous cediranib and rifampicin drug–drug interaction study, the geometric mean ratio of steady‐state AUC of cediranib with coadministration of rifampicin (600 mg/day for 7 days to steady‐state) to without coadministration of rifampicin was 0.61 (0.57–0.66) 23. Given the narrow range of the ratio and that cediranib exposure at steady‐state induction of rifampicin is of pertinent clinical interest, the effect on steady‐state cediranib exposure with coadministration of rifampicin was simulated by including a factor of increasing mean cediranib Cl/F of 1.64 (1/0.61). Comparisons of cediranib exposure in Cmax,ss and AUCss for cediranib 15 and 20 mg administered alone vs. cediranib 20 and 30 mg coadministered with rifampicin 600 mg qd, respectively, were plotted as box‐and‐whisker plots with an overlay of the predicted exposures.
Results
Data
The final dataset consisted of 625 patients and 7011 cediranib plasma samples. The majority of patients were initially administered cediranib doses of 20 mg (24%), 30 mg (31%), or 45 mg (42%). Accordingly, the majority of the 7011 plasma samples were from patients who received cediranib 20 mg (20%), 30 mg (35%) or 45 mg (34%). A summary of continuous and categorical covariates is shown in Table 1. Given the limited number of African American patients and patients with race labelled as Other in the dataset, these were pooled with Caucasian patients in the covariate analysis, resulting in only two categories for race, namely, Asian and non‐Asian patients.
Table 1.
Summary of baseline demographic and laboratory covariates
| Continuous covariate | Median (min, max) |
|---|---|
| Body weight, kg | 73 (35, 150) |
| Age, years | 59 (19, 89) |
| Aspartate aminotransferase, IU l –1 | 26 (6, 284) |
| Alanine aminotransferase, IU l –1 | 23 (4, 356) |
| Alkaline phosphatase, IU l –1 | 107 (32, 2638) |
| Total bilirubin, μmol l –1 | 8.6 (1, 62) |
| Albumin, g l –1 | 38 (18, 51) |
| Creatinine clearance, ml min –1 | 91.9 (28.9, 273.2) |
| Categorical | n (%) |
|---|---|
| Sex | |
| Male | 362 (58) |
| Female | 263 (42) |
| Race | |
| Caucasian | 532 (85) |
| Asian | 79 (13) |
| African American | 12 (92) |
| Other | 2 (0.2) |
| Platinum‐containing chemotherapy | |
| Yes | 42 (7) |
| No | 587 (93) |
Model development
Base model
The final base model was a two‐compartment disposition model with sequential zero‐ and first‐order absorption characterized by the relative bioavailability (F1), duration of zero‐order absorption (D1), first‐order absorption rate constant (Ka), Cl/F, Vc/F, apparent inter‐compartmental clearance (Q/F), and apparent volume of distribution of the peripheral compartment (Vp/F). The IIVs were included for Cl/F, Vc/F and Ka, and an IOV for F1 was included between intensive plasma sampling occasions, with the sparse plasma sampling occasion being fixed to the reference value (F = 1). Attempts to include IIV in more parameters (D1, Vp/F and Q/F) resulted in unstable models, suggesting that the data did not support estimation of more IIV parameters. A single additive residual error model at the log‐transformed cediranib concentration (proportional error when back‐transformed to normal scale) with separate residual errors for rich and sparse sampling occasions was supported by the data.
Covariate model
No strong correlations were observed among all covariates (r ≤ 0.34); therefore, all covariates were evaluated in the PPK analysis. The final covariate model included WT impact on Cl/F and Vc/F, as well as age on Cl/F, with the following power relationship:
Inclusion of these covariate effects reduced the IIV in Cl/F from 55.7 to 53.7 percentage coefficient of variation (CV%), and the IIV in Vc/F from 63.1 to 61.5 CV%. Parameter estimates for the final model are shown in Table 2.
Table 2.
