

Abstract
Aim
The aim of the present study was to investigate whether increasing the bosentan dosing frequency from 2 mg kg–1 twice daily (b.i.d.) to 2 mg kg–1 three times daily (t.i.d.) in children with pulmonary arterial hypertension (PAH) (from ≥3 months to <12 years of age) would increase exposure.
Methods
An open‐label, prospective, randomized, multicentre, multiple‐dose, phase III study was conducted. Patients (n = 64) were randomized 1:1 to receive oral doses of bosentan of 2 mg kg–1 b.i.d. or t.i.d. The main pharmacokinetic endpoint was the daily exposure to bosentan over 24 h corrected to the 2 mg kg–1 dose (AUC0–24C). The maximum plasma concentration corrected to the 2 mg kg–1 dose (CmaxC), the time to reach the maximum plasma concentration (tmax) and safety endpoints were also assessed.
Results
The geometric mean [95% confidence interval (CI)] for AUC0–24C was 8535 h.ng ml–1 (6936, 10 504) and 7275 h.ng ml–1 (5468, 9679) for 2 mg kg–1 b.i.d. and t.i.d., respectively [geometric mean ratio (95% CI) 0.85 (0.61, 1.20)]. The geometric mean (95% CI) for CmaxC was 743 ng ml–1 (573, 963) and 528 ng ml–1 (386, 722) for 2 mg kg–1 b.i.d. and t.i.d., respectively [geometric mean ratio (95% CI) 0.71 (0.48, 1.05)]. The median (range) for tmax was 3.0 h (0.0–7.5) and 3.0 h (1.0–8.0) for 2 mg kg–1 b.i.d. and t.i.d., respectively. The proportions of patients who experienced ≥1 adverse event were similar in the b.i.d. (66.7%) and t.i.d. (67.7%) groups.
Conclusions
There appeared to be no clinically relevant difference in exposure to bosentan, or in safety, when increasing the frequency of bosentan dosing from b.i.d. to t.i.d. Therefore, the present study provides no indication that the dosing recommendation should be changed, and 2 mg kg–1 b.i.d. remains the recommended dosing regimen for bosentan in paediatric PAH patients.
Keywords: paediatrics, pharmacokinetics, vascular disease
What is Already Known about this Subject
- In paediatric pulmonary arterial hypertension patients:
-
◯Bosentan exposure following 2 mg kg–1 twice‐daily (b.i.d.) dosing is approximately half that observed in adults with bosentan 125 mg b.i.d., whereas the efficacy seems to be similar.
-
◯Increasing the dose to 4 mg kg–1 b.i.d. does not increase exposure.
-
◯Bosentan has been shown to be well tolerated at both doses.
-
◯
What this Study Adds
The present study used a new bosentan dosing regimen, 2 mg kg–1 t.i.d., to investigate whether exposure could be increased in paediatric pulmonary arterial hypertension patients.
The study also provided the first information on bosentan safety and pharmacokinetics in patients below 2 years of age.
Tables of Links
| TARGETS |
|---|
| G protein‐coupled receptors |
| ETA receptor |
| ETB receptor |
| LIGANDS |
|---|
| Bosentan |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease caused by proliferation and remodelling of the pulmonary vasculature, resulting in right ventricular failure and death 3, 4. With regard to definition, pathophysiology, symptoms and response to PAH‐specific therapies, there are important similarities between PAH in children and in adults 5.
The use of PAH‐specific therapies in paediatric patients is generally based on evidence from randomized controlled trials in adults. Data to support their use in children are limited mainly to uncontrolled studies and clinical experience 6, 7, 8, 9, 10. There is also a general lack of information with regard to safety and optimal dosing strategies in paediatric PAH patients 7. The European Medicines Agency (EMA) guidelines for the conduct of paediatric PAH studies require comprehensive pharmacokinetic (PK) data and long‐term safety data to be provided 7. Therefore, there is a clear need for long‐term trials in paediatric PAH, with an emphasis on the collection of both PK and safety data, as it is difficult to extrapolate from adult studies 11.
Bosentan is an oral dual endothelin receptor antagonist (ERA) that is approved for the treatment of PAH 12. Studies have been conducted to support the effectiveness of bosentan using haemodynamic variables in adult 13, 14, 15, 16 and paediatric patients 9. Similar haemodynamic improvements were shown in both populations after administration of the film‐coated (adult) tablet formulation 9, 13, 14, 15, 16. In the Bosentan Randomised Trial of Endothelin Antagonist Therapy‐3 (BREATHE‐3), clinically relevant improvements from baseline in the exploratory endpoints mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR) index were observed after 12 weeks of treatment, even though bosentan plasma concentrations in paediatric patients did not reach those previously observed in adult PAH patients (approximately 50% lower) 8, 9, 17. Several observational studies have also indicated that bosentan may be effective in the treatment of paediatric PAH 18, 19, 20, 21. One study demonstrated that bosentan improved or maintained World Health Organization functional class (WHO FC) in 90% of patients over a median treatment duration of 14 months; decreases in mPAP and PVR were also observed 18.
In the Paediatric FormUlation of bosenTan in pUlmonary arterial hypeRtEnsion (FUTURE‐1) open‐label, uncontrolled study, an oral, dispersible paediatric formulation of bosentan with a quadrisecting score line was investigated, which provided more flexible dosing and increased acceptance for paediatric patients 8. A systemic exposure plateau was observed at a dose of 2 mg kg–1 twice daily (b.i.d.) in paediatric patients, with administration of bosentan at 4 mg kg–1 b.i.d. not resulting in higher plasma concentrations. As a result, the level of exposure to bosentan observed in adult PAH patients could not be reached in paediatric patients with b.i.d. dosing 8. Nevertheless, observations in bosentan‐naïve patients suggested a beneficial therapeutic effect following initiation of bosentan 8. FUTURE‐2, an open‐label, uncontrolled extension of FUTURE‐1 in which patients were allowed additional PAH‐specific therapy in the case of PAH worsening, demonstrated an improvement in different efficacy parameters over long‐term treatment 6. The paediatric formulation of bosentan was well‐tolerated in both FUTURE studies 6, 8. Bosentan 32 mg dispersible tablets were approved in the European Union, based on the results of the paediatric clinical development programme that included the studies listed above.
