

Abstract
Aims
The safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of single and multiple doses of a novel mGlu2 agonist were assessed in healthy males.
Methods
In two, Phase 1 investigator‐ and subject‐blind, placebo‐controlled studies, oral doses of prodrug LY2979165 were evaluated: single doses (20–150 mg, N = 30) and multiple once‐daily (QD) doses (20–400 mg; N = 84), using a titration regimen. The plasma and urine PK of LY2979165 and active moiety, 2812223, were measured. Cerebrospinal fluid (CSF) was collected to determine PK and neurotransmitter levels. Safety parameters were assessed throughout.
Results
Nausea and vomiting were dose limiting following single doses; dose titration allowed higher doses to be tested over 14 days. The most common adverse events related to LY2979165 were dizziness, vomiting, nausea, somnolence and headache. The plasma PK of 2812223 were approximately linear with minimal accumulation with QD dosing. Conversion of LY2979165 to 2812223 was extensive, with minimal LY2979165 measurable in plasma. There was no effect of food on the PK of LY2979165 and 2812223. After 60 mg LY2979165 single‐dose, 2812223 exposure in CSF was approximately 2–6% and plasma exposure and peak concentrations were approximately four‐fold higher than the mGlu2 agonist in vitro EC50 value. No consistent effects were observed on CSF neurotransmitter levels.
Conclusions
Oral doses of LY2979165 up to 60 mg as a single dose and up to 400 mg given as multiple QD doses, using a titration regimen, were well tolerated with linear PK. Overall, these data support further clinical evaluation of LY2979165.
Keywords: 2812223, CSF, LY2979165, pharmacodynamics, pharmacokinetics, safety
What is Already Known about this Subject
Glutamate is the principal excitatory neurotransmitter in the mammalian central nervous system.
LY2979165 is the alanine prodrug of 2812223, a selective orthosteric mGlu2 receptor agonist, which suppresses glutamate release at presynaptic receptors during periods of enhanced, abnormal, glutamate activity and has potential for use in treating psychiatric and neurologic disorders.
What this Study Adds
First clinical safety, PK and PD data for selective mGlu2 agonist, 2812223, supporting titration regimen to overcome tolerability limitations (e.g. vomiting).
Plasma PK of 2812223 was approximately linear, and there was minimal accumulation with once‐daily dosing.
The CSF levels of 2812223 exceeded mGlu2 agonist in vitro EC50 at LY2979165 doses of 60 and 150 mg.
Table of Links
| TARGETS |
|---|
| G protein‐coupled receptors 2 |
| Metabotropic Glutamate Receptor http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2534 |
Introduction
Glutamate serves as the principal excitatory neurotransmitter in the mammalian central nervous system 3, 4, and aberrant glutamate neurotransmission has been implicated in both psychiatric and neurologic disorders 3, 5, 6. Metabotropic glutamate (mGlu) receptors are plasma membrane‐associated G‐protein‐coupled proteins that function via presynaptic, postsynaptic, and glial mechanisms to modulate neuronal excitability 4, 7. The mGlu2 and mGlu3 receptors are heavily distributed within cortical and limbic structures of the brain (e.g. prefrontal cortex, hippocampus and amygdala) and show a predominant, though not exclusive, cellular localization on presynaptic nerve terminals 4, 8, 9. Activation of mGlu2 and mGlu3 receptors by glutamate or by other exogenous agonists leads to the inhibition of synaptic glutamate release, thereby dampening downstream postsynaptic excitation 3. Consequently, mGlu2/3 receptor agonists have demonstrated efficacy in animal models of hyper‐glutamatergic tone (e.g. states associated with stress/anxiety or psychosis) 7. Clinical evaluation of two balanced mGlu2/3 receptor agonists has been reported, one in generalized anxiety disorder and one in schizophrenia, respectively 10, 11. However, a recent report showed a lack of efficacy with pomaglumetad methionil (a methionine prodrug of LY404039, an mGlu2/3 agonist) for the treatment of schizophrenia in two clinical studies assessing the efficacy in reducing psychopathology either as a monotherapy in patients with an acute psychotic exacerbation (HBBM) or as an add‐on therapy in patients with prominent negative symptoms (HBCO) 12.
LY2979165 (Figure 1A) is the alanine prodrug of 2812223 (Figure 1B), a selective and potent orthosteric mGlu2 receptor agonist 7. A GTP‐γ‐[35S] functional binding assay was used to evaluate 2812223 mGlu2 pharmacology in human cortical brain tissue samples where it was found to be a partial mGlu2 agonist with an in vitro EC50 value of 7.57 nM (unpublished data on file, Eli Lilly and Company 13, Indianapolis, IN, USA). It has shown activity in the phencyclidine‐evoked ambulation model in rats, an animal model of psychosis known to be sensitive to mGlu2‐receptor (and not to mGlu3‐receptor) activation 7. Two clinical studies were conducted to assess the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of LY2979165 and 2812223 following single and multiple doses of oral LY2979165 in healthy subjects to support future studies in patients with psychiatric conditions. The measurement of biogenic amines was included as an exploratory biomarker of central PD activity based on preclinical changes observed with LY2979165 and LY404039 in rats 14, 15 and prior translatable effects in rats and humans with the balanced mGlu2/3 agonist pomeglumetad methionil 16.
Figure 1.
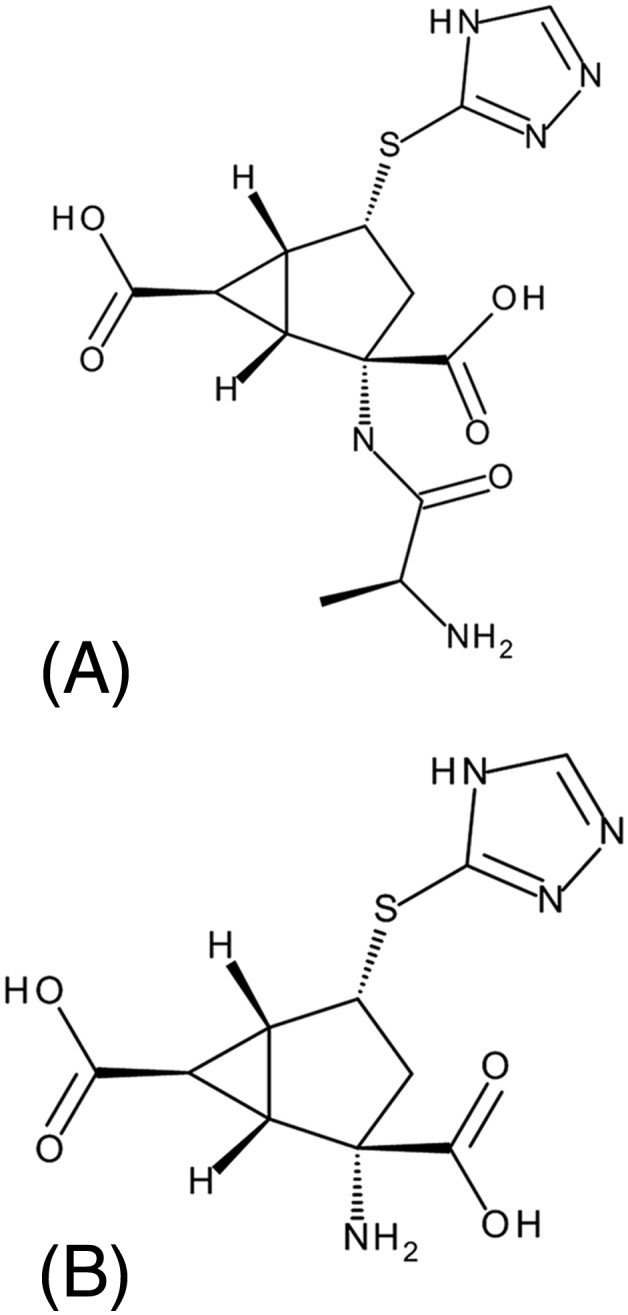
Chemical structures for (A) LY2979165 and (B) 2812223
Method
Study designs
The single ascending dose (SAD) study was a Phase 1, single‐centre, two‐part, investigator‐ and subject‐blind, randomized, placebo‐controlled study in healthy male subjects, exploring a dose range of 20–150 mg LY2979165 (Figure 2). Part A was a four‐period crossover (minimum 7‐day washout), dose‐escalation design, involving two alternating cohorts, each consisting of eight randomized subjects (six LY2979165 and two placebo at each dose level). Nausea and vomiting led to early termination of the dose escalation; therefore, a food effect evaluation was conducted during period 4, whereby LY2979165 was administered after a standardized breakfast. Subjects received either three doses of LY2979165 and one dose of placebo or four doses of LY2979165 across the four periods (if placebo was not administered in periods 1–3). Prior to dose escalation, all available safety data (adverse events [AEs], safety laboratory parameters, electrocardiograms [ECGs] and vital signs) were reviewed to determine the appropriate dose for the next study period. Part B was a parallel design involving a single cohort of 12 subjects, to assess the time course of plasma and CSF PK and CSF PD effects (neurotransmitter levels) of LY2979165 and 2812223. Subjects were randomized to a single dose of 60 mg LY2979165 (n = 8) or placebo (n = 4); a second permitted cohort was not enrolled. Safety and tolerability data were collected throughout the study.