Parameter estimates for the final model
| Parameter | Estimates | RSE (%) | 95% CI |
|---|---|---|---|
| Cl/F, l h –1 | 26.3 | 2.48 | 25.0, 27.6 |
| Vc/F, l | 489 | 2.79 | 462, 516 |
| Q/F, l h –1 | 11.8 | 11.4 | 9.18, 14.4 |
| Vp/F, l | 213 | 7.14 | 183, 243 |
| Ka, h –1 | 2.70 | 5.65 | 2.40, 3.00 |
| D1, h | 1.68 | 3.88 | 1.55, 1.81 |
| F1 (fixed for sparse sample) | 1.00 | ||
| Age effect on Cl/F (AGECL): ~(Age/59) AGECL | –0.409 | 13.2 | –0.303, –0.515 |
| WT effect on Cl/F (WTCL): ~(WT/73) WTCL | 0.517 | 17.5 | 0.34, 0.694 |
| WT effect on Vc/F (WTVc): ~(WT/73) WTVc | 0.65 | 17.6 | 0.425, 0.874 |
| IIV in Cl/F, CV% | 53.7 | 3.71 | 49.8, 57.6 |
| IIV in Vc/F, CV% | 61.5 | 4.53 | 56, 66.9 |
| Correlation between IIVs in Cl/F and Vc/F | 0.839 | 2.67 | 0.795, 0.883 |
| IIV in Ka, CV% | 151 | 4.35 | 138, 164 |
| IOV in F1, CV% | 44.6 | 7.99 | 37.6, 51.6 |
| Residual variability for rich profile, CV% | 26.5 | 2.97 | 25.0, 28.1 |
| Residual variability for sparse profile, CV% | 47.3 | 3.67 | 43.9, 50.7 |
CI, confidence interval; CV%, coefficient of variation; IIV, inter‐individual variability; IOV, inter‐occasion variability; RSE, relative standard error
Model evaluation
The final covariate model was evaluated using standard diagnostic plots. All diagnostic plots (not shown, except for the prediction‐corrected visual predictive check of the final model, which is shown in the supplementary material) demonstrated that the final model described the observed data and satisfied the model assumptions well. These model evaluations supported the view that the model adequately described cediranib concentration–time courses and was suitable for exposure simulation studies.
Model application
Impact of covariate relations on cediranib exposure
Forest plots for age and WT covariate effect on cediranib AUC and Cmax are shown in Figures 1 and 2, respectively. The effects of age and WT on exposure, as assessed for the ratio of the exposure at the 5th and 95th percentiles of the covariate over that at the median value, were within values of 0.74–1.28.
Figure 1.
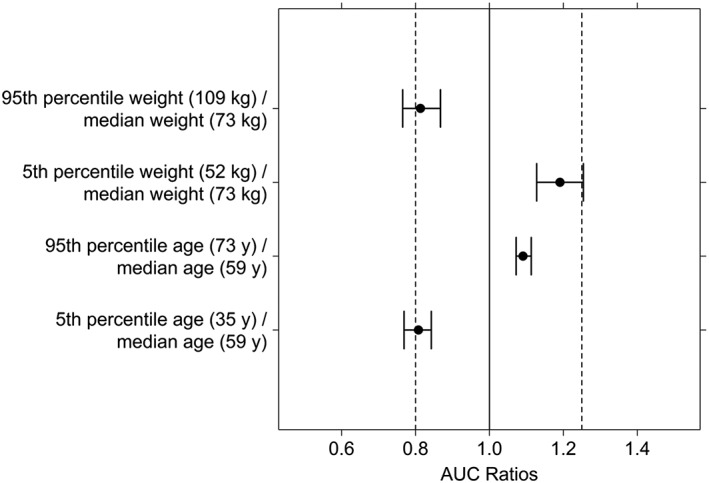
Forest plot of the effect of age and body weight on the AUC of cediranib. Filled circles and error bars represent median and 90% confidence interval for the ratio of AUC at the 5th or 95th percentile of the covariate over that at the median value of the covariate
Figure 2.
