

Abstract
Aims
Rituximab is a monoclonal antibody directed against CD20, which is approved in rheumatoid arthritis (RA). This study aimed at assessing the influence of CD19+ cell counts as target‐antigen amount, and of immunoglobulin G (IgG) serum concentrations on rituximab pharmacokinetics in RA patients.
Methods
In a cohort of 64 RA patients who had received repetitive courses of rituximab, the influence of CD19+ cell count, IgG serum concentration, body surface area, sex and disease activity score in 28 joints on rituximab pharmacokinetic parameters was assessed using a population pharmacokinetic analysis.
Results
A two‐compartment model, with first‐order distribution and elimination best described the data. The volume of distribution of central compartment and clearance of rituximab were estimated at 4.7 l and 0.56 l day–1, respectively. Distribution and elimination half‐lives were 0.9 days and 17.3 days, respectively. As expected, the central volume of distribution increased with body surface area (P = 0.012) and was higher in male than in female (P = 0.004). We found that the elimination rate constant (k10) increased with CD19+ count (P = 0.00022) and IgG concentration (P = 7.4 × 10−8), and that k10 decreased with time (P = 0.00015), partly explained by a change in target‐antigen amount.
Conclusions
The association between CD19+ count and k10 may be explained by target‐mediated drug disposition, while the association between IgG serum concentration and k10 may be explained by a saturation of the neonatal Fc receptor at high IgG concentrations, resulting in decreased recycling of rituximab.
Keywords: elimination rate constant, immunoglobulin, pharmacokinetics, rheumatoid arthritis, rituximab
What is Already Known about this Subject
Rituximab is effective in rheumatoid arthritis but clinical response and time to relapse vary between patients.
Rituximab concentrations can be studied using a two‐compartment pharmacokinetic model.
In rheumatoid arthritis patients, the volume of distribution of rituximab increases with body weight and is higher in males.
What this Study Adds
As previously reported in haematological malignancies, target‐antigen amount influences rituximab pharmacokinetics in rheumatoid arthritis.
Rituximab elimination rate constant increases with CD19 + cell number and pretherapeutic immunoglobulin G serum concentration.
Tables of Links
| LIGANDS |
|---|
| rituximab |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by synovitis, joint destruction, and bone erosions leading to disability. Conventional disease‐modifying antirheumatic drugs (DMARDs), such as methotrexate, and biological DMARDs targeting proinflammatory cytokines, such as tumour necrosis factor α, interleukin‐1, interleukin‐6, or the adaptive immune system, including B‐cells or T‐cells costimulation, are nowadays the cornerstone of the therapeutic management of RA 3. Among them, rituximab is a chimeric immunoglobulin G (IgG) monoclonal antibody that targets CD20 and promotes a rapid and prolonged peripheral B‐cell depletion 4. Rituximab is approved as a second line treatment in active RA patients in combination with methotrexate 5, 6. However, there is a large difference in clinical response between patients. Different factors have been proposed to influence response to rituximab, including a high baseline disease activity score in 28 joints (DAS28), fewer previous tumour necrosis factor inhibitors anti‐drug antibody, rheumatoid factor positive, and anticitrullinated protein antibodies in controlled or uncontrolled studies. Moreover, the optimal dose and interval for retreatment remain uncertain. To assess this gap of knowledge specific pharmacokinetics studies are needed 7.
As rituximab is also approved in CD20 lymphomas and chronic lymphocytic leukaemia, previous studies in oncology–haematology suggested that rituximab pharmacokinetics may be influenced by target‐antigen burden: in patients with low‐grade follicular lymphomas, high tumour volume and lymphocyte count at baseline were associated with low rituximab concentrations 8. In a murine model of lymphoma expressing human CD20, rituximab elimination increased with tumour volume 9. Finally, rituximab elimination rate was shown to decrease with time, an observation which was attributed to a decrease in lymphocyte burden with time in chronic lymphocytic leukaemia 9.
In addition, the influence of pretherapeutic IgG serum concentration on the response of RA patients to rituximab has previously been reported with contradictory results. Increased IgG concentration was associated with a poorer response to rituximab in a single centre prospective study of 67 RA patients 10, whereas it was associated with a better response in a much larger multicentre randomized study of 205 RA patients 11. One hypothesis may be that the association of IgG concentration with clinical response may be due to its influence on rituximab pharmacokinetics.