Figure 2.
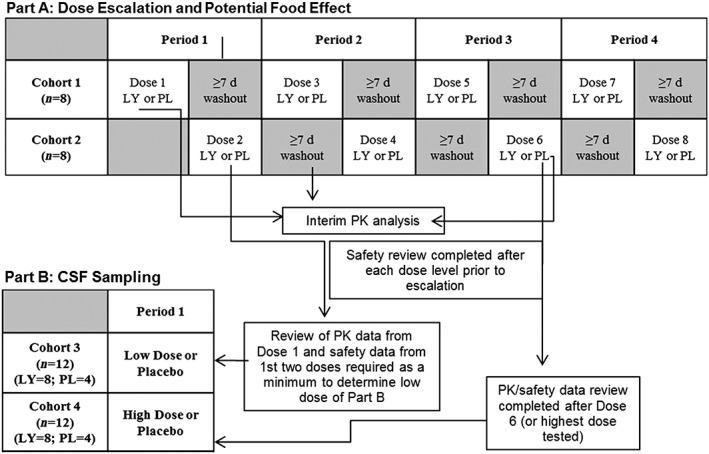
Study design: single ascending dose (SAD) study. d, days; LY, LY2979165; PK, pharmacokinetic; PL, placebo
The multiple ascending dose (MAD) study was a Phase 1, single‐centre, two‐part, investigator‐ and subject‐blind, randomized, placebo‐controlled, parallel design in healthy subjects exploring doses of 20–400 mg LY2979165 QD, using a dose titration regimen for doses of 60–400 mg to mitigate nausea and vomiting (Figure 3). The titration increments were 20, 60, 100, 150, 250 and 400 mg, with titration occurring at the cohort level. All subjects participated in a single 14‐day study period with daily dosing. Part A, the dose‐escalation phase, enrolled six cohorts (cohorts 1–4, 6 and 7), each consisting of 12 subjects (nine LY2979165, three placebo). Prior to dose escalation, all available safety data were reviewed to determine the appropriate dose for the next cohort. Part B enrolled a single cohort of 12 subjects (cohort 5) randomized to 150 mg QD LY2979165 (n = 9) or placebo (n = 3) who underwent a single lumbar puncture at baseline and on day 14 to explore CSF PK and PD. Safety and tolerability data were collected throughout the study.
Figure 3.
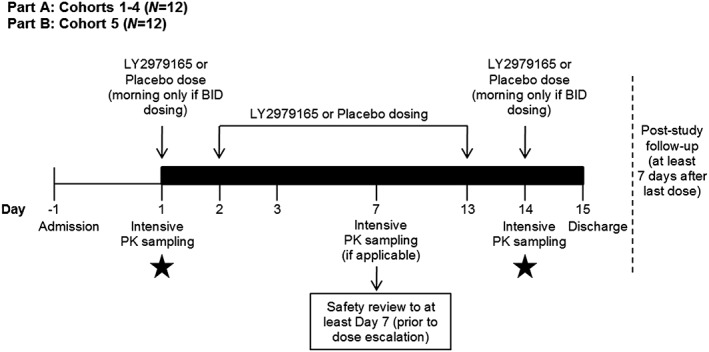
Study design: multiple ascending dose (MAD) study. LY2979165 to placebo 3:1. Black bar represents inpatient stay. In Part B, subjects may be permitted to leave the clinical research unit and return for dosing on an outpatient basis between days 4 and 13 (at the discretion of the investigator and sponsor). Black stars represent collection of CSF sample (Part B). BID, twice daily; n, number of subjects; PK, pharmacokinetic; CSF, cerebrospinal fluid
The sample sizes for both studies are customary for Phase 1 studies evaluating safety and PK parameters and are not powered on the basis of statistical hypothesis testing.
Both studies were conducted in Singapore. They were approved by the National Healthcare Group Domain Specific Review Board (NCT01248052, NCT01383967), and were conducted in accordance with all applicable regulatory and Good Clinical Practice guidelines and the Declaration of Helsinki. All subjects provided written informed consent prior to study participation.
Participants
Eligible subjects were overtly healthy males, between the ages of 21 and 65 years, with a body mass index ranging from 18.5 to 29.9 kg m−2, inclusive. The recruitment of subjects was based on medical history, screening evaluations (including ECG, clinical laboratory blood tests and blood pressure assessment) and physical examination.
Key exclusion criteria included any medical condition that may constitute a risk when taking the study medication or of interfering with the interpretation of data; history of significant allergy; increased risk of seizures; evidence of significant active neuropsychiatric disease; and use of concomitant medications. In addition, for part B of both studies, any back problems or contraindications to lumbar puncture, bleeding disorders, or allergy to local anaesthetics were pre‐specified exclusion criteria.
Study procedures
LY2979165 and placebo were administered as matching blue size 0 capsules in both studies. To maintain the blind, subjects receiving placebo received the same number of capsules as those receiving LY2979165 on the same day/period. All capsules were administered after an overnight fast of at least 8 h with approximately 200 ml water (except in period 4 of the SAD study). Subjects in period 4, received a standardized breakfast after an overnight fast, which was completed in 30 min or less, and LY2979165 (no placebo) was administered approximately 30 min after the start of breakfast.
Blood and CSF sampling
In the SAD study, plasma samples for PK analysis were taken pre‐dose and 0.25, 0.5, 1, 2, 3, 4, 6, 9, 12, 16, 24, 36 and 48 h post‐dose and urine was collected for the first 24 h post‐dose. In the MAD study, plasma samples were taken pre‐dose and 0.25, 0.5, 1, 2, 3, 4, 6, 9, 12, 16 and 24 h after dosing on days 1, 7 (cohort 1 only – no titration), and 14 (to 48 h) with some additional trough and peak samples on intermediate days.