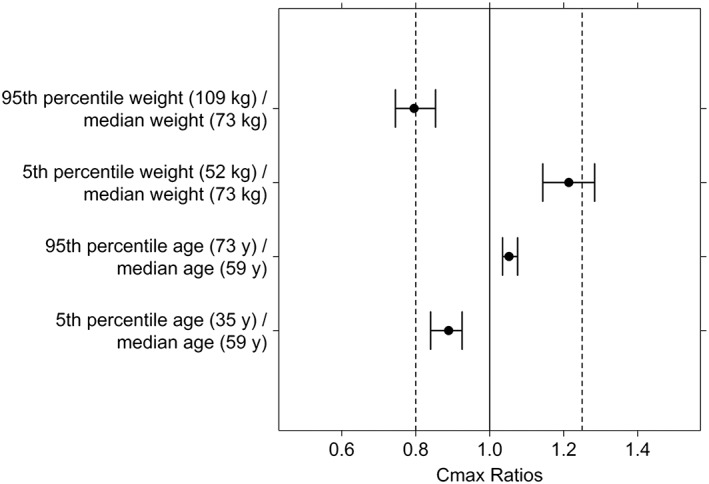
Forest plot of the effect of age and body weight on the Cmax of cediranib. Filled circles and error bars represent median and 90% confidence interval for the ratio of Cmax at the 5th or 95th percentile of the covariate over that at the median value of the covariate
Exposure comparison between ovarian cancer patients and other cancer patients
Exposure comparisons in Cmax,ss, Cmin,ss and AUCss between ovarian (n = 17) and other cancer patients (n = 608) at steady state, assuming the same repeated daily cediranib 20 mg administration, are shown in Figure 3.
Figure 3.
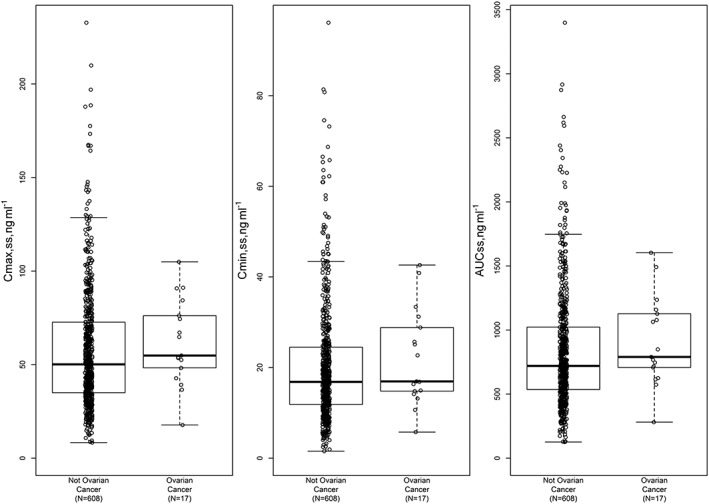
Comparison of estimated Cmax,ss, Cmin,ss and AUCss (normalized to 20 mg dose level) between ovarian and nonovarian cancer patients
Exposure comparison between patients with and without antihypertensive medication
Exposure comparisons in Cmax,ss, Cmin,ss and AUCss between patients taking antihypertensive medication (n = 232) vs. no antihypertensive medication (n = 393), also assuming the same repeated daily cediranib 20 mg administration, are shown in Figure 4.
Figure 4.
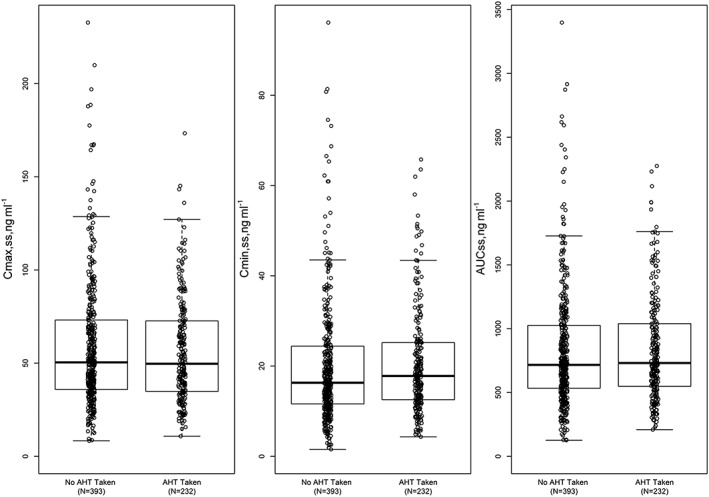
Comparison of estimated Cmax,ss, Cmin,ss and AUCss (normalized to 20 mg dose level) between patients taking antihypertensive drugs and those not. AHT, antihypertensive
Simulations of cediranib plasma profiles and target coverage
The simulated median unbound cediranib concentration after cediranib 15 or 20 mg repeat‐dose administration, together with in vitro concentrations required to obtain IC50 of VEGFR‐1, ‐2 and ‐3 5, are shown in Figure 5.
Figure 5.