FUTURE‐3 aimed to complement the understanding of bosentan dosing in paediatric patients by investigating whether increasing the dosing frequency from b.i.d. to three times daily (t.i.d.) would result in increased daily systemic exposure. The main objective of FUTURE‐3 was to investigate the PK of the paediatric formulation of bosentan at doses of 2 mg kg–1 b.i.d. and t.i.d. in children with PAH from ≥3 months to <2 years of age and from 2 years to <12 years of age. Other main objectives of the study were to evaluate the tolerability and safety of bosentan in children with PAH.
Methods
Study design
FUTURE‐3 was an open‐label, prospective, randomized, multicentre, multiple‐dose (two dose regimens), phase III study in a paediatric population of PAH patients (clinicaltrials.gov identifier: NCT01223352). It was predominantly designed as a PK study, and patients were enrolled in 30 centres in Europe, North America, Latin America, Australia, Asia and Africa.
FUTURE‐3 consisted of a screening period (maximum 4 weeks), followed by a 24‐week treatment period and a 7‐day adverse event (AE) follow‐up period. Patients were randomized 1:1 to receive oral doses of bosentan of 2 mg kg–1 b.i.d. (4 mg kg–1 total daily) or t.i.d. (6 mg kg–1 total daily), stratified for baseline PAH‐specific treatment [treatment‐naïve, bosentan, prostanoid, phosphodiesterase type‐5 (PDE‐5) inhibitor or combinations thereof]. Patients were randomized according to a randomization code generated using Parexel® Informatics, Waltham, MA, USA. Randomized treatment assignment was centralized via an interactive voice/web response system in order to avoid investigator bias in the allocation of treatment. Patients who prematurely discontinued study treatment had to complete an end‐of‐study (EOS) assessment. Patients completing the study had the option of participation in a 1‐year follow‐up extension study. Those patients who did not enter the 1‐year follow‐up extension had a 60‐day post‐treatment follow‐up of serious adverse events (SAEs) and deaths.
Patients
The study population included male and female PAH patients (≥3 months to <12 years of age) with idiopathic PAH (IPAH), heritable PAH (HPAH) or associated PAH (APAH) persisting after complete repair of a congenital heart defect, or PAH–congenital heart disease (CHD) associated with systemic‐to‐pulmonary shunts or Eisenmenger syndrome. Patients with small defects and co‐incidental APAH–CHD were also permitted to enrol in the study. Patients were in WHO FC I‐III and diagnosed by right heart catheterization (RHC). Baseline PAH therapy (calcium channel blocker, bosentan, PDE‐5 inhibitor, prostanoid), if present, had to be stable for at least 3 months prior to screening. Exclusion criteria included aspartate transaminase (AST) or alanine aminotransferase (ALT) levels >1.5 times the upper limit of normal range, moderate to severe hepatic impairment and haemoglobin and/or haematocrit levels <75% of the lower limit of normal range. Patients who required additional or an increased dose of PAH‐specific therapy had to discontinue the study, with the option of joining the 1‐year follow‐up extension study.
The protocol was approved by the Independent Ethics Committee or Institutional Review Board at each participating centre (Table S1) and the study was performed in accordance with the principles of the Declaration of Helsinki and within the regulations for each country. Written informed consent was obtained from every child's parents or legal representatives (and assent was obtained from each child if applicable) prior to any study procedure.
Outcome measures
The main PK endpoint was defined as the daily exposure to bosentan – i.e., the area under the concentration–time curve (AUC) over a period of 24 h corrected to the 2 mg kg–1 target dose (AUC0–24C), for b.i.d. or t.i.d. dosing (refer to PK and statistical analyses section for details). Dose‐corrected values were used as the smallest dose unit was 8 mg (a quarter of a tablet), so it was not possible to achieve the exact target dose in all patients.
Other PK endpoints were the maximum plasma concentration corrected to the 2 mg kg–1 dose (CmaxC) of bosentan, time to reach the maximum plasma concentration (tmax) of bosentan, and metabolite to parent AUC0–24C ratio for the metabolites Ro 47–8634, Ro 48–5033, and Ro 64–1056. The PK of the three metabolites was assessed in order to understand whether a potential difference in exposure to bosentan could be explained by the activity of the cytochrome P450 (CYP)3A4 and/or CYP2C9 enzymes responsible for the formation of these metabolites. Safety endpoints included occurrence of treatment‐emergent AEs, SAEs, AEs leading to treatment discontinuation, death, laboratory abnormalities, change from baseline in vital signs and 12‐lead electrocardiogram (ECG), and growth.
Trial procedures and analytical methods
Patient characteristics and demographic data were collected at baseline. PK assessments were performed at week 4, after at least 2 weeks of stable study treatment. Blood samples were collected into ethylene diamine tetra‐acetic acid‐containing tubes by venepuncture or from an indwelling catheter in an arm vein. For the 2 mg kg–1 b.i.d. dosing regimen, samples were collected immediately before administration of the study medication dose (predose), and at 0.5, 1, 3, 7.5 and 12 h postdose. For the 2 mg kg–1 t.i.d. dosing regimen, samples were collected predose and at 0.5, 1, 3, 5 and 8 h postdose. Concentrations of bosentan and its metabolites in plasma were determined as previously described 22 or by using liquid chromatography–tandem‐mass spectrometry following protein precipitation with methanol. The lower limit of quantification for bosentan varied between 1.00 ng ml–1 and 2.00 ng ml–1, and for Ro 47–8634, Ro 48–5033 and Ro 64–1056 it was 2.00 ng ml–1. The day‐to‐day precision for both methods varied between 1.2% and 15.1% for bosentan and its metabolites, and accuracy varied between 85.1% and 112.9%. Dried blood spot samples were also collected and compared with the plasma samples 23. Safety and tolerability were evaluated by monitoring safety variables, including laboratory abnormalities, AEs, SAEs, death, vital signs, 12‐lead ECG and growth.
PK and statistical analyses
The sample size was based on the expectation that recruitment of at least 64 patients would lead to at least 50 evaluable patients; for a sample size of 25 patients in each dosing regimen, and assuming that the standard deviation (SD) of the log‐transformed data is 0.650 8, an increased daily exposure of at least 45% would be necessary to obtain a 95% confidence interval (CI) that does not include 1.00 (i.e., statistically significant).