In part B of both studies, subjects had CSF samples collected. In the SAD study, a lumbar catheter allowed sampling for pre‐dose (−2, −1 and 0), and 1, 2, 3, 4, 6, 9, 12, 16 and 24 h post‐dose, and in the MAD study, a single lumbar puncture was conducted at pre‐dose day 1 and on day 14 at the time of approximate maximum observed drug concentration (C max) in CSF from the SAD study. Samples were used to measure concentrations of LY2979165, 2812223, and PD (DOPAC [3,4‐dihydroxyphenylacetic acid], HVA [homovanillic acid], 5‐HIAA [5‐hydroxyindoleacetic acid], dopamine, l‐DOPA [l‐3,4‐dihydroxyphenylalanine], MHPG [3‐methoxy‐4‐hydroxyphenylglycol], 5‐HT [5 hydroxytryptamine] and VMA [vanillylmandelic acid]).
Blood samples were taken for safety laboratory assessments (haematology and clinical chemistry) at screening, pre‐dose day 1 throughout the clinical phase of the study, and at follow‐up (at least 7 days after final dose of study drug).
Bioanalytical methods
Plasma and urine PK
Plasma samples were analysed at Advion BioServices, Inc. (Advion, Ithaca, NY, USA). The samples were quantified for LY2979165 and 2812223 using a validated liquid chromatography/tandem mass spectrometry (LC/MS/MS) method.
The lower limit of quantitation (LLOQ) was 1 nM and the upper limit of quantitation (ULOQ) was 200 nM for both analytes in plasma, and the LLOQ was 200 nM and the ULOQ was 50 000 nM for both analytes in urine. Samples above the ULOQ were diluted to yield results within the calibrated range. For the initial 3‐day validation in plasma, the inter‐assay accuracy (% relative error) during validation ranged from −5.93% to 0.00% for LY2979165 and −7.74% to −4.00% for 2812223. The inter‐assay precision (% relative standard deviation) during validation was ≤7.00% for LY2979165 and ≤15.63% for 2812223. In urine the inter‐assay accuracy ranged from 4.1% to 11.5% for LY2979165 and −0.9% to 2.1% for 2812223. The inter‐assay precision (% relative standard deviation) was ≤5.1% for LY2979165 and ≤7.6% for 2812223.
CSF PK
The CSF PK samples were analysed using the same assay procedures as plasma. The LLOQ was 0.5 nM and the ULOQ was 50 nM for both analytes. For the initial 3‐day validation, the inter‐assay accuracy ranged from −1.13% to 10.20% for LY2979165 and 0.95% to 11.80% for 2812223. The inter‐assay precision was ≤6.17% for LY2979165 and ≤8.05% for 2812223.
CSF PD
DOPAC, HVA, 5‐HIAA, dopamine, L‐DOPA, MHPG, 5‐HT and VMA samples from LY2979165‐ and placebo‐treated subjects were analysed at Advion (Ithaca, NY, USA) using a validated method (Pharmanet TM.748). The method applied was adapted from the published work of Eckstein et al. 17 and involved gas chromatography/tandem mass spectrometry (GC/MS/MS) analysis.
Data analysis
Safety data
Safety parameters, including safety labs, vital signs and ECG parameters, were summarized descriptively.
Pharmacokinetics
PK parameters for LY2979165 and 2812223 were calculated by standard noncompartmental methods of analysis using WinNonlin version 5.3 and included area under the concentration–time curve from zero to time t last, where t last is the last time point with a measurable concentration (AUC(0–tlast)), area under the concentration curve from zero to infinity (AUC(0‐∞)) or area under the concentration–time curve during one dosing interval at steady state (AUC(τ,ss)), and C max after single or repeated doses. Concentrations of LY2979165 were low and only measurable at early time points; therefore, only limited PK parameters were calculated. Other parameters, such as time of maximal concentration (t max), apparent clearance, apparent volume of distribution, elimination half‐life, accumulation ratio, and linearity index were calculated, data permitting. Subjects who vomited on the PK assessment day were excluded from the analysis.
Statistics
Pharmacokinetics
An exploratory analysis of dose proportionality of plasma 2812223 PK was conducted for C max and AUC(0–∞)/AUCτ,ss using a power model 18 fitting loge (PK parameter) against loge (dose) and subject as a random effect. Predicted ratios of dose‐normalized means, starting with the highest dose/lowest dose tested and corresponding 95% confidence intervals (CIs), were estimated.
Food effect
In an exploratory analysis, the effect of food on dose‐normalized plasma 2812223 AUC(0‐∞) and C max was estimated by combining parameters for the 20 and 60 mg doses in the SAD study. Data were loge‐transformed and analysed using a mixed effect model, with fed/fasted condition as a fixed effect and subject as a random effect. Point estimates and corresponding 95% CIs were constructed for the comparisons of LY2979165 fed:fasted, and back‐transformed to provide point estimates and 95% CIs for the geometric mean ratios fed:fasted.
Pharmacodynamics
Statistical analysis of CSF PD concentrations was conducted using an analysis of covariance, comparing the overall mean difference in the concentration of neurotransmitters for each dose of LY2979165 to placebo using a 95% CI. The response was change from baseline (pre‐dose) in CSF PD and included a fixed‐effect term of treatment (placebo, LY2979165). For the MAD study, there was no covariate for time (only a single post‐dose time point collected); for the SAD study, time and the interaction of time with treatment were added as fixed effects. An unstructured correlation matrix was used. There were no adjustments for multiple testing. Note that CSF concentrations of 5‐HT were not analysed because concentrations were either below the limit of quantification or no valid result was determined for almost all samples.
Results
Disposition
The SAD study enrolled 30 subjects: 18 in part A (17 received at least one dose of LY2979165); 12 in part B (LY2979165 [n = 8], placebo [n = 4]). Twenty‐four subjects completed the study with six early withdrawals from part A. One subject was withdrawn due to nodal rhythm on ECG after receiving a single dose of placebo in period 1. Other withdrawals were due to subject decision (unavailable to complete study; n = 2), physician decision (n = 2; ventricular ectopics pre‐dose and elevated liver enzymes pre‐dose), and AE (vomiting following 150 mg LY2979165).
The MAD study enrolled 84 subjects, all of whom completed the study: 72 in part A (LY2979165 [n = 54], placebo [n = 18]) and 12 in part B (LY2979165 [n = 9], placebo [n = 3]).
Demographics
The demographics of each study population were well balanced between cohorts and treatment groups (Table 1). The mean age for the SAD and MAD study subjects was 37 (range 23–65) and 32 (range 22–59) years, respectively; mean body mass index was 22.9 (range 18.9–27.8) and 23.7 (range 18.8–29.2) kg m−2, respectively. The majority of subjects in each study were Asian (96.7% [29/30] SAD; 96.4% [81/84] MAD).
Table 1.
Subject demographics and clinical characteristics in the SAD and MAD studies
| SAD study | MAD study | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | (B) | (A&B) | (A) | (A) | (A) | (A) | (B) | (A) | (A) | ||||
| Cohort 1 (n = 9) | Cohort 2 (n = 9) | Cohort 3 (n = 12) | Overall (n = 30) | PL (n = 21) | 20 mg LY (n = 9) | 60 mg LY (n = 9) | 100 mg LY (n = 9) | 150 mg LY (n = 9) | 150 mg LY (n = 9) | 250 mg LY (n = 9) | 400 mg LY (n = 9) | Overall (n = 84) | |
| Age, years | 35.0 (11.7) | 32.6 (6.9) | 41.3 (13.5) | 36.8 (11.6) | 33.4 (7.0) | 32.9 (6.4) | 38.1 (10.5) | 29.1 (7.2) | 30.0 (9.1) | 34.9 (3.4) | 29.7 (6.1) | 27.1 (6.6) | 32.1 (7.6) |
| mean (SD) | |||||||||||||
| BMI, kg m −2 | 22.3 (3.0) | 23.2 (2.9) | 23.2 (1.8) | 22.9 (2.5) | 23.8 (2.8) | 25.1 (3.2) | 23.7 (2.9) | 22.6 (1.7) | 23.0 (1.6) | 25.5 (2.1) | 23.8 (2.5) | 22.1 (1.7) | 23.7 (2.6) |
| mean (SD) | |||||||||||||
| Race, % | |||||||||||||
| Asian | 100.0 | 100.0 | 91.7 | 96.7 | 95.2 | 88.9 | 100.0 | 100.0 | 100.0 | 100.0 | 88.9 | 100.0 | 96.4 |
| White | 0.0 | 0.0 | 8.3 | 3.3 | 4.8 | 11.1 | 0.0 | 0.0 | 0.0 | 0.0 | 11.1 | 0.0 | 3.6 |
(A), part A; (B), part B; BMI, body mass index; LY, LY2979165; MAD, multiple ascending dose; PL, placebo; SAD, single ascending dose; SD, standard deviation.