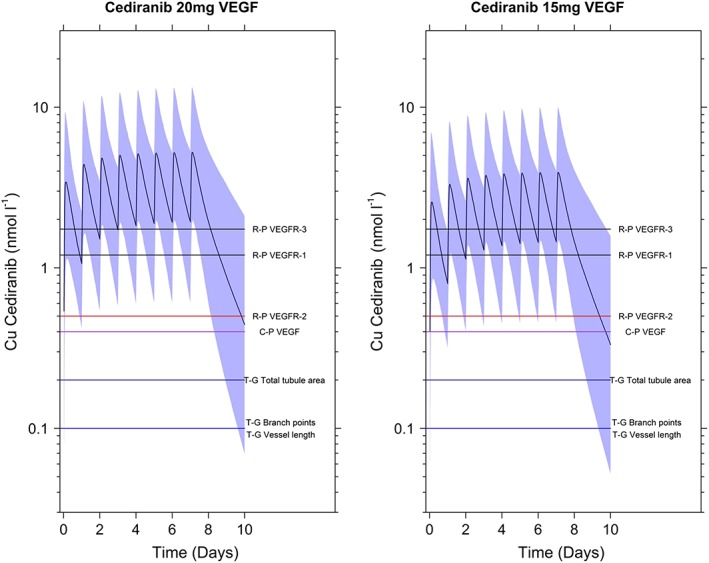
Simulated median (black solid oscillation line) and 90% prediction interval (shaded area) of free cediranib plasma concentration–time profile following once‐daily dose administration of cediranib 20 and 15 mg for 8 days, compared with in vitro IC50 (horizontal lines) for VEGFR inhibition. C‐P, estimate for inhibition of growth‐factor‐stimulated cellular proliferation; R‐P, estimate for inhibition of growth‐factor‐stimulated receptor phosphorylation; T‐G, estimate for inhibition of in vitro tubule growth
Simulations of cediranib exposure in the presence of strong CYP3A4/UGT/Pgp inducers
Box‐and‐whisker plots overlaying the different steady‐state cediranib exposures for cediranib 15 or 20 mg without coadministration of rifampicin, and for cediranib 20 or 30 mg with coadministration of rifampicin 600 mg qd, are shown in Figure 6. The simulations suggest that cediranib 20 or 30 mg, when coadministered with rifampicin, would have similar exposure to that of cediranib 15 or 20 mg administered alone.
Figure 6.
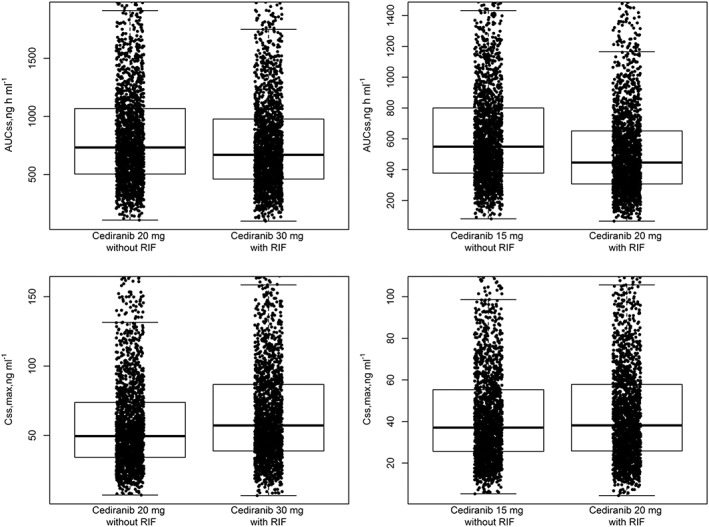
Simulated AUCss and Cmax,ss for 2000 individuals for cediranib with and without coadministered rifampicin. Left panels, AUCss (top) and Cmax,ss (bottom) after daily administration of cediranib 20 mg without rifampicin (left box‐and‐whisker plot) and daily doses of cediranib 30 mg with rifampicin (right box‐and‐whisker plot). Right panels, same simulation for AUCss and Cmax,ss but for cediranib 15 mg without rifampicin and cediranib 20 mg with rifampicin, respectively. RIF, rifampicin 600 mg qd
Discussion
This analysis is the first comprehensive investigation of cediranib PPK in patients with a variety of cancer types. The final cediranib PPK model included a two‐compartment disposition model with sequential zero‐ and first‐order absorption. The disposition model is consistent with biphasic behaviour of the semi‐log cediranib concentration–time courses observed in multiple studies with rich cediranib concentration sampling. Different absorption models were examined, and sequential zero‐ and first‐order absorption was included in the final model because of a significant drop in OFV (283 units) and an improvement in the goodness of fit to the observed cediranib concentrations. For a typical individual of 73 kg and 59 years, the final PPK model predicted cediranib Cl/F of 26.3 l h–1, Vss/F of 702 l (derived from Vc/F + Vp/F = 489 + 213), and a half‐life of 24 h. These results are in agreement with previously reported values from noncompartmental analyses 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32.