Analyses of PK parameters were performed for the overall population and for age subgroups <2 years and ≥2 years. Statistical analyses of PK parameters were performed on the PK set, which included patients who received at least one dose of study drug, and provided at least five of the six blood samples (including the predose and the 8 h or 12 h postdose samples) and who did not violate the protocol in a way that might affect the evaluation of the main PK endpoint. The PK parameters were determined using a noncompartmental analysis using the Phoenix 64 6.3 software package (Certara L.P., St Louis, MO, USA) on the basis of scheduled time points if there was not more than 5% deviation from the actual ones. AUC0–24C was calculated as a multiple of the exposure over a dosing interval (AUC) (3 × AUC and 2 × AUC for t.i.d. and b.i.d., respectively). AUC0–24 and Cmax were corrected to the target dose of 2 mg kg–1 bosentan (AUC0–24C and CmaxC) by dividing these values by the actual dose received and multiplying by 2. All AUC and Cmax values were assumed to be log‐normally distributed.
The ratios of geometric means of AUC0–24C and CmaxC of bosentan were calculated [test treatment (t.i.d.) : reference treatment (b.i.d.)] and a linear fixed‐effects model, with dosing regimen as fixed effect, was used to estimate the 95% CIs. Geometric mean ratios (90% CI) of the metabolite to bosentan AUC0–24C values were also calculated. The influence of baseline characteristics (WHO FC, body weight, gender, and age at study entry) on AUC0–24C was assessed in post hoc sensitivity analyses using the linear fixed‐effects model to produce baseline covariate‐adjusted geometric mean ratios. One interim safety analysis was planned for the study, to identify the need for any dose change. There were no changes in the planned dosing due to this analysis. Safety data were analysed descriptively for the all‐treated set, which included all randomized patients who received at least one dose of study drug. AEs and SAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 16.0. Upper respiratory tract infection AEs were analysed based on Actelion internal MedDRA queries (nasopharyngitis, influenza, laryngitis, pharyngitis, tonsillitis, bacterial upper respiratory tract infection, viral pharyngitis, and viral rhinitis). Statistical Analysis System (SAS) software, version 9.3 (SAS Institute, Cary, NC, USA) was used for all statistical analyses.
Results
Patient characteristics
In FUTURE‐3, 64 patients were randomized to bosentan 2 mg kg–1 b.i.d. (n = 33) or 2 mg kg–1 t.i.d. (n = 31) (Figure 1). Of the randomized patients, 21 were <2 years old (10 treated with 2 mg kg–1 b.i.d. and 11 t.i.d.). All patients who were randomized received the study treatment. The PK set included 58 patients (90.6% of the all‐randomized set). Among all‐randomized patients, demographic and baseline characteristics were generally similar in the 2 mg kg–1 b.i.d. and t.i.d. groups, although there was a smaller proportion both of females and FC III patients in the 2 mg kg–1 t.i.d. group compared with b.i.d. (female: 32.3% vs. 54.5%, respectively; FC III: 19.4% vs. 36.4%, respectively) (Table 1). The percentage of patients on background therapy was consistent across dosing regimens, with 60.6% and 67.7% of patients in the 2 mg kg–1 b.i.d. and t.i.d. groups, respectively, on background therapy in the overall population. The majority of patients were receiving either bosentan or a PDE‐5 inhibitor at baseline (Table 1). The mean duration of bosentan treatment (weeks ± SD) was 23.6 ± 3.7 weeks in the 2 mg kg–1 b.i.d. group and 23.3 ± 5.0 weeks in the 2 mg kg–1 t.i.d. group.
Figure 1.
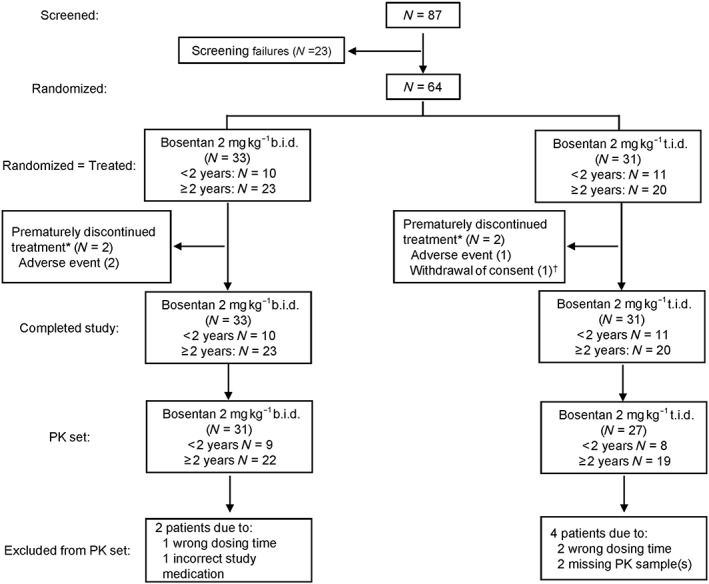
Patient disposition. *Patients who prematurely discontinued treatment were considered to have completed the study, per protocol, as they provided a valid end‐of‐study assessment. †This patient did not provide postbaseline laboratory data. b.i.d., twice daily; PK, pharmacokinetic; t.i.d., three times daily
Table 1.