Safety
A summary of AEs is provided in Table 2. The SAD study results indicate that single oral doses of up to 60 mg LY2979165 were well tolerated; nausea and vomiting were dose limiting at higher doses. In the MAD study, doses up to 400 mg were well tolerated over 14 days, using a titration regimen. Dizziness, vomiting, nausea, headache and somnolence were the most commonly reported treatment‐emergent AEs across both studies. The majority of AEs (92% in SAD and 100% in MAD) were rated as mild in intensity; the moderate AEs were predominantly from part B of the SAD study and related to the indwelling lumbar catheter, with two events of vomiting in one subject in part A. In the MAD study, nausea and vomiting accommodated during titration, with the majority of events occurring on day 1. Dizziness did not show accommodation and appeared to increase with dose, particularly at the 150 and 250 mg dose levels. However, these data are presented at the cohort target dose rather than the actual dose received on the day of the event (during titration). Dizziness events presented by dose at the time of event rather than the cohort target dose suggest that there is less of a dose‐related effect, with similar incidences (4–15%) across placebo and 20, 60, 100, 150 and 400 mg LY2979165 (Table 3). The 250 mg dose stands out as an anomaly, suggesting a possible cohort effect of event reporting.
Table 2.
Summary of adverse events for the SAD and MAD studies
| SAD study | MAD study a | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | (A) | (A) | (A) | (A) | (A&B) | (A) | (A) | (A) | (A) | (B) | (A) | (A) | ||
| MedDRA preferred term | PL (n = 10) | 20 mg LY Fasted (n = 6) | 20 mg LY Fed (n = 6) | 60 mg LY Fasted (n = 5) | 60 mg LY Fed (n = 6) | PL (n = 21) | 20 mg LY (n = 9) | 60 mg LY (n = 9) | 100 mg LY (n = 9) | 150 mg LY (n = 9) | 150 mg LY (n = 9) | 250 mg LY (n = 9) | 400 mg LY (n = 9) | Overall (n = 84) |
| Dizziness | 2 (2) | 3 (3) | 2 (2) | 2 (2) | ‐ | 5 (3) | 4 (3) | 1 (1) | 3 (2) | 12 (4) | 11 (5) | 17 (8) | 3 (2) | 56 (28) |
| Procedure site reaction | 1 (1) | ‐ | ‐ | ‐ | 3 (2) | 10 (8) | 3 (3) | 4 (4) | 6 (3) | 6 (4) | 13 (6) | 5 (5) | 1 (1) | 48 (34) |
| Nausea | ‐ | 1 (1) | 1 (1) | 1 (1) | 2 (2) | 1 (1) | 4 (3) | 1 (1) | 2 (1) | 2 (1) | 10 (5) | 2 (2) | 1 (1) | 23 (15) |
| Headache | ‐ | ‐ | ‐ | ‐ | ‐ | 1 (1) | 3 (3) | ‐ | ‐ | ‐ | 4 (3) | 3 (2) | 1 (1) | 12 (10) |
| Somnolence | ‐ | ‐ | ‐ | ‐ | ‐ | 4 (3) | 3 (3) | ‐ | ‐ | 1 (1) | ‐ | 2 (1) | 2 (2) | 12 (10) |
| Vessel puncture site haematoma | ‐ | ‐ | ‐ | ‐ | 2 (1) | 2 (2) | ‐ | 1 (1) | 1 (1) | ‐ | 1 (1) | 4 (2) | 3 (1) | 12 (8) |
| Vomiting | ‐ | ‐ | ‐ | ‐ | 3 (2) | ‐ | 1 (1) | ‐ | 5 (2) | 2 (1) | 2 (2) | 2 (2) | ‐ | 12 (8) |
| Catheter site pain/swelling | ‐ | ‐ | 1 (1) | ‐ | 1 (1) | 2 (2) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | ‐ | 1 (1) | 8 (8) |
| Catheter site reaction | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 (1) | 2 (2) | 3 (2) | 2 (1) | ‐ | ‐ | 8 (6) |
| Cough | ‐ | ‐ | ‐ | ‐ | 1 (1) | 2 (2) | 2 (2) | 1 (1) | ‐ | ‐ | 1 (1) | 1 (1) | ‐ | 7 (7) |
| Back pain | ‐ | ‐ | ‐ | ‐ | ‐ | 1 (1) | ‐ | ‐ | 1 (1) | ‐ | 4 (4) | ‐ | ‐ | 6 (6) |
| Balance disorder | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 (1) | ‐ | 1 (1) | 3 (3) | 5 (5) |
| Dry throat | ‐ | ‐ | ‐ | ‐ | ‐ | 2 (2) | 3 (2) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 5 (4) |
| Feeling hot/pyrexia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 (1) | 1 (1) | ‐ | 2 (2) | 4 (4) |
| Oropharyngeal pain | ‐ | ‐ | ‐ | ‐ | ‐ | 2 (2) | ‐ | 1 (1) | ‐ | ‐ | ‐ | 1 (1) | ‐ | 4 (4) |
| Hypoaesthesia | ‐ | ‐ | 2 (1) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
(A), part A; (B), part B, LY, LY2979165; MAD, multiple ascending dose; MeDRA, Medical Dictionary for Regulatory Activities; PL, placebo; SAD, single ascending dose.
Data are reported as number of adverse events (number of subjects with adverse event).
In ≥2 subjects.
Table 3.
Dizziness events presented by dose level (MAD study)
| Number of events (number of subjects) | Number of subjects exposed (% reporting dizziness) | ||
|---|---|---|---|
| LY2979165 dose | By cohort target dose | By actual dose | By actual dose |
| PL | 3 (2) | 3 (2) | 18 (11%) |
| 20 mg | 4 (3) | 9 (7) | 54 (13%) |
| 60 mg | 1 (1) | 3 (2) | 45 (4.4%) |
| 100 mg | 3 (2) | 2 (2) | 36 (5.6%) |
| 150 mg | 12 (4) | 8 (4) | 27 (15%) |
| 250 mg | 17 (8) | 18 (9) | 18 (50%) |
| 400 mg | 3 (2) | 1 (1) | 9 (11%) |
MAD, multiple ascending dose; PL, placebo.
There were no serious adverse events (SAEs); one AE led to withdrawal following LY2979165 – vomiting (three episodes) following 150 mg in the SAD study. This subject had previously received 20 mg LY2979165, which resulted in AEs of nausea and dizziness.
During the course of both studies, there were no clinically relevant alterations in haematology, clinical chemistry, vital signs, urinalysis or ECG measurements.