The IIVs were estimated to be 53.7 and 61.5 CV% for Cl/F and Vc/F, respectively, with high correlation (0.839), while the IIV for Ka was estimated to be larger (> 150 CV%). Many factors may contribute to the large IIV in Ka, for example, unknown or inaccurate dosing times (assuming dose in relation to nominal sampling). An IOV of 44.6 CV% was estimated for relative bioavailability, based on the rich sampling occasions. Since absolute bioavailability was not identifiable for oral dose administration, the IOV accounts for random variability in relative bioavailability between different rich sampling occasions and could also include any nonrecorded dosing error or lack of compliance, as well as potential variability in first‐pass elimination (gut and liver) or fraction absorbed between occasions. The separate proportional residual error for sparse and trough sampling was estimated to be 47% and was larger than that for the rich profiles (27%). The larger proportional error for sparse plasma sampling may be a result of frequent imputation of dosing times prior to trough cediranib measurements to 09:00, or of potential IOV among different predose sparse data that were not estimated in the PPK analysis.
Among the covariates evaluated, only age and WT were identified as significant covariates. However, age and WT covariates explained only a very limited part of the overall variability in Cl/F and Vc/F and had limited effects on AUC and Cmax exposure. The AUC or Cmax ratios at the 5th and 95th percentiles of the covariate distribution over that at the median (50th percentile) were near or within normal bioequivalence criteria (0.8–1.25), suggesting that no dose adjustments are warranted in terms of either WT or age. It is also expected that neither liver nor renal function would impact cediranib PK 22, 28.
Cediranib is under development in combination with platinum‐based chemotherapy, followed by cediranib maintenance monotherapy, for the treatment of PSR ovarian cancer. Since there was no dedicated Phase I study to characterize cediranib PK specifically in ovarian cancer patients, model‐based simulations were conducted to predict the PK profiles and derive the steady‐state exposure parameters in these patients. The simulation demonstrated that cediranib exposures between ovarian and other cancer patients are comparable overall, as shown in Figure 3: individual predicted Cmax,ss, Cmin,ss and AUCss from the 17 ovarian cancer patients are completely within the exposure ranges of the non‐ovarian cancer patients.
As a VEGFR inhibitor, cediranib may increase blood pressure; hence, antihypertensive comedication for cediranib‐treated cancer patients may be necessary at times. Differences in cediranib exposures (Cmax,ss, Cmin,ss and AUCss) between cancer patients taking antihypertensive medication and those not were evaluated using box‐and‐whisker plots. Figure 4 indicates that exposures in 232 patients who received antihypertensive medication were completely within the exposure ranges for patients who had not received such medication. Co‐administration with antihypertensives appears to have no clinically relevant influence on the PK of cediranib.
A cediranib 15 mg tablet is also available for patients who cannot tolerate the 20 mg daily dose. Effective cediranib exposure following cediranib 15 or 20 mg qd was examined via a simulation analysis. Figure 5 indicates that the simulated steady‐state median concentration–time course of unbound cediranib following a 15 or 20 mg qd dose exceeded the in vitro estimated IC50 required to inhibit VEGFR‐1, ‐2 and ‐3 5, regardless of the methods used to calculate these IC50 values. Furthermore, the lower bound of the 90% predictive interval of this simulated steady‐state mean concentration–time course (unbound cediranib) following a 15 or 20 mg qd dose also exceeded the IC50 for VEGFR‐2, the major target of cediranib, as well as the IC50 for the inhibition of growth factor‐stimulated receptor phosphorylation and growth factor‐stimulated cellular proliferation. These results suggest that, overall, steady‐state cediranib concentrations following repeat qd administration of 15 or 20 mg doses are predicted to cover the primary biomarker targets for cediranib for antitumour effects. Cediranib 20 or 15 mg (if not well tolerated) was studied in a randomized, double‐blind, placebo‐controlled, Phase III trial 16 comparing cediranib against placebo in combination with carboplatin and paclitaxel in platinum‐sensitive recurrent ovarian cancer patients. A total of 456 women have been randomized to receive standard carboplatin and paclitaxel plus placebo followed by placebo as maintenance therapy, or standard carboplatin and paclitaxel plus cediranib 20 mg/day followed by placebo or cediranib 20 mg/day as maintenance therapy. The combination of carboplatin and paclitaxel with cediranib followed by cediranib 20 or 15 mg qd as maintenance therapy improved PFS (11.4 vs 9.4 months; HR 0.68; P = 0.002) and resulted in a significant increase in median overall survival (20.3 vs 17.6 months; HR 0.70; P = 0.042), compared with the other two treatment regimens.