Summary of baseline demographics and characteristics by dosing regimen and age group
| 2 mg kg –1 b.i.d. | 2 mg kg –1 t.i.d. | |||||
|---|---|---|---|---|---|---|
| <2 years n = 10 | ≥2 years n = 23 | Overall n = 33 | <2 years n = 11 | ≥2 years n = 20 | Overall n = 31 | |
| Gender, n (%) | ||||||
| Males | 1 (10.0) | 14 (60.9) | 15 (45.5) | 10 (90.9) | 11 (55.0) | 21 (67.7) |
| Females | 9 (90.0) | 9 (39.1) | 18 (54.5) | 1 (9.1) | 9 (45.0) | 10 (32.3) |
| Age (years), mean ± SD | 1.3 ± 0.50 | 5.9 ± 3.07 | 4.5 ± 3.35 | 1.1 ± 0.51 | 7.5 ± 2.74 | 5.2 ± 3.81 |
| Baseline PAH‐specific treatment, (consolidated stratification factor) a n (%) | ||||||
| Treatment‐naïve | 4 (40.0) | 9 (39.1) | 13 (39.4) | 3 (27.3) | 7 (35.0) | 10 (32.3) |
| Bosentan | 3 (30.0) | 7 (30.4) | 10 (30.3) | 3 (27.3) | 5 (25.0) | 8 (25.8) |
| Prostanoid | – | – | – | – | 1 (5.0) | 1 (3.2) |
| PDE‐5 inhibitor | 3 (30.0) | 7 (30.4) | 10 (30.3) | 5 (45.5) | 7 (35.0) | 12 (38.7) |
| Aetiology for PAH, n (%) b | ||||||
| IPAH | 3 (30.0) | 11 (47.8) | 14 (42.4) | 5 (45.5) | 10 (52.6) | 15 (50.0) |
| HPAH | 1 (10.0) | 1 (4.3) | 2 (6.1) | – | – | – |
| APAH c | 1 (10.0) | 10 (43.5) | 11 (33.3) | 5 (45.5) | 8 (42.1) | 13 (43.3) |
| PAH–CHD associated with systemic‐to‐pulmonary shunts or Eisenmenger syndrome | 5 (50.0) | 1 (4.3) | 6 (18.2) | 1 (9.1) | 1 (5.3) | 2 (6.7) |
| Time from first observed/assumed PAH symptoms d (days e ), mean ± SD | 320.0 ± 218.98 | 796.4 ± 902.59 | 601.5 ± 735.71 | 283.0 ± 200.12 | 1058.5 ± 1053.42 | 800 ± 933.44 |
| WHO FC, n (%) | ||||||
| I | 2 (20.0) | 6 (26.1) | 8 (24.2) | 3 (27.3) | 5 (25.0) | 8 (25.8) |
| II | 3 (30.0) | 10 (43.5) | 13 (39.4) | 4 (36.4) | 13 (65.0) | 17 (54.8) |
| III | 5 (50.0) | 7 (30.4) | 12 (36.4) | 4 (36.4) | 2 (10.0) | 6 (19.4) |
In the case of a combination of PAH‐specific medications, the following hierarchy was applied: bosentan > prostanoid > PDE‐5 inhibitor
One patient from the ≥2‐years t.i.d. dosing group had pulmonary hypertension associated with a congenital diaphragmatic hernia (nontargeted aetiology), which was clarified after randomization.
Persisting after complete repair of a congenital heart defect (PAH had to be persistent for at least 6 months after surgery)
Time from PAH symptoms excludes patients with an APAH aetiology
Calculated in relation to the date of screening
All‐randomized set. APAH, associated PAH; b.i.d., twice daily; HPAH, heritable PAH; IPAH, idiopathic PAH; PAH, pulmonary arterial hypertension; PAH–CHD, PAH with congenital heart disease; PDE‐5, phosphodiesterase‐type 5; SD, standard deviation; t.i.d., three times daily; WHO FC, World Health Organization functional class
PK parameters in the overall population
The PK parameters of bosentan for the 2 mg kg–1 b.i.d. and t.i.d. dosing regimens are described in Table 2. In the overall population, the main PK endpoint of AUC0–24C was lower for 2 mg kg–1 t.i.d. [geometric mean (95% CI): 7275 h.ng ml–1 (5468, 9679)] compared with 2 mg kg–1 b.i.d. [geometric mean (95% CI): 8535 h.ng ml–1 (6936, 10 504)]; however, there was high interindividual variability in AUC0–24C for both dosing regimens (Figure S1). The geometric mean ratio (95% CI) was 0.85 (0.61, 1.20) and as the CI between the two dose groups included 1.00, no statistically significant difference could be demonstrated, suggesting that AUC0–24C was comparable between the two regimens.
Table 2.
Summary of bosentan pharmacokinetic parameters by dosing regimen and age group
| PK parameter | Age group | 2 mg kg –1 b.i.d. | 2 mg kg –1 t.i.d. | Geometric mean ratio (t.i.d./b.i.d.) (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Geometric mean (95% CI) | n | Geometric mean (95% CI) | |||
| AUC 0–24C (h*ng ml –1 ) | Overall | 31 | 8535 (6936, 10 504) | 27 | 7275 (5468, 9679) | 0.85 (0.61, 1.20) |
| <2 years | 9 | 7879 (4783, 12 979) | 8 | 6756 (3761, 12 135) | 0.86 (0.43, 1.72) | |
| ≥2 years | 22 | 8820 (6939, 11 210) | 19 | 7506 (5236, 10 759) | 0.85 (0.57, 1.28) | |
| C maxC (ng ml –1 ) | Overall | 31 | 743 (573, 963) | 27 | 528 (386, 722) | 0.71 (0.48, 1.05) |
| <2 years | 9 | 622 (350, 1106) | 8 | 487 (262, 905) | 0.78 (0.36, 1.69) | |
| ≥2 years | 22 | 799 (587, 1087) | 19 | 546 (366, 814) | 0.68 (0.42, 1.11) | |
| t max a (h) | Overall | 31 | 3.0 (0.0, 7.5) | 27 | 3.0 (1.0, 8.0) | |
| <2 years | 9 | 3.0 (0.0, 3.0) | 8 | 4.0 (1.0, 8.0) | ||
| ≥2 years | 22 | 3.0 (0.0, 7.5) | 19 | 3.0 (1.0, 8.0) | ||
Median (range)
PK set. AUC0–24C, area under the concentration–time curve from 0 to 24 h; b.i.d., twice daily; CI, confidence interval; CmaxC, maximum plasma concentration; tmax, time to maximum plasma concentration; PK, pharmacokinetic; t.i.d., three times daily
In the overall population, the geometric mean (95% CI) for CmaxC of bosentan in patients who were dosed 2 mg kg–1 t.i.d. was 29% lower than for patients who were dosed 2 mg kg–1 b.i.d. [528 ng ml–1 (386, 722) and 743 ng ml–1 (573, 963), respectively]. However, as for AUC0–24C, the 95% CI around the geometric mean ratio [0.71 (0.48, 1.05)] showed high variability and included 1.00, indicating that CmaxC was also comparable for both dosing regimens (Table 2). The PK profiles of bosentan for both dosing regimens were characterized by rapid absorption, with a median tmax of 3 h (range 0.0–7.5 and 1.0–8.0 for 2 mg kg–1 b.i.d. and t.i.d., respectively) (Figure 2, Table 2).
Figure 2.