Pharmacokinetics
Mean 2812223 plasma concentration–time profiles after single and multiple once‐daily doses are presented in Figure 4. Systemic uptake of prodrug, LY2979165, and conversion to 2812223 was rapid and extensive, as demonstrated by measurable 2812223 plasma concentrations within 30 min of dosing for the majority of subjects. Low concentrations of LY2979165 were detectable in plasma (<1% of 2812223 exposure). Plasma concentrations of 2812223 reached a maximum, on average 3–4 h after dosing, and declined thereafter in a bi‐exponential manner, with the majority of drug eliminated within the first 24 h.
Figure 4.
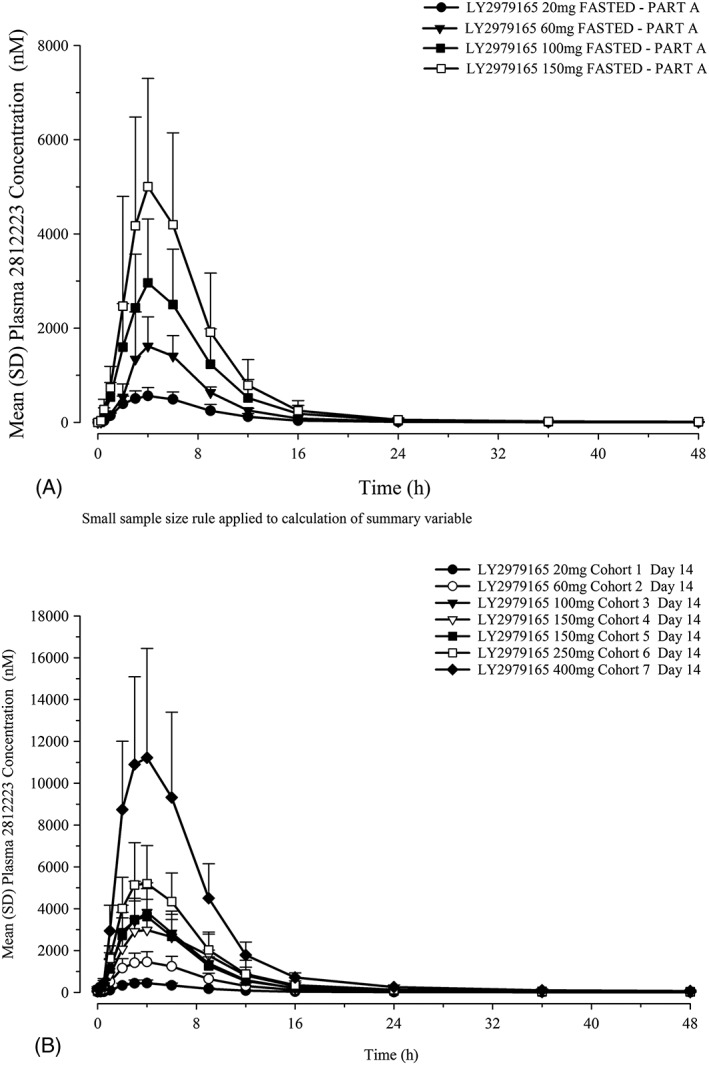
Mean 2812223 plasma concentration–time profiles: (A) SAD study with single doses of LY2979165 administered under fasted conditions and (B) MAD study on day 14. MAD, multiple ascending dose; SAD, single ascending dose; SD, standard deviation
A summary of 2812223 plasma PK parameters (Table 4) on day 1 (SAD) and day 14 (MAD) and the statistical analysis results (Table 5) are presented. In general, 2812223 exposure (C max and AUC) increased as dose increased and did not show a large departure from linearity over the dose range studied, with ratios of dose‐normalized geometric means close to 1. Minimal accumulation of 2812223 was observed with repeated once‐daily dosing. The plasma PK parameters for 2812223 were not substantially different when LY2979165 was dosed fed or fasted (Table 4). The mean fraction of the dose excreted (fe) as 2812223 in the urine over 24 h was 44–62% across the dose groups, with an overall geometric mean of 53% in the SAD study.
Table 4.
Summary of 2812223 plasma pharmacokinetic parameters after single dose (SAD study, day 1) and multiple dose (MAD study, day 14) LY2979165
| SAD study a | MAD study | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasted | Fasted | Fasted | Fasted | Fed | Fed | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | |
| 20 mg LY (n = 6) | 60 mg LY (n = 5) | 100 mg LY (n = 6) | 150 mg LY (n = 7) | 20 mg LY (n = 6) | 60 mg LY (n = 4) | 20 mg LY (n = 9) | 60 mg LY (n = 9) | 100 mg LY (n = 9) | 150 mg LY (n = 9) | 150 mg LY (n = 9) | 250 mg LY (n = 9) | 400 mg LY (n = 9) | |
| t max b (h) | 4.00 (3.00–6.00) | 4.00 (3.00–6.23) | 4.00 (3.00–6.00) | 4.00 (2.00–6.00) | 4.00 (3.00–6.00) | 5.00 (4.00–6.00) | 4.00 (3.00–4.08) | 4.00 (2.00–6.00) | 4.00 (3.00–4.00) | 4.00 (3.00–9.00) | 4.00 (3.00–4.00) | 3.00 (3.00–6.00) | 4.00 (3.00–6.00) |
| C max (nM) | 588 (27) | 1580 (46) | 2790 (44) | 4600 (57) | 621 (43) | 1800 (21) | 433 (31) | 1500 (26) | 3640 (39) | 3260 (44) | 3440 (48) | 5340 (30) | 10 900 (42) |
| AUC c (nM h −1 ) | 4340 (31) | 10 900 (28) | 20 200 (47) | 31 700 (54) | 3960 (54) | 11 200 (14) | 3240 (26) | 11 300 (34) | 25 400 (40) | 25 000 (36) | 24 100 (41) | 38 600 (28) | 80 100 (36) |
| t 1/2 (h) | 9.91 (47) | 12.5 (68) | 17.8 (44) | 14.1 (46) | 8.42 (47) | 8.83 (25) | 11.0 (35)d | 10.6 (24) | 9.0 (12) | 10.0 (24) | 10.9 (21)e | 11.4 (3.8) | 10.9 (28) |
AUC, area under the concentration–time curve; C max, maximum observed drug concentration; CV, coefficient of variation; LY, LY2979165; MAD, multiple ascending dose; SAD, single ascending dose; t 1/2, terminal elimination half‐life; t max, time of maximum observed drug concentration.
Data are presented as geometric means (CV%).
Subjects who vomited are excluded.
Median (minimum − maximum).
AUC(0‐∞), area under the concentration versus time curve from zero to infinity reported for the SAD study; AUCτ,ss, area under the concentration versus time curve during 1 dosing interval at steady state reported for the MAD study.
n = 7, Subjects 104 and 105 are not included in calculation of summary statistics as % AUC extrapolated exceeded 20%.
n = 8, Subject 502 is not included in calculation of summary statistics as % AUC extrapolated exceeded 20%.
Table 5.