Cediranib is a Pgp substrate and its oxidative metabolism appears to be mediated by the flavin‐containing monooxygenase (FMO) enzymes (FMO1 and FMO3), while phase 2 metabolism is mediated primarily via glucuronidation by UGT1A4. The final cediranib PPK model was used to simulate cediranib exposure for potential dose adjustments, if cediranib was to be coadministered with a strong inducer of the UGT/Pgp transporter, rifampicin (Figure 6). Differences in predicted cediranib exposures in AUCss or Cmax,ss between cediranib 20 mg alone vs. cediranib 30 mg coadministered with rifampicin 600 mg, or between cediranib 15 mg alone vs. cediranib 20 mg coadministered with rifampicin 600 mg, were <20%; moreover, these exposures largely overlapped with each other. These results suggest that the 15 or 20 mg qd dose may require an increase to 20 or 30 mg qd, respectively, when cediranib is coadministered with a potent inducer of UGT/Pgp such as rifampicin.
Conclusions
Cediranib concentration–time courses are well described by a two‐compartment disposition and a sequential zero‐ and first‐order absorption model. No covariate was identified requiring dose adjustment for cediranib in patients with cancer. However, an increase in cediranib dose, either from 15 to 20 mg or from 20 to 30 mg, may be needed when cediranib is coadministered with a strong UGT/Pgp inducer such as rifampicin. Cediranib exposures following repeated, once‐daily 15 or 20 mg administration are adequate overall with respect to target coverage for the inhibition of in vitro estimated VEGFR‐1, ‐2 and ‐3 activities.
Competing Interests
All authors have completed the unified Competing Interest Form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: J.L., W.T., N.A.‐H. and E.M. are employed by the study sponsor (AstraZeneca) and own stock in AstraZeneca; A.H. is a consultant from qPharmetra, LLC and has received payment from AstraZeneca.
The authors would like to thank their colleague, Gabriel Helmlinger, PhD, who assisted with the preparation of the manuscript. They also thank Clara Tan, from Mudskipper Business Ltd, who provided editorial assistance funded by AstraZeneca.
Supporting information
Table S1 Description of studies included in the population pharmacokinetic analysis
Figure S1 Prediction‐corrected visual predictive check for the final cediranib pharmacokinetic model
Li, J. , Al‐Huniti, N. , Henningsson, A. , Tang, W. , and Masson, E. (2017) Population pharmacokinetic and exposure simulation analysis for cediranib (AZD2171) in pooled Phase I/II studies in patients with cancer. Br J Clin Pharmacol, 83: 1723–1733. doi: 10.1111/bcp.13266.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29. [DOI] [PubMed] [Google Scholar]
- 4. Doufekas K, Olaitan A. Clinical epidemiology of epithelial ovarian cancer in the UK. Int J Wom Health 2014; 6: 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double‐blind, placebo‐controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum‐sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 2012; 30: 2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lieu CH, Tran H, Jiang Z‐Q, Mao M, Overman MJ, Lin E. The association of alternate VEGF ligands with resistance to anti‐VEGF therapy in metastatic colorectal cancer. PLoS One 2013; 8: e77117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor‐2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res 2005; 65: 4389–4400. [DOI] [PubMed] [Google Scholar]
- 8. Jeltsch M, Karpanen T, Strandin T, Aho K, Lankinen H, Alitalo K. Vascular endothelial growth factor (VEGF)/VEGF‐C mosaic molecules reveal specificity determinants and feature novel receptor binding patterns. J Biol Chem 2006; 281: 12187–12195. [DOI] [PubMed] [Google Scholar]