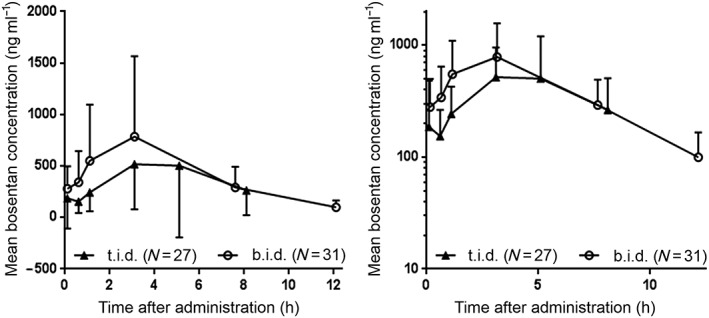
Arithmetic mean dose‐corrected plasma concentration (± standard deviation) vs. time profiles of bosentan on a linear and semi‐logarithmic scale. Overall age group; PK set. b.i.d., twice daily; PK, pharmacokinetic; t.i.d., three times daily
Post hoc sensitivity analyses were performed to explore the influence of specific baseline covariates on AUC0–24C and CmaxC between the two dosing regimens. After adjusting for these covariates, the results were consistent with the unadjusted analyses (Table 3).
Table 3.
Geometric mean ratios between treatment groups for bosentan pharmacokinetic parameters, with and without adjustment for baseline covariates
| Estimation method | Geometric mean ratio between treatment groups (t.i.d./b.i.d.) (95% CI) | |
|---|---|---|
| AUC 0–24C (h*ng ml –1 ) | C maxC (ng ml –1 ) | |
| Unadjusted | 0.85 (0.61, 1.20) | 0.71 (0.48, 1.05) |
| Adjusted by weight at baseline | 0.84 (0.59, 1.18) | 0.70 (0.47, 1.05) |
| Adjusted by WHO FC at baseline | 0.91 (0.66, 1.27) | 0.74 (0.50, 1.11) |
| Adjusted by age at study entry | 0.83 (0.59, 1.17) | 0.70 (0.47, 1.05) |
| Adjusted by gender | 0.89 (0.63, 1.26) | 0.75 (0.50, 1.12) |
Overall age group, PK set
n numbers are as follows: 2 mg kg–1 t.i.d.: (Overall: 27; <2 years: 8; ≥2 years: 19); 2 mg kg–1 b.i.d.: (Overall: 31; <2 years: 9; ≥2 years: 22)
AUC0–24C, area under the concentration time curve from 0 to 24 h; b.i.d., twice daily; CI, confidence interval; CmaxC, maximum plasma concentration; PK, pharmacokinetic; t.i.d., three times daily; WHO FC: World Health Organization functional class
The metabolite to parent AUC0–24C ratios for the 2 mg kg–1 b.i.d. and t.i.d. dosing regimens were comparable (Table 4).
Table 4.
Summary of metabolite to bosentan AUC0–24C geometric mean ratios
| Treatment group | n | Geometric mean ratio (metabolite/parent) (90% CI) | ||
|---|---|---|---|---|
| Ro 47–8634/bosentan | Ro 48–5033/bosentan | Ro 64–1056/bosentan | ||
| Overall | ||||
| b.i.d. | 31 | 0.02 (0.02, 0.03) | 0.16 (0.14, 0.18) | 0.12 (0.10, 0.15) |
| t.i.d. | 27 | 0.03 (0.02, 0.03) | 0.13 (0.12, 0.15) | 0.10 (0.09, 0.12) |
| <2 years | ||||
| b.i.d. | 9 | 0.02 (0.02, 0.03) | 0.17 (0.14, 0.19) | 0.13 (0.09, 0.18) |
| t.i.d. | 8 | 0.02 (0.02, 0.03) | 0.15 (0.11, 0.21) | 0.11 (0.08, 0.16) |
| ≥2 years | ||||
| b.i.d. | 22 | 0.03 (0.02, 0.03) | 0.16 (0.13, 0.18) | 0.12 (0.09, 0.15) |
| t.i.d. | 19 | 0.03 (0.02, 0.03) | 0.13 (0.11, 0.14) | 0.10 (0.08, 0.11) |
PK set. AUC0–24C, area under the concentration time curve from 0 to 24 h; b.i.d., twice daily; CI, confidence interval; PK, pharmacokinetic; t.i.d., three times daily
PK parameters by age group
Overall, PK parameters for b.i.d. and t.i.d. dosing were comparable in both age groups, as indicated by the 95% CI of the geometric mean of AUC0–24C and CmaxC, which largely overlapped for both dosing regimens in both age groups (Table 2). The median tmax was identical (3 h) for patients ≥2 years for both dosing regimens. In patients <2 years, the median tmax occurred later for t.i.d. dosing (4 h) compared with b.i.d. dosing (3 h). The dosing regimen had no effect on the metabolite to parent AUC0–24C ratios of Ro 47–8634, Ro 48–5033, or Ro 64–1056 across the age groups (Table 4). PK parameters of bosentan and the metabolite to bosentan AUC0–24C ratio in patients <2 years were consistent with those in patients ≥2 years (Table 2, Table 4).
Safety and tolerability
In the overall population, the proportions of patients who experienced ≥1 AE were similar in the b.i.d. (66.7%) and t.i.d. (67.7%) groups (Table 5), with events denoting upper respiratory tract infections being the most frequent and occurring at a similar frequency in both groups (36.4%, b.i.d.; 45.2%, t.i.d.; Table S2). The proportions of patients with ≥1 AE in the b.i.d. and t.i.d. regimens were comparable across age groups (Table 5). Overall, there was a slightly higher proportion of SAEs in the t.i.d. dosing regimen (19.4%) compared with b.i.d. (12.1%) (Table 5) and all SAEs were assessed by the investigator as unrelated to study drug administration. A total of three (4.7%) patients [two patients (6.1%), b.i.d.; one patient (3.2%), t.i.d.] permanently discontinued study treatment owing to AEs that indicated a worsening of PAH, which in one case was associated with bronchopneumonia and in another with infection in the context of a congenital metabolic disease. All AEs that led to study treatment discontinuation were SAEs. Two patients (3.1%) died during the study [up to 60 days after study drug discontinuation; one patient (3.0%), b.i.d.; one patient (3.2%), t.i.d.]. In general, changes in laboratory variables were balanced between the b.i.d. and t.i.d. groups. Elevations in ALT and/or AST >3 times the upper limit of normal were reported for two patients (3.1%), both of whom were in the t.i.d. group. In both patients, the ALT/AST levels returned to normal levels, following permanent discontinuation of bosentan treatment in one case and treatment interruption in the other. Decreases in haemoglobin values to <100 g l–1 were reported in four patients (6.3%) [three patients (9.1%), b.i.d.; one patient (3.2%), t.i.d.]. There were no relevant differences between dosing regimens in the changes in vital signs, ECG parameters or growth variables.