Statistical results for dose proportionality assessment of plasma 2812223 in single (SAD study) and multiple dose (MAD study) LY2979165
| Dose range (mg) | PK parameter | Mean slope | Ratio of dose‐normalized geometric means (95% CI) | CV (%) |
|---|---|---|---|---|
| SAD study | ||||
| 20 to 150a | AUC(0‐∞), nM h−1 | 1.09 | 1.19 (0.92, 1.55) | 14.0 |
| C max, nM | 1.11 | 1.25 (0.92, 1.69) | 16.5 | |
| 20 to 100 | AUC(0‐∞), nM h−1 | 1.08 | 1.13 (1.07, 1.19) | 1.7 |
| C max, nM | 1.09 | 1.15 (0.99, 1.34) | 5.1 | |
| Fed (n = 10) vs Fasted (n = 11) | AUC(0‐∞), nM h−1 | — | 1.05 (0.88, 1.26) | — |
| Fed (n = 10) vs Fasted (n = 11) | C max, nM | — | 1.21 (1.00, 1.47) | — |
| MAD study | ||||
| 20 to 400 | AUCτ,ss, nM h−1 | 1.01 | 1.04 (0.77, 1.40) | 37.6b |
| C max, nM | 1.02 | 1.07 (0.77, 1.48) | 40.9b | |
AUC(0‐∞), area under the concentration–time curve from zero to infinity; AUCτ,ss, area under the concentration–time curve during one dosing interval at steady state; CI, confidence interval; C max, maximum observed concentration; CV, coefficient of variation (within subject variability only); MAD, multiple ascending dose; PK, pharmacokinetic; SAD, single ascending dose.
Model for dose proportionality assessment: log(PK) = log(dose) + subject. Model for fed vs. fasted comparison: Model: log(pk) = condition(fed or fasted) + subject; note: fed/fasted model includes dose‐normalized PK parameters for 20 mg and 60 mg doses.
Dose range (20–150 mg) is the complete dose range studied in the fasted condition.
Between subject.
In the SAD study, CSF concentrations of 2812223 were detectable for the duration of the 24‐h sampling period after a 60 mg dose, with mean C max of 29.8 nM (coefficient of variation [CV] 98%) (Figure 5). Exposure to 2812223 in CSF was lower than in plasma, with the ratio of CSF:plasma ranging from approximately 0.02 to 0.06 (Table 6). In the MAD study, at approximately 7–8 h post‐dose on day 14, the mean 2812223 CSF concentration was 87 nM (range: 38–141 nM) and was moderately variable (CV 49%).
Figure 5.
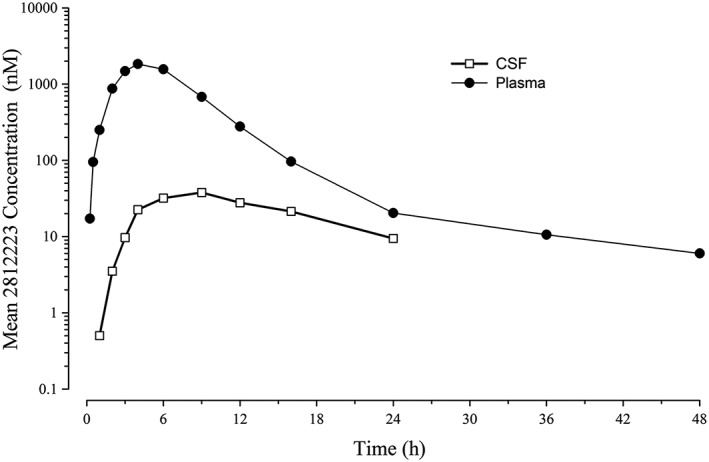
2812223 arithmetic mean plasma and CSF concentration‐time profiles following administration of a single 60 mg dose of LY2979165 under fasted conditions (SAD study). CSF, cerebrospinal fluid; SAD, single ascending dose
Table 6.
Summary of 2812223 plasma and CSF PK parameters following administration of a single 60 mg dose of LY2979165 under fasted conditions (SAD study) and following 150 mg QD dosing on day 14 (MAD study)
| Plasma | CSF | |
|---|---|---|
| SAD study | ||
| n | 6 | 6 |
| C max , nM | 1740 (57) | 29.8 (98) |
| AUC (0–24) , nM hr −1 | 11 400 (54) | 342 (86)a |
| t max b , h | 3.5 (3.0–6.0) | 9.1 (6.1–12.1) |
| CSF:Plasma ratio | 0.02:0.06 | |
| MAD study | ||
| n | 8 | 8 |
| Single concentration on day 14 (7–8 h post‐dose), nM c | 2808 (39.9) | 87.1 (49.3) |
AUC(0–24), area under the concentration–time curve from time zero to 24 h; C max, maximum observed drug concentration; CSF, cerebrospinal fluid; CV, coefficient of variation; MAD, multiple ascending dose; n, number of subjects; PK, pharmacokinetic; QD, once daily; SAD, single ascending dose; t max, time of maximum observed drug concentration.
Data are provided as geometric mean (CV%).
n = 5, Subject 303 is not included in calculation of summary statistics.
Median (minimum − maximum).
Plasma PK samples were taken at approximately 6 h post‐dose, the nearest time point to the CSF sampling time for the majority of subjects.
Pharmacodynamics
In the SAD study, there was no strong evidence of a differential drug effect on DOPAC and HVA up to 24 h post‐dose (Table 7). Significant (P < 0.2) differences in l‐DOPA concentrations (0.7–2.2 ng ml−1) were seen between LY2979165 60 mg and placebo from 4 to 24 h post‐dose. Significant (P < 0.2) reductions were seen in VMA concentrations for LY2979165 60 mg compared to placebo at 1–2 and 12–24 h post‐dose (−0.19–0.04 ng ml−1). Sensitivity analysis, including the two subjects who vomited, did not show any statistically significant changes on VMA concentrations when dosing with LY2979165. In the MAD study, only VMA showed a trend in the individual data between baseline and day 14 (Table 7). This was consistent with the SAD study as described above – in general, placebo subjects demonstrated an increase and LY2979165 subjects demonstrated a decrease.
Table 7.
Statistical analysis of change from baseline in neurotransmitter levels in CSF on day 1 (SAD study) and day 14 (MAD study)
| SAD study | MAD study | |||||||
|---|---|---|---|---|---|---|---|---|
| Day 1 a | Day 14 | |||||||
| Neurotransmitter (ng ml −1 ) | Test LS mean | Reference LS mean placebo | Estimate of the difference (95% CI) | P‐value | Test LS mean 150 mg | Reference LS mean placebo | Estimate of the difference (95% CI) | P‐value |
| 5‐HIAA | 2.631 | ‐1.170 | 3.80 (−5.373, 12.975) | 0.3960 | ‐0.740 | 0.082 | ‐0.822 (−4.887,3.243) | 0.6535 |
| DOPAC | 0.044 | 0.000 | 0.04 (−0.127, 0.214) | 0.6055 | ‐0.049 | ‐0.035 | ‐0.014 (−0.329,0.302) | 0.9211 |
| Dopamine | ‐0.189 | ‐0.464 | 0.28 (−0.035, 0.586) | 0.0783 | ‐0.249 | ‐0.293 | 0.044 (−0.104, 0.192) | 0.5105 |
| HVA | 7.251 | 0.881 | 6.37 (−17.82, 30.563) | 0.5867 | ‐4.179 | ‐0.987 | ‐3.191 (−13.943, 7.561) | 0.5130 |
| L‐DOPA | ‐0.283 | ‐2.333 | 2.05 (0.096, 4.004) | 0.0407 | ‐0.846 | ‐0.140 | ‐0.705 (−5.354, 3.943) | 0.7354 |
| MHPG | ‐8.889 | ‐2.109 | ‐6.78 (−31.77, 18.211) | 0.5761 | NC | NC | NC | NC |
| VMA | ‐0.062 | ‐0.004 | ‐0.06 (−0.324, 0.207) | 0.6586 | ‐0.042 | 0.077 | ‐0.119 (−0.206, −0.032)b | 0.0134 |
CI, confidence interval; CSF, cerebrospinal fluid; DOPAC, 3,4‐dihydroxyphenylacetic acid; 5‐HIAA, 5‐hydroxyindoleacetic acid; HVA, homovanillic acid; L‐DOPA, 3,4‐dihydroxyphenylalanine; LS, least squares; MHPG, 3‐methoxy‐4‐hydroxyphenylglycol; MAD, multiple ascending dose; NC, not calculated; SAD, single ascending dose; VMA, vanillylmandelic acid.