- 9. Heckman CA, Holopainen T, Wirzenius M, Keskitalo S, Jeltsch M, Ylä‐Herttuala S, et al. The tyrosine kinase inhibitor cediranib blocks ligand‐induced vascular endothelial growth factor receptor‐3 activity and lymphangiogenesis. Cancer Res 2008; 68: 4754–4762. [DOI] [PubMed] [Google Scholar]
- 10. Leppänen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, et al. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc Natl Acad Sci U S A 2010; 107: 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brave SR, Ratcliffe K, Wilson Z, James NH, Ashton S, Wainwright A, et al. Assessing the activity of cediranib, a VEGFR‐2/3 tyrosine kinase inhibitor, against VEGFR‐1 and members of the structurally related PDGFR family. Mol Cancer Ther 2011; 10: 861–873. [DOI] [PubMed] [Google Scholar]
- 12. Decio A, Taraboletti G, Patton V, Alzani R, Perego P, Fruscio R, et al. Vascular endothelial growth factor c promotes ovarian carcinoma progression through paracrine and autocrine mechanisms. Am J Pathol 2014; 184: 1050–1061. [DOI] [PubMed] [Google Scholar]
- 13. Decio A, Cesca M, Bizzaro F, Belotti D, Giavazzi R. Cediranib affects tumor progression and survival of mice bearing patient derived ovarian carcinoma xenografts (EOC‐PDX). Cancer Res 2014; 74 (19 Suppl): 2994. [Google Scholar]
- 14. Raja FA, Griffin CL, Qian W, Hirte H, Parmar MK, Swart AM, et al. Initial toxicity assessment of ICON6: a randomised trial of cediranib plus chemotherapy in platinum‐sensitive relapsed ovarian cancer. Br J Cancer 2011; 105: 884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruscito I, Gasparri ML, Marchetti C, Medici CD, Bracchi C, Palaia I, et al. Cediranib in ovarian cancer: state of the art and future perspectives. Tumour Biol 2016; 37: 2833–2839. [DOI] [PubMed] [Google Scholar]
- 16. Ledermann JA, Embleton AC, Raja F, Perren TJ, Jayson GC, Rustin GJS, et al. Cediranib in patients with relapsed platinum‐sensitive ovarian cancer (ICON6): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2016; 387: 1066–1074. [DOI] [PubMed] [Google Scholar]
- 17. Drevs J, Siegert P, Medinger M, Mross K, Strecker R, Zirrgiebel U, et al. Phase I clinical study of AZD2171, an oral vascularendothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol 2007; 25: 3045–3054. [DOI] [PubMed] [Google Scholar]
- 18. Ryan CR, Stadler WM, Roth B, Hutcheon D, Conry S, Puchalski T, et al. Phase I dose escalation and pharmacokinetic study of AZD2171, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinase, in patients with hormone refractory prostate cancer (HRPC). Invest New Drugs 2007; 25: 445–451. [DOI] [PubMed] [Google Scholar]
- 19. van Cruijsen H, Voest EE, Punt CJA, Hoekman K, Witteveen PO, Meijerink MR, et al. Phase I evaluation of cediranib, a selective VEGFR signaling inhibitor, in combination with gefitinib in patients with advanced tumours. Eur J Cancer 2010; 46: 901–911. [DOI] [PubMed] [Google Scholar]
- 20. LoRusso P, Shields AF, Gadgeel S, Vaishampayan U, Guthrie T, Puchalski T, et al. Cediranib in combination with various anticancer regimens: results of a phase I multi‐cohort study. Invest New Drugs 2011; 29: 1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laurie SA, Gauthier I, Arnold A, Shepherd FA, Ellis PM, Chen E, et al. Phase I and pharmacokinetic study of daily oral AZD2171, an inhibitor of vascular endothelial growth factor tyrosine kinases, in combination with carboplatin and paclitaxel in patients with advanced non–small‐cell lung cancer: the National Cancer Institute of Canada clinical trials group. J Clin Oncol 2008; 26: 1871–1878. [DOI] [PubMed] [Google Scholar]
- 22. Reid A, Tang A, Spicer J, Gallerani E, Mears D, Shaw H, et al. An exploration of the ability of DCE‐CT scans to evaluate blood flow in an open, pharmacokinetic (PK) and mass balance study of 14[C]‐cediranib. AACR‐NCI‐EORTC International Conference on Molecular Targets and Cancer Therapeutics. San Francisco, CA, USA, 22–26 October 2007; Abstract B259.
- 23. Lassen U, Miller WH, Hotte S, Evans TRJ, Kollmansberger C, Adamson D, et al. Phase I evaluation of the effects of ketoconazole and rifampicin on cediranib pharmacokinetics in patients with solid tumours. Cancer Chemother Pharmacol 2013; 71: 543–549. [DOI] [PubMed] [Google Scholar]
- 24. Mitchell CL, O'Connor JPB, Roberts C, Watson Y, Jackson A, Cheung S, et al. A two‐part phase II study of cediranib in patients with advanced solid tumours: the effect of food on single‐dose pharmacokinetics and an evaluation of safety, efficacy and imaging pharmacodynamics. Cancer Chemother Pharmacol 2011; 68: 631–641. [DOI] [PubMed] [Google Scholar]
- 25. Goss G, Shepherd FA, Laurie S, Gauthier I, Leighl N, Chen E, et al. A phase I and pharmacokinetic study of daily oral cediranib, an inhibitor of vascular endothelial growth factor tyrosine kinases, in combination with cisplatin and gemcitabine in patients with advanced non‐small cell lung cancer: a study of the National Cancer Institute of Canada clinical trials group. Eur J Cancer 2009; 45: 782–788. [DOI] [PubMed] [Google Scholar]
- 26. Yamamoto N, Tamura T, Yamamoto N, Yamada K, Yamada Y, Nokihara H, et al. Phase I, dose escalation and pharmacokinetic study of cediranib (RECENTIN™), a highly potent and selective VEGFR signaling inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 2009; 64: 1165–1172. [DOI] [PubMed] [Google Scholar]
- 27. Mulders P, Hawkins R, Nathan P, de Jong I, Osanto S, Porfiri E, et al. Cediranib monotherapy in patients with advanced renal cell carcinoma: results of a randomised phase II study. Eur J Cancer 2012; 48: 527–537. [DOI] [PubMed] [Google Scholar]
- 28. van Herpen CML, Lassen U, Desar IME, Brown KH, Marotti M, de Jonge MJA. Pharmacokinetics and tolerability of cediranib, a potent VEGF signalling inhibitor, in cancer patients with hepatic impairment. Anticancer Drugs 2013; 24: 204–211. [DOI] [PubMed] [Google Scholar]
- 29. Langenberg MHG, van Herpen CML, De Bono J, Schellens JHM, Unger C, Hoekman K, et al. Effective strategies for management of hypertension after vascular endothelial growth factor signaling inhibition therapy: results from a phase II randomized, factorial, double‐blind study of cediranib in patients with advanced solid tumors. J Clin Oncol 2009; 27: 6152–6159. [DOI] [PubMed] [Google Scholar]
- 30. Satoh T, Yamaguchi K, Boku N, Okamoto W, Shimamura T, Yamazaki K, et al. Phase I results from a two‐part phase I/II study of cediranib in combination with mFOLFOX6 in Japanese patients with metastatic colorectal cancer. Invest New Drugs 2012; 30: 1511–1518. [DOI] [PubMed] [Google Scholar]
- 31. Judson I, Scurr M, Gardner K, Barquin E, Marotti M, Collins B, et al. Phase II study of cediranib in patients with advanced gastrointestinal stromal tumors or soft‐tissue sarcoma. Clin Cancer Res 2014; 20: 3603–3612. [DOI] [PubMed] [Google Scholar]
- 32. Satoh T, Yamada Y, Muro K, Hayashi H, Shimada Y, Takahari D, et al. Phase I study of cediranib in combination with cisplatin plus fluoropyrimidine (S‐1 or capecitabine) in Japanese patients with previously untreated advanced gastric cancer. Cancer Chemother Pharmacol 2012; 69: 439–446. [DOI] [PubMed] [Google Scholar]
- 33. Lindbom L, Ribbing J, Jonsson E. Perl‐speaks‐NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed 2004; 75: 85–94. [DOI] [PubMed] [Google Scholar]
- 34. Menon‐Andersen D, Yu B, Madabushi R, Bhattaram V, Hao W, Uppoor RS, et al. Essential pharmacokinetic information for drug dosage decisions: a concise visual presentation in the drug label. Clin Pharmacol Ther 2011; 90: 471–474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Description of studies included in the population pharmacokinetic analysis
Figure S1 Prediction‐corrected visual predictive check for the final cediranib pharmacokinetic model