Table 5.
Summary of adverse events, serious adverse events and adverse events leading to discontinuationa
| 2 mg kg –1 b.i.d. | 2 mg kg –1 t.i.d. | |||||
|---|---|---|---|---|---|---|
| <2 years n = 10 | ≥2 years n = 23 | Overall n = 33 | <2 years n = 11 | ≥2 years n = 20 | Overall n = 31 | |
| Treatment exposure, weeks, mean ± SD | 22.8 ± 6.0 | 24.0 ± 2.2 | 23.6 ± 3.7 | 21.8 ± 8.3 | 24.1 ± 1.4 | 23.3 ± 5.0 |
| Patients with ≥1 AE, n (%) | 6 (60.0) | 16 (69.6) | 22 (66.7) | 8 (72.7) | 13 (65.0) | 21 (67.7) |
| Total no. of AEs | 19 | 43 | 62 | 31 | 61 | 92 |
| Patients with ≥1 SAE, n (%) | 2 (20.0) | 2 (8.7) | 4 (12.1) | 4 (36.4) | 2 (10.0) | 6 (19.4) |
| Total no. of SAEs | 5 | 3 | 8 | 8 | 2 | 10 |
| Patients with ≥1 AE leading to treatment discontinuation, n (%) | 1 (10.0) | 1 (4.3) | 2 (6.1) | 1 (9.1) | – | 1 (3.2) |
| No. of AEs leading to treatment discontinuation | 1 | 1 | 2 | 2b | – | 2b |
Up to end of treatment +7 days; all‐treated set
One patient experienced two separate AEs leading to treatment discontinuation (PAH worsening and bronchopneumonia)
AE, adverse event; b.i.d., twice daily; PAH, pulmonary arterial hypertension; SAE: serious adverse event; SD, standard deviation; t.i.d., three times daily
One patient with a congenital diaphragmatic hernia, already on chronic bosentan therapy, did not meet the aetiology inclusion criteria and was erroneously included in the study. The condition of the patient was not expected to affect the safety profile and therefore the patient remained in the study. Furthermore, no impact on the bosentan PK was expected as a result of this condition. The patient was on t.i.d. treatment and had an exposure of 6126 h.ng ml–1, where the geometric mean (95% CI) of AUC0–24C in the t.i.d. group was 7275 h.ng ml–1 (5468, 9679). This exposure value would therefore not have affected a mean performed with 27 patients in the PK set. For the age group analysis, this patient was part of the group ≥2 years. As for the overall population, this exposure value would not have affected the mean calculated for the 19 patients in the PK set. No specific safety issues were noted for this patient.
Discussion
In the present study, the PK and safety profile of an oral paediatric formulation of bosentan were compared following 2 mg kg–1 b.i.d. or t.i.d. dosing in paediatric PAH patients. Although AUC0–24C and CmaxC differed slightly between the b.i.d. and t.i.d. dosing regimens, given the high variability of bosentan PK, these differences were small and not considered clinically relevant, and statistical analysis indicated that increasing the dosing frequency from b.i.d. to t.i.d. did not alter these parameters in a significant manner. Baseline factors of age, gender, body weight or FC did not influence the comparison of AUC0–24C or CmaxC between the two dosing regimens. Other PK endpoints (tmax for bosentan and metabolite to bosentan exposure ratios) were also comparable between the dosing regimens, with the latter indicating that the dosing regimen did not influence the extent of metabolism of bosentan. There was no effect of dosing regimen on the PK parameters of bosentan or the metabolite to parent exposure ratios across the age groups (<2 years and ≥2 years), and PK parameters in patients <2 years were consistent with those in patients ≥2 years.
Previous studies have demonstrated that bosentan plasma concentrations in paediatrics are lower than those in adult PAH patients, both with the adult film‐coated 9 and the paediatric dispersible formulation tablets 8. FUTURE‐1 showed that increasing the dose of bosentan above 2 mg kg–1 did not increase exposure in paediatric PAH patients in a b.i.d. regimen 8. Therefore, the t.i.d. dosing regimen was explored in the present study. The data from FUTURE‐3 show that – across the paediatric age groups – dosing with bosentan more frequently than twice daily is unlikely to result in increased exposure. A small decrease in exposure was seen with t.i.d. dosing, although this was not considered to be clinically relevant and, overall, the exposure after both t.i.d. and b.i.d. dosing was similar to that observed with bosentan 2 mg kg–1 or 4 mg kg–1 b.i.d. in previous paediatric studies 8, 9. The median tmax for both dosing regimens in the study was also similar to values observed previously in healthy adults 17, 24 and paediatric populations 8, 9. The results of the present study support those previously seen in FUTURE‐1, where it was suggested that paediatric PAH patients reach a bosentan exposure plateau at a lower dose than adults 8. One possible explanation for this could be the smaller intestinal surface area and/or different absorption characteristics in paediatric patients 8. In addition, in BREATHE‐3, paediatric patients experienced auto‐induction of the CYP3A4 and 2C9 enzymes following bosentan administration, similarly to adults 9. It is not known if increasing the dosing frequency may have had an impact on the level of auto‐induction seen in paediatrics, and whether this affected the exposure to bosentan after t.i.d. dosing. In adult patients, it is usual practice for safety reasons to initiate bosentan at 62.5 mg b.i.d. and increase to the maintenance dose of 125 mg b.i.d. after 4 weeks 12. However, this is not required in paediatric patients as children have a lower exposure to bosentan compared with adults and therefore already have an adequate safety margin. Thus, in the present study, no dose increase was performed, in line with the current prescribing information for bosentan in paediatric patients 12. In previous studies, 2 mg kg–1 b.i.d. bosentan has been shown to be effective in improving haemodynamics in adults 13, 14, 15 and children 9, and its long‐term safety profile has been assessed 6. The present study provided no indication that the dosing recommendation should be changed, and 2 mg kg–1 b.i.d. remains the recommended dosing regimen for bosentan in paediatric PAH patients.
Bosentan induces CYP3A4 and CYP2C9; these isozymes catalyse the formation of the three bosentan metabolites 12. PK parameters of bosentan and its metabolite to parent exposure ratio in patients <2 years were consistent with those in patients ≥2 years. These data indicate that the enzyme activity levels are similar in both age groups. This is an important finding as this was the first paediatric study of bosentan to investigate PK in a population <2 years of age. Although ERAs, PDE‐5 inhibitors and prostanoids are all used for the treatment of paediatric PAH and have improved outcomes 25, the evidence base for their use in paediatric patients <2 years is limited 7, 8, 9, 25, 26.