Excluding outliers (defined as standardized residual <−3.5 or >3.5) and vomiters (n = 2).
Confidence interval excludes zero.
Discussion
These studies provide safety, tolerability, PK and PD data for a novel mGlu2 agonist, 2812223 in healthy male subjects following administration of single and multiple doses of the prodrug, LY2979165. No safety signals were identified in these studies; dose escalation in the SAD study was restricted beyond 150 mg by tolerability, specifically vomiting. The maximum tolerated dose was not identified in the MAD study; the highest dose, 400 mg, was generally well tolerated using a dose titration regimen starting at 20 mg LY2979165.
Dizziness, vomiting, nausea, headache and somnolence were the most commonly reported TEAEs across the studies, which is consistent with the pattern seen with mGlu2/3 agonists (e.g. LY2140023 19) administered to healthy subjects. When vomiting occurred, it was typically following the first dose and did not recur with continued daily dosing. The time of vomiting relative to dosing was not consistent among subjects. Several subjects vomited within an hour of LY2979165 administration when 2812223 concentrations were low or unquantifiable, which could suggest a direct effect on the gastrointestinal tract rather than a centrally mediated effect. However, it was also accompanied by dizziness, with a similar onset time, in most subjects. In other subjects, the time of vomiting was close to the time of maximal 2812223 concentrations and could be suggestive of a centrally mediated drug effect. Vomiting on day 1 in the MAD study occurred in 7 of the 63 subjects (11%) with the 20 mg dose and was mostly a single episode (one subject vomited twice on day 1 and once on day 2, but there was no further recurrence with continued dose titration). Again, exposure to 2812223 did not show a consistent relationship to vomiting; some who vomited had lower exposure and others with similar exposure did not vomit. Together, these data suggest that vomiting is typically a first‐dose effect, independent of drug exposure; however, they do not address any potential dose effect as all subjects started titration at 20 mg LY2979165. It is not possible to predict whether the incidence of vomiting after a starting dose of 20 mg LY2979165 in this study would be maintained if a higher starting dose is used in future studies, as there is no clear relationship to 2812223 exposure. From the data generated in these studies, no clear association or predictor has been identified for the nausea and vomiting. Importantly, the use of a titration regimen allows the nausea and vomiting to be overcome, thus permitting exploration of an appropriate dose range for any future studies.
In the SAD study, administration of LY2979165 in the fed state did not appear to improve the tolerability, and in some cases, the tolerability following 60 mg LY2979165 was worse in the fed state (no subjects vomited after 60 mg in the fasted state, but two vomited when 60 mg was administered in the fed state). However, this could be an impact of timing and prior doses of LY2979165 because the fed assessment was in period 4 when all subjects had received LY2979165 in at least two previous periods. For example, some subjects in the SAD study did not vomit after 150 mg during period 2 but vomited after 100 mg LY2979165 during period 3.
The reporting of dizziness in the MAD study increased with dose, particularly at 250 mg LY2979165; however, when the data are presented according to the actual dose taken on the day of the event, the incidences across the doses are in a similar range (4–15%) with the exception of the 250 mg dose. This suggests a potential cohort effect rather than a true dose effect. For cohort 6 (250 mg), eight of the nine subjects who received LY2979165 reported dizziness after reaching the 250 mg dose, whereas in cohort 7 (400 mg), one subject reported dizziness after 250 mg and one subject after 400 mg.
Systemic uptake of LY2979165 and conversion into the active compound, 2812223, was rapid, with measurable 2812223 plasma concentrations within 30 min of dosing in the majority of subjects. Conversion of LY2979165 to 2812223 was extensive, as very little prodrug (generally <1% of 2812223) was detected in the plasma. After single doses or 14 days daily dosing, plasma concentrations of 2812223 reached a maximum on average 3–4 h after dosing and declined thereafter in a bi‐exponential manner, with the majority of drug eliminated within the first 24 h post‐dose. Variability in exposure was moderate‐to‐high and mean half‐life estimates ranged from 9 to 17 h across dose groups and studies. Exposure increased as dose increased and was dose proportional across the entire dose range (20–400 mg) in the MAD study. In the SAD study over a smaller dose range, exposure increased as dose increased, but did not meet statistical criteria for dose proportionality. The ranges of dose‐normalized C max and AUC estimates overlapped for each dose group without a consistent increase or decrease in the mean estimates as dose increased, suggesting there is no large departure from linearity from 20 to 150 mg. Overall, the data suggest that the PK of 2812223 are approximately dose proportional from 20 to 400 mg.
A preliminary assessment of the effect of food on the PK of LY2979165 and 2812223 was made at doses of 20 and 60 mg in the SAD study. Concentrations of LY2979165 remained low under fed conditions. Mean 2812223 exposures were similar, and there was no delay in t max when LY2979165 was administered under fed conditions, suggesting that food does not have a large impact on the PK of 2812223.
CSF exposure to 2812223 was achieved in all subjects in the SAD study (and the MAD study single time point), with concentrations still quantifiable at the last time point (24 h post‐dose) in all subjects. The time to achieve maximal concentrations was longer than in plasma (6–12 h vs. 3–6 h), although the relatively flat CSF profile shows that good exposure levels were achieved within 3–4 h for the majority of subjects. The ratio of 2812223 in CSF compared with plasma was approximately 2 to 6%. In the SAD study after a 60 mg dose of LY2979165, mean maximal CSF concentrations exceeded the in vitro EC50 (7.57 nM) by approximately four‐fold and mean concentrations exceeded the EC50 for the entire 24‐hour measurement period. In the MAD study after a 150 mg dose of LY2979165, the mean CSF concentration of 2812223 was approximately 11‐fold higher than the in vitro EC50 value. LY2979165 was essentially not quantifiable in the CSF. Overall, the PK data for 2812223 following administration of LY2979165 support administration of once‐daily LY2979165 in future clinical studies.
There were no statistically significant changes in the levels of DOPAC and HVA when the 60 mg LY2979165 dose group was compared to placebo in the SAD study. These findings are in contrast to the findings with the balanced mGlu2/3 agonist pomeglumetad methionil, where significant increases were seen for DOPAC, HVA, 5‐HIAA and MHPG in the CSF of subjects receiving pomeglumetad, but not placebo 16. However, variability in placebo data was high, and combined with the small numbers of subjects studied and the lack of intra‐subject comparisons, may have precluded determination of a statistical response. In the SAD study, potential trends were observed in CSF concentrations of l‐DOPA and VMA; however, there was no strong evidence of a differential drug effect for l‐DOPA at steady state. Glutamate and glutamine were not measured in CSF in these studies, but there are published studies showing that ketamine increases glutamate and/or glutamine levels in the human brain in response to a ketamine challenge 20, 21, and one could hypothesize that treatment with an mGlu agonist would normalize this (as shown in rodent models using microdialysis) 22. A clinical study using a ketamine‐challenge pharmacological magnetic resonance imaging paradigm in healthy volunteers has been conducted with LY2979165 and pomaglumetad methionil and is the subject of a manuscript that will be submitted in the near future 23.