A previous study described rituximab pharmacokinetics in RA patients using population pharmacokinetic modelling, but this study investigated neither the influence of antigenic burden nor of IgG serum concentration 13. Since both lymphocytes‐B count and IgG serum concentration may alter the pharmacokinetics of rituximab, the present study aimed at assessing and quantifying the influence of these factors on rituximab pharmacokinetic in RA patients.
Methods
Patient cohort and study design
We retrospectively analysed data from 64 consecutive RA patients who started rituximab treatment in the routine clinical practice of the department of rheumatology of the University Hospital of Tours between July 2007 and October 2010, who fulfilled American College of Rheumatology criteria, and received one or more courses of rituximab. A course was defined as the standard regimen of rituximab infusion of 1000 mg on days 1 and 15. Follow‐up visits and evaluation for the recurrence of symptoms between each course were scheduled at weeks 12, 24 and 48 as described previously 14. The follow‐up period was completed in November 2012. Decisions about retreatment and treatment intervals were based on clinical response to the previous cycle and symptoms of relapse after 24 weeks (as‐needed basis) 15. As individual results for serum rituximab concentrations were sent to the prescriber within the framework of a routine therapeutic drug monitoring service and were discussed during clinic‐biological rounds, ethical approval and informed consent was not sought. The study protocol was in accordance with the guidelines of the French Society of Rheumatology 5.
Demographic data
Prior to rituximab treatment, data were collected on sex, age, weight, height, body surface area (BSA), duration of the disease, initial DAS28 and previous history of treatment with methotrexate, corticosteroids or biological DMARDs.
Biological data
Erythrocyte sedimentation rate, C‐reactive protein, anti‐citrullinated protein antibodies were measured at baseline and presence of rheumatoid factor was detected. B‐cell number was assessed by CD19 counts at each visit, prior to each rituximab infusion using flow cytometry, as previously described and without additional sampling 14. Pre‐therapeutic serum concentrations of IgG were measured by nephelometry (BNII nephelometer; Siemens Healthcare Ltd, Camberley. Surrey. UK) at the Immunology Laboratory, University Hospital of Tours prior to each rituximab infusion.
Rituximab concentrations
Rituximab concentrations were determined by a standardized and fully validated ELISA derived from Blasco et al. 16. Microtitre plates were sensitized overnight at 4°C with 1 μg/mL of anti‐rituximab antibody (AbD Serotec, Kidlington, Oxfordshire, UK), blocked with phosphate buffered saline/1% (w/v) bovine serum albumin, followed by the addition of standard curve, control or patient sera, and detection using anti‐human IgG–horseradish peroxidase (Sigma–Aldrich, St. Quentin Fallavier, France) and FAST o‐phenylene diamine dihydrochloride (Sigma–Aldrich). The colorimetric reaction was recorded on a Biotek EL 808 plate reader at wavelengths 490 and 630 nm and the results were analysed using GEN5 software. Measurements of the rituximab concentrations in patients were made after a minimal 1/100 dilution of samples phosphate buffered saline/1% (w/v) bovine serum albumin/0.05% Tween. The criteria for data acceptability included accuracy of no more than 20% deviation from the actual value and imprecision <20% 16. Limit of detection, lower limit of quantitation, and upper limit of quantitation of the assay were 0.061, 0.20 and 9.0 mg l–1, respectively. Coefficients of variation from intraday variability were 8.4, 5.4 and 6.4% and from interday variability were 4.6, 6.8 and 5.3%, for 0.2, 3.0 and 7.0 mg l–1 quality controls, respectively.
Because 27.9% of rituximab concentrations were below the quantitation limit (BQL = 0.061 mg l–1), BQL concentrations were interval‐censored as being superior to 0 and inferior to 0.061 mg l–1. Indeed, censoring BQL data was shown to be the best strategy to provide unbiased parameter estimates in the presence of data containing BQL concentrations 17.
Pharmacokinetic analysis
Population pharmacokinetic analysis of rituximab concentration‐time data was performed using the nonlinear mixed‐effects program MONOLIX 4.3.2 software (Lixoft, Saclay, France). Many iterations were performed to reach the best stochastic approximation of the expectation–maximization convergence (K1 = 700 and K2 = 300, where K1 and K2 are the ‘iteration kernels’ 1 and 2 in MONOLIX). Two Markov chains were used, and simulated annealing was applied to improve the convergence of the stochastic approximation of the expectation–maximization algorithm towards the global maximum likelihood. The Fisher information matrix and likelihood were computed using stochastic approximation and importance sampling, respectively. The random seed was changed between each run.
Structural pharmacokinetics model design
Rituximab concentrations were described using compartmental pharmacokinetics models. One‐, two‐ and three‐compartmental mammillary models with first‐order distribution and elimination rate constants were tested. Structural models were compared using Akaike's information criterion (AIC). AIC was defined as: AIC = OFV + 2.p, where the objective function value (OFV) is the −2 log‐likelihood (−2LL) and p is the number of model parameters to be estimated. The structural model with lowest OFV and AIC was chosen.
Interindividual, intercourse and residual models
Interindividual variability (IIV) refers to differences in pharmacokinetics parameter values between individuals, whereas intercycle variability refers to interoccasion variability (IOV; i.e. differences in pharmacokinetics parameter values between courses/cycles). In this study, a course is considered as a rituximab treatment cycle of 2‐weekly 1000 mg day–1. Both IIV and IOV of pharmacokinetic parameters were described using an exponential model: θi = θTV × exp(ηi) × exp(κj) for i th patient and j th course, respectively, where θi is the estimated individual parameter, θTV is the typical value of the parameter, and ηi and κj are the random effects for the i th patient and j th course, which were assumed to be normally distributed with zero mean and variances ω2 and γ2, respectively. Additive, proportional and mixed additive‐proportional residual error models were tested. For example, the combined additive‐proportional model was implemented as follows: Yo,ij = Yp,ij × (1+ Ɛprop,ij) + Ɛadd,ij, where Yo,ij and Yp,ij are observed and predicted values of j th measurement for the i th patient, respectively, Ɛprop,ij and Ɛadd,ij are additive and proportional errors with zero mean and variance σadd and σprop, respectively. Intercourse variability of k10 and central volume of distribution (Vc) are estimated by taking the first course as a reference.
Covariates
The influence of both continuous (COV) and categorical (CAT) covariates was analysed using power functions of pharmacokinetics parameters. The continuous covariates were age, BSA, DAS28, CD19 counts and IgG serum concentrations. All the continuous covariates were centred on their median and were power transformed as follows: θi = θ0 × (COV/med(COV))β COV , where θ0 is the θ value for a median subject, βCOV quantifies the influence of COV on θ and med (COV) is the median value of COV. The categorical covariates were sex, corticosteroid, methotrexate cotreatment and the number of rituximab treatment courses (RTC). The influence of categorical covariate on θTV was determined using Ln(θTV) = Ln(θCAT=0) + βCAT=1, where θCAT=0 is the value of θ for the reference category and βCAT=1 is a parameter which provides the θTV value for the other category. Among these covariates, CD19 counts, serum IgG concentrations and RTC were tested on both IIV and IOV. As a categorical covariate, RTC categories were 1, 2, 3, 4 and 5. Since only one patient had five rituximab courses, fourth and fifth rituximab courses were gathered. The influence of RTCCAT was determined as follows: Ln(θRTC) = Ln(θRTC=0) + βRTC=1 + βRTC=2 + βRTC=3 + βRTC=4–5. The first rituximab course was taken as a reference. Moreover, the RTC was also tested as continuous covariate. As a continuous covariate, the influence of RTC power was transformed and centred on the first rituximab course.
Model comparison and covariate selection
The OFV and AIC of the interindividual, interoccasion, residual and covariate models were compared using likelihood ratio test (LRT), and the model with the lowest OFV was chosen. The difference in OFV (∆OFV) between two models was assumed to follow χ2 distribution. The influence of the selected covariates was assessed using stepwise approaches i.e., univariate and multivariate. Univariate step was intended to assess the influence of each covariate on the pharmacokinetic parameters associated with IIV. During this step, the covariates were added individually to the base model. The covariates that exhibit significant influence (α < 0.1) were included in the full model. Multivariable step (backward stepwise) analysis involves the elimination of covariates from the full model one‐by‐one, until a statistically significant increase in the OFV (α < 0.01) was noticed. The remaining covariates were retained in the final model. The goodness of covariate description was inspected by visual inspection of random effects (i.e., ηi vs. covariate plots).
Model goodness of fit and evaluation
The goodness of fit for each model was assessed by plotting population‐predicted and individual‐predicted rituximab concentrations vs. observed rituximab concentrations, and individual‐predicted and observed rituximab concentrations vs. time. Residual distribution was evaluated by graphical inspection of population‐weighted residual and individual‐weighted residual distributions, and normalized prediction distribution errors 18.
Simulations
Simulations were made after the modelling step with estimated pharmacokinetics parameters. Pharmacokinetic profiles with five CD19 count values (10, 50, 100, 200 and 500 μl−1), five serum IgG concentration values (5, 10, 15, 20 and 25 g l–1, respectively) and five rituximab treatment courses with typical parameters for a median BSA patient (1.8 m2) were used to analyse the quantitative influence of (i) CD19 count and (ii) serum IgG concentrations on elimination, and (ii) of RTC and CD19 count on volume of distribution and elimination. Dosing regimen was 1000 mg doses at weeks 0 and 2.
Results
A total of 64 patients, with a number of courses ranging from one to five per patient (125 rituximab courses), were included retrospectively (Table 1). The pharmacokinetics analysis was based on 674 blood samples. Rituximab concentrations were best described using a two‐compartment model with first‐order distribution and elimination constants (one‐, two‐ and three‐compartment AIC were 8435.4, 4524.3 and 5203.2, respectively). Parameterization as first‐order transfer and elimination rate constants led to reduced AIC compared to volume/clearance parameterization (4524.4 vs. 4645.5), lower shrinkages (Shk10 = 31% vs. ShCL = 49%, respectively) and decreased correlation of estimates between Vc and elimination parameter (rk10 = 0.15 and rCL = 0.32, respectively) and was therefore chosen.
Table 1.
Summary of patient's characteristics
| Characteristics | Rituximab course | Patients (n = 64) |
|---|---|---|
| Women, n (%) | 53 (82.8) | |
| Age, median (range), years | 59 (38–84) | |
| BSA, median (range), m 2 | 1.8 (1.35–2.29) | |
| Disease duration, median (range), years | 1.4 (0.27–4.6) | |
| Initial DAS28, median (range) | 5.24 (2.1–8.35) | |
| CRP, median (range), mg l –1 | 17 (1–148.6) | |
| Rheumatoid factor positive, n (%) | 44 (68.8) | |
| Anti‐citrullinated protein antibody positive, n (%) | 53 (82.9) | |
| Past anti‐TNF use, n (%) | 51 (79.6) | |
| Corticosteroids, n (%) | 48 (75) | |
| Methotrexate, n (%) | 31 (48.4) | |
| Rituximab cycles, n (%) | 1 | 64 (100) |
| 2 | 31 (48.4) | |
| 3 | 21 (32.8) | |
| 4 | 9 (14.1) | |
| 5 | 2 (3.1) | |
| CD19 count, median (range), mm 3 | 1 | 214 (2–706) |
| 2 | 44 (0–478) | |
| 3 | 33 (0–437) | |
| 4 | 126 (2–276) | |
| 5 | 142 (2–282) | |
| Serum IgG concentrations, median (range), g l –1 | 1 | 10.6 (5.0–25.1) |
| 2 | 10.4 (4.6–16.3) | |
| 3 | 9.2 (4.3–18.8) | |
| 4 | 8.9 (6.12–10.4) | |
| 5 | 10.2 (9.9–10.6) | |
BSA, body surface area; DAS28, disease activity score in 28 joints; CRP, C‐reactive protein concentration; IgG: immunoglobulins; TNF, tumour necrosis factor.
Population, individual residuals and normalized prediction distribution error plots showed that there was no obvious model misspecification (Figure 1 and Table 2). The plot of observed vs. model‐predicted population concentration showed a trend towards under‐prediction of high concentrations is due to inter‐course variability. The estimation of IOV for both Vc and k10 significantly improved the model (∆OFV = 56.3). IIV of the distribution parameters from central‐to‐peripheral compartment (k12) and from peripheral‐to‐central compartment (k21) were poorly identifiable and therefore were set to 0. The best residual model was mixed additive–proportional. Pharmacokinetic parameters were estimated with good accuracy. Estimated parameters of the base model [interindividual standard deviation]: were (Vc) = 4.7 l [0.28], elimination rate constant (k10) = 0.12 day−1 [0.30], central‐to‐peripheral (k12) = 0.43 day−1 and peripheral‐to‐central (k21) = 0.26 day−1 rate constants (Table 2). Secondary parameters [interindividual standard deviation] were: clearance (CL) = 0.56 l day–1 [0.29], peripheral volume of distribution (VP) = 7.8 l and intercompartment clearance (Q) = 2.0 l day–1. Distribution (T½‐α) and elimination (T½‐β) half‐lives were 0.9 days and 17.3 days, respectively. Plots of predicted vs. observed concentrations showed that the pharmacokinetics model described the data satisfactorily (Figure 1).
Figure 1.
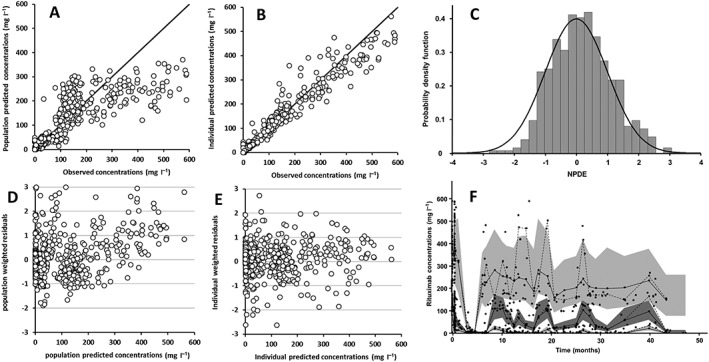
Diagnostic plots of the pharmacokinetic model: (A) observed vs. population model‐predicted rituximab concentrations; (B) observed vs. individual model‐predicted rituximab concentrations; (C) normalized prediction distribution errors (NPDE) vs. gaussian law; (D) population weighted residuals vs. population predicted rituximab concentrations; (E) individual weighted residuals vs. individual predicted rituximab concentrations; (F) prediction‐corrected visual predictive check; observed concentrations (black circles), theoretical (dashed bold lines) and empirical (continuous thin lines) percentiles (from bottom to top: 10%, 50% and 90% percentiles) and prediction interval (from bottom to top: 10%, 50% and 90% prediction intervals)
Table 2.
Estimated pharmacokinetic parameters
| Model | Base | Final | ||
|---|---|---|---|---|
| parameter (unit) | estimate | RSE (%) | estimate | RSE (%) |
| V 1 (L) | 4.0 | 6 | 4.1 | 7 |
| BSA on V 1 | – | – | 1.0 | 38 |
| Sex on V 1 | – | – | 0.37 | 34 |
| RTC on V 1 | – | – | 0.41 | 19 |
| k 10 (day −1) | 0.12 | 8 | 0.11 | 8 |
| IgG on k 10 | – | – | 0.50 | 19 |
| CD19 on k 10 | – | – | 0.035 | 27 |
| k 12 (day −1 ) | 0.43 | 5 | 0.42 | 6 |
| k 21 (day −1 ) | 0.26 | 5 | 0.25 | 4 |
| ω V1 | 0.28 | 21 | 0.22 | 25 |
| ω k10 | 0.30 | 12 | 0.24 | 13 |
| ω k12 | – | – | – | – |
| ω k21 | – | – | – | – |
| γ V1 | 0.45 | 12 | 0.33 | 12 |
| γ k10 | 0.11 | 18 | 0.08 | 23 |
| γ k12 | – | – | – | – |
| γ k21 | – | – | – | – |
| σ add (mg l –1 ) | 0.31 | 4 | 0.31 | 4 |
| σ prop (%) | 0.27 | 2 | 0.27 | 2 |
V1, volume of distribution; BSA, body surface area; RTC, rituximab treatment course; k, elimination rate constant; IgG, immunoglobulin G serum concentration (g l–1); CD19, CD19+ (B‐cell) count (mm−3); RSE, residual standard error (standard error/parameter value); ω, interindividual standard deviation; γ, interoccasion standard deviation; σadd, additive residual standard deviation; σprop, proportional residual standard deviation.
During univariate analysis, sex and BSA were found to influence Vc. RTC was found to influence IOV of Vc. Counts of CD19, serum IgG concentrations and RCT were found to influence both IIV and IOV of k10. Notably, the RTC parameterization as continuous covariate led to lower AIC than as categorical covariate (5236.1 vs. 5252.5) and was therefore chosen. No significant influence of either methotrexate use or baseline DAS28 on pharmacokinetic parameters was observed. As expected, in the multivariable analysis, Vc was found to increase with BSA (∆OFV = 6.9, P = 0.0084) and to be higher in men than in women (LRT = 8.7, P = 0.0031); there was an increase in Vc (∆OFV = 28.7, P = 8.3 10−8) with RTC (Figure 2). The k10 increased with increasing CD19 counts (LRT = 13.6, P = 0.00022) as well as with serum IgG concentrations (LRT = 28.9, P = 7.4 10−8) in multivariable analysis. The significant decrease of k10 with RTC found in univariate step disappeared when CD19 counts were added.
Figure 2.
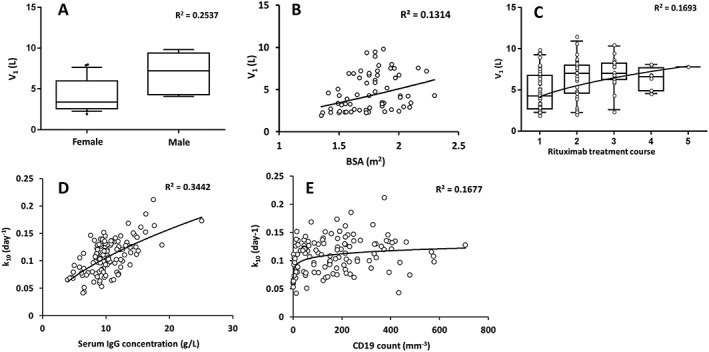
Individual volume of distribution (V1, top) and elimination rate constant (k10, bottom) estimates vs. covariates: (A) sex, (B) body surface area and (C) rituximab treatment course on V1, and (D) serum IgG concentrations and (E) CD19 counts on k10. Open circles are observed values, lines are correlation lines, Horizontal lines of boxplots represent, from bottom to top, , 5th, 25th, 50th, 75th and 95th percentiles of pharmacokinetics parameters
Our simulations of rituximab concentrations in typical patients with five different CD19+ counts showed that T½‐β decreased with increasing values of CD19+ count. Similarly, T½‐β decreased with increasing values of baseline IgG concentration: T½‐β was 28 days and 11 days for the minimum and the maximum IgG concentrations, respectively. Between the first and the fifth rituximab treatment course, Vc increased by 104% and k10 decreased by 24%, leading to an increase in T½‐β of rituximab from 18 to 23 days (Figure 3).
Figure 3.
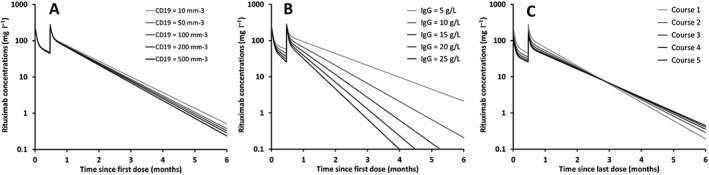
Simulations of five rituximab pharmacokinetic profiles using typical pharmacokinetics parameters, (A) increasing CD19 counts (10 to 500 mm−3), (B) increasing serum IgG concentrations (5 to 25 g l–1), and (C) for rituximab courses 1–5 showing time‐dependence of rituximab pharmacokinetics
Discussion
To our knowledge, the present study is the first to describe the influence of the