The majority of AEs were those that would be expected within a paediatric population (e.g. respiratory tract infections) 6, or were consistent with the known safety profile of bosentan in paediatrics 8. The safety profile showed no clinically relevant differences between t.i.d. or b.i.d. dosing regimens. FUTURE‐3 provided important safety data for bosentan in this group of paediatric PAH patients and there were no new safety risks identified in patients aged 3 months to <2 years.
Limitations of the study included a small sample size of patients below 2 years, which reflects the challenges in recruiting for paediatric PAH studies due to the rare and heterogeneous nature of paediatric PAH 7, 10. In addition, no placebo or active comparator was used, which may have been a limitation for the safety analysis of bosentan. However, the use of placebo was not considered appropriate due to the widespread use of bosentan and/or other PAH‐specific medications in the paediatric PAH population. Although the target dose of bosentan was 2 mg kg–1, the smallest dose unit was 8 mg (a quarter of a tablet). It was therefore not possible to achieve the exact target dose in all patients. This imprecision could have affected the PK parameters to a small extent, particularly for children with low body weights. Dose‐corrected values were used during the analysis, to control for this imprecision.
Overall, there appeared to be no difference in daily exposure, and no clinically relevant differences in safety, when increasing the frequency of bosentan dosing from 2 mg kg–1 b.i.d. to 2 mg kg–1 t.i.d. In addition, the PK of bosentan in patients <2 years of age were consistent with those in patients ≥2 years. Therefore, the present study provided no indication that the dosing recommendation should be changed, and it is concluded that 2 mg kg–1 b.i.d. remains the recommended dosing regimen for bosentan in paediatric PAH patients.
Competing Interest
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (http://www.icmje.org/coi_disclosure.pdf) (available on request from the corresponding author) and declare:
R.B. reports nonfinancial support from Actelion Pharmaceuticals Ltd during the conduct of the study and the University Medical Center Groningen contracts with Actelion Pharmaceuticals Ltd, and has received fees for consultancy activities. Outside of the submitted work, the University Medical Center Groningen has contracted with and received consultancy fees from Eli Lilly, Bayer, Pfizer and GSK. M.G. is an employee of Actelion Pharmaceuticals Ltd. M.B. reports nonfinancial support and personal fees from Actelion Pharmaceuticals Ltd during the conduct of the study, and grants and personal fees from Actelion Pharmaceuticals Ltd outside the submitted work. Outside of the submitted work, M.B. has received grants and fees from Bayer, and consultancy fees from Eli Lilly, Pfizer and GSK. D.I. reports nonfinancial support from Actelion Pharmaceuticals Ltd during the conduct of the study, and consultancy fees through a contract between the University of Colorado and Actelion Pharmaceuticals Ltd. Outside of the submitted work, the University of Colorado contracts with and receives fees from Eli Lilly, United Therapeutics, Bayer and GSK for D.I. to be a consultant. A.K.P. is an employee of Actelion Pharmaceuticals Ltd and holds stock/stock options in Actelion. P.C. reports nonfinancial support and personal fees from Actelion Pharmaceuticals Ltd during the conduct of the study. S.G. is an employee of Actelion Pharmaceuticals Ltd. D.B. reports nonfinancial support and personal fees from Actelion Pharmaceuticals Ltd during the conduct of the study, and personal fees from Bayer Healthcare and Eli Lilly outside the submitted work.
The FUTURE‐3 investigators who contributed to the recruitment of patients in the studies are listed by country. Australia: Robert Weintraub MD, Suraj Varma MD: Royal Children's Hospital, Vic. Belarus: Jouri Chesnov MD: RSPC ‘Cardiology’, Minsk. China: Zhuoming Xu MD: Shanghai Children's Medical Center, Shanghai; Zhiwei Zhang MD: Guangdong General Hospital, Guangzhou; Xin Wu MD: Fuwai Hospital, Beijing. Czech Republic: Václav Chaloupecký MD: Faculty Hospital Motol Prague, Prague. France: Damien Bonnet MD: Hospital Necker Cardiology, Paris; Yves Dulac MD: Hospital Paediatric, Toulouse. Germany: Christian Apitz MD: Universitätsklinikum Giessen, Giessen; Johannes Breuer MD: Uniklinik Bonn, Bonn. Hungary: András Szatmári MD: Gottsegen Gyorgy National Cardiology Institute, Budapest; Márta Katona MD: University of Szeged Clinical Center, Szeged. India: Nageswara Rao Koneti MD: Care Hospital, Hyderabad. Israel: Einat Birk MD: Schneider Child Medical Center Institute, Petach Tikvah. Italy: Giacomo Pongiglione MD: Hospital Bambino Gesu, Rome. Mexico: Tomás René Pulido Zamudio MD: Instituto Nacional de Cardiologia, Mexico City. Poland: Robert Sabiniewicz MD: Cardiology Paediatric Gdańsk University, Gdańsk; Małgorzata Raś MD: Cardiology Paediatric Wrocław State Hospital, Wrocław. Russian Federation: Anna Galustyan MD: University Hospital Clinical Diagnostics Department, St Petersburg; Olga Moiseeva MD: University Hospital Cardiosurgery Department, St Petersburg; Irina Leontieva MD: University Hospital Congenital & Inherited Heart Diseases Department, Moscow. Serbia: Vojislav Parezanović MD: University Children's Hospital, Belgrade; Vladislav Vukomanović MD: Mother and Child Health Care Institute, Belgrade. South Africa: Stephen Brown MD: UFS Paediatric Cardiology, Bloemfontein; Luthando Adams MD: Paediatric Cardiology Steve Biko Academic Hospital, Pretoria. Spain: Maria Jesús Del Cerro MD: Hospital Uni La Paz, Madrid; Antonio Moreno MD: Hospital Uni Vall d'Hebron, Barcelona. USA: Erika Rosenzweig MD: Columbia Medical Center Children's Hospital of New York, New York; Dunbar Ivy MD: Children's Hospital Colorado, Aurora. Ukraine: Vladimir Zhovnir MD: Gover Ins‐Scienti Practcl, Cardiosur Paediatric Center, Kiev.