An adequate positron emission tomography tracer does not exist for orthosteric mGlu2(3) receptor agonists, resulting in target engagement for this mechanism relying upon measurements of central PD effects 16. Clinical experience with mGlu2(3) agonists suggests that central exposures (i.e. CSF drug levels) need to at least achieve the mGlu2 EC50 for therapeutic signals to be observed (for LY544344 in GAD patients 10 and for pomaglumetad methionil in schizophrenia patients 24). In conclusion, oral doses of prodrug LY2979165, up to 60 mg as a single dose and 400 mg as multiple doses, using a titration regimen, were well tolerated. The dose‐limiting effect of single doses of LY2979165 was vomiting, which showed accommodation to titration of multiple doses. LY2979165 dosed in the fed state did not appear to improve the tolerability profile. There were no SAEs. Systemic uptake of prodrug LY2979165 and conversion into 2812223 was rapid and extensive; only low concentrations of the prodrug were detectable in plasma. Plasma 2812223 PK were approximately linear, with minimal accumulation following once‐daily dosing; 2812223 was detectable in CSF after a single dose of 60 mg. Of note, the studies enrolled only males and predominantly Asian subjects, so any potential effect of gender and race has not been explored, specifically regarding tolerability. Nevertheless, overall, these data support further clinical evaluation of LY2979165.
Competing Interests
The study was sponsored by Eli Lilly and Company. All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (http://www.icmje.org/coi_disclosure.pdf) (available on request from the corresponding author) and declare: J.M., C.B., S.S., S.T.‐W., J.F., D.S. and K.J. are employees of the study sponsor, Eli Lilly and Company, and stockholders. At the time of the study, D.S., S. Swanson and S.T.‐W. were former employees of Eli Lilly and Company.
The authors thank the staff of the Lilly NUS Clinical Pharmacology Unit for conducting the study, staff in the Lilly Laboratory for Experimental Medicine for supporting the biomarker evaluations, and Gina Moore, MS, inVentiv Health Clinical, for technical support.
Contributors
Study design: J.M., C.B., K.J. Study conduct: J.M. Data collection: J.M., C.B., K.J. Data analysis: C.B., S.S., K.J. Data interpretation: J.M., C.B., S.S., K.J. Drafting manuscript: J.M., C.B., K.J. Revising manuscript content: J.M., C.B., S.S., S. Swanson, S.T.‐W., J.F., D.S., K.J.
McColm, J. , Brittain, C. , Suriyapperuma, S. , Swanson, S. , Tauscher‐Wisniewski, S. , Foster, J. , Soon, D. , and Jackson, K. (2017) Evaluation of single and multiple doses of a novel mGlu2 agonist, a potential antipsychotic therapy, in healthy subjects. Br J Clin Pharmacol, 83: 1654–1667. doi: 10.1111/bcp.13252.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 2008; 7: 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre‐ and postsynaptic sites. Neuroscience 2001; 106: 481–503. [DOI] [PubMed] [Google Scholar]
- 5. Chaki S. Group II metabotropic glutamate receptor agonists as a potential drug for schizophrenia. Eur J Pharmacol 2010; 639: 59–66. [DOI] [PubMed] [Google Scholar]
- 6. Stevenson JM, Reilly JL, Harris MS, Patel SR, Weiden PJ, Prasad KM, et al. Antipsychotic pharmacogenomics in first episode psychosis: a role for glutamate genes. Transl Psychiatry 2016; 6: e739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Monn JA, Prieto L, Taboada L, Hao J, Reinhard MR, Henry SS, et al. Synthesis and pharmacological characterization of C4‐(thiotriazolyl)‐substituted‐2‐aminobicyclo[3.1.0]hexane‐2,6‐dicarboxylates. Identification of (1R,2S,4R,5R,6R)‐2‐amino‐4‐(1H‐1,2,4‐triazol‐3‐ylsulfanyl)bicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid (LY2812223), a highly potent, functionally selective mGlu2 receptor agonist. J Med Chem 2015; 58: 7526–7548. [DOI] [PubMed] [Google Scholar]
- 8. Ster J, Mateos JM, Grewe BF, Coiret G, Corti C, Corsi M, et al. Enhancement of CA3 hippocampal network activity by activation of group II metabotropic glutamate receptors. Proc Natl Acad Sci U S A 2011; 108: 9993–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, et al. Metabotropic glutamate receptor 3 activation is required for long‐term depression in medial prefrontal cortex and fear extinction. Proc Natl Acad Sci U S A 2015; 112: 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology 2008; 33: 1603–1610. [DOI] [PubMed] [Google Scholar]
- 11. Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med 2007; 13: 1102–1107. [DOI] [PubMed] [Google Scholar]
- 12. Kinon BJ, Millen BA, Downing AC, Zhang L, Stauffer VL, Andersen SW, et al. LY2140023 monohydrate in the treatment of patients with schizophrenia: results of 2 clinical trials assessing efficacy in treating acutely ill patients and those with prominent negative symptoms. Schizophr Bull 2013; 39: S338. [Google Scholar]
- 13. Eli Lilly and Company . A study of LY2140023 in patients with schizophrenia. In: ClinicalTrials.gov [online]. Bethesda, MD: National Library of Medicine. 2013. Available at http://clinicaltrials.gov/show/NCT01307800 (last accessed 20 December 2016).
- 14. McKinzie D, Johnson B, Thompson L, Seidel W, Edgar D, Wafford K, et al. Use of pharmacodynamic measures to demonstrate functional central activity of a novel group II metabotropic glutamate receptor agonist Prodrug [abstract]. Curr Neuropharmacol 2011; 9 (Suppl 1): 41. [Google Scholar]
- 15. Rorick‐Kehn LM, Johnson BG, Knitowski KM, Salhoff CR, Witkin JM, Perry KW, et al. In vivo pharmacological characterization of the structurally novel, potent, selective mGlu2/3 receptor agonist LY404039 in animal models of psychiatric disorders. Psychopharmacology (Berl) 2007; 193: 121–136. [DOI] [PubMed] [Google Scholar]
- 16. Lowe S, Dean R, Ackermann B, Jackson K, Natanegara F, Anderson S, et al. Effects of a novel mGlu2/3 receptor agonist prodrug, LY2140023 monohydrate, on central monoamine turnover as determined in human and rat cerebrospinal fluid. Psychopharmacology (Berl) 2016; 219: 959–970. [DOI] [PubMed] [Google Scholar]
- 17. Eckstein JA, Ammerman GM, Reveles JM, Ackermann BL. Simultaneous profiling of multiple neurochemical pathways from single cerebrospinal fluid sample using GC/MS/MS with electron capture detection. J Mass Spectrom 2008; 43: 782–790. [DOI] [PubMed] [Google Scholar]
- 18. Ayan‐Oshodi M, Wondmagegnehu ET, Lowe SL, Kryzhanovskaya L, Walker DJ, Kinon BJ. Adverse events in healthy subjects exposed to single and multiple doses of LY2140023 monohydrate: pooled results from 10 phase 1 studies. J Clin Psychopharmacol 2012; 32: 408–411. [DOI] [PubMed] [Google Scholar]
- 19. Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res 2000; 17: 1278–1283. [DOI] [PubMed] [Google Scholar]
- 20. Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on the anterior cingulate glutamate metabolism in healthy humans: a 4‐T proton MRS study. Am J Psychiatry 2005; 162: 394–396. [DOI] [PubMed] [Google Scholar]
- 21. Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H‐MRS: relationship to ketamine‐induced psychopathology. Mol Psychiatry 2012; 17: 664–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998; 281: 1349–1352. [DOI] [PubMed] [Google Scholar]
- 23. Schmechtig A, McColm J, Jackson K, Brittain C, Gonzales C, Suriyapperuma S, et al. Functional brain imaging of experimental group II metabotropic glutamate receptor (mGluR) agonists LY2979165 and LY2140023 in healthy human subjects [abstract]. Curr Neuropharmacol 2014; 12 (Suppl 1): 54–55. [Google Scholar]
- 24. Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry 2015; 78: 754–762. [DOI] [PubMed] [Google Scholar]