We thank Jasper Dingemanse PhD, PharmD, FCP (Actelion Pharmaceuticals Ltd) and Laurent Nicolas PhD for their valuable contribution to the study design and for study oversight. Actelion Pharmaceuticals Ltd funded the study and participated in the design of the study, data analysis, interpretation and preparation of the manuscript. Inovalab, Reinach, Switzerland performed some of the bioanalytical analyses. Medical writing assistance was provided by Stephanie Carter of nspm ltd, Meggen, Switzerland and funded by Actelion Pharmaceuticals Ltd.
Contributors
All authors contributed to the study design and/or data analysis and interpretation, critical review and final approval of the manuscript and have agreed to be accountable for the accuracy and integrity of all aspects of the work in the manuscript.
Supporting information
Figure S1 Comparison of bosentan exposures per treatment group. Overall age group; pharmacokinetic set. Geometric means of each dataset are shown as horizontal bars. AUC0–24C, area under the concentration–time curve from 0 to 24 h; b.i.d., twice daily; t.i.d., three times daily
{kind=link}
Table S1 List of independent ethics committees and institutional review boards
Table S2 Summary of adverse events up to the end of treatment +7 days
Berger, R. M. F. , Gehin, M. , Beghetti, M. , Ivy, D. , Kusic‐Pajic, A. , Cornelisse, P. , Grill, S. , Bonnet, D. , and the FUTURE‐3 investigators (2017) A bosentan pharmacokinetic study to investigate dosing regimens in paediatric patients with pulmonary arterial hypertension: FUTURE‐3. Br J Clin Pharmacol, 83: 1734–1744. doi: 10.1111/bcp.13267.
Steering Committee FUTURE‐3: M. Beghetti, R. M. F. Berger, D. Bonnet and D. Ivy
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44: D1054–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simonneau G, Gatzoulis M, Adatia I. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62: D34–D41. [DOI] [PubMed] [Google Scholar]
- 4. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004; 351: 1655–1665. [DOI] [PubMed] [Google Scholar]
- 5. Barst RJ, Ertel SI, Beghetti M, Ivy DD. Pulmonary arterial hypertension: a comparison between children and adults. Eur Respir J 2011; 37: 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berger RM, Haworth SG, Bonnet D, Dulac Y, Fraisse A, Galiè N, et al. FUTURE‐2: results from an open‐label, long‐term safety and tolerability extension study using the pediatric FormUlation of bosenTan in pUlmonary arterial hypeRtEnsion. Int J Cardiol 2016; 202: 52–58. [DOI] [PubMed] [Google Scholar]
- 7. Beghetti M, Berger RM. The challenges in paediatric pulmonary arterial hypertension. Eur Respir Rev 2014; 23: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beghetti M, Haworth SG, Bonnet D, Barst RJ, Acar P, Fraisse A, et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE‐1 study. Br J Clin Pharmacol 2009; 68: 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 2003; 73: 372–382. [DOI] [PubMed] [Google Scholar]
- 10. Berger RM. Pulmonary hypertension: smaller kids, smaller steps. Lancet Respir Med 2014; 2: 348–350. [DOI] [PubMed] [Google Scholar]
- 11. Vorhies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Paediatr Drugs 2013; 16: 43–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tracleer® (bosentan) . Summary of product characteristics, February 2016.
- 13. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin‐receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo‐controlled study. Lancet 2001; 358: 1119–1123. [DOI] [PubMed] [Google Scholar]
- 14. Galiè N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double‐blind, randomized, placebo‐controlled study. Circulation 2006; 114: 48–54. [DOI] [PubMed] [Google Scholar]
- 15. Humbert M, Barst RJ, Robbins IM, Channick RN, Galiè N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE‐2. Eur Respir J 2004; 24: 353–359. [DOI] [PubMed] [Google Scholar]
- 16. Galiè N, Rubin L, Hoeper M, Jansa P, Al‐Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double‐blind, randomised controlled trial. Lancet 2008; 371: 2093–2100. [DOI] [PubMed] [Google Scholar]
- 17. Dingemanse J, van Giersbergen PL. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin Pharmacokinet 2004; 43: 1089–1115. [DOI] [PubMed] [Google Scholar]
- 18. Rosenzweig EB, Ivy DD, Widlitz A, Doran A, Claussen LR, Yung D, et al. Effects of long‐term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol 2005; 46: 697–704. [DOI] [PubMed] [Google Scholar]
- 19. Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart 2006; 92: 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hislop AA, Moledina S, Foster H, Schulze‐Neick I, Haworth SG. Long‐term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur Respir J 2011; 38: 70–77. [DOI] [PubMed] [Google Scholar]
- 21. van Loon RL, Hoendermis ES, Duffels MG, Vonk‐Noordegraaf A, Mulder BJ, Hillege HL, et al. Long‐term effect of bosentan in adults versus children with pulmonary arterial hypertension associated with systemic‐to‐pulmonary shunt: does the beneficial effect persist? Am Heart J 2007; 154: 776–782. [DOI] [PubMed] [Google Scholar]
- 22. Dell D, Lausecker B, Hopfgartner G, van Giersbergen P, Dingemanse J. Evolving bioanalytical methods for the cardiovascular drug bosentan. Chromatographia 2002; 55 (Suppl): S115–S119. [Google Scholar]
- 23. Gehin M, Sidharta PN, Dingemanse J. Bosentan pharmacokinetics in pediatric patients with pulmonary arterial hypertension: comparison of dried blood spot and plasma analysis. Pharmacology 2016; 98: 111–114. [DOI] [PubMed] [Google Scholar]
- 24. Gutierrez MM, Nicolas LB, Donazzolo Y, Dingemanse J. Relative bioavailability of a newly developed pediatric formulation of bosentan vs. the adult formulation. Int J Clin Pharmacol Ther 2013; 51: 529–536. [DOI] [PubMed] [Google Scholar]
- 25. Ivy D, Abman SH, Barst RJ. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62: D117–D126. [DOI] [PubMed] [Google Scholar]
- 26. Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, et al. A randomized, double‐blind, placebo‐controlled, dose‐ranging study of oral sildenafil citrate in treatment‐naive children with pulmonary arterial hypertension. Circulation 2012; 125: 324–334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of bosentan exposures per treatment group. Overall age group; pharmacokinetic set. Geometric means of each dataset are shown as horizontal bars. AUC0–24C, area under the concentration–time curve from 0 to 24 h; b.i.d., twice daily; t.i.d., three times daily
Table S1 List of independent ethics committees and institutional review boards
Table S2 Summary of adverse events up to the end of treatment +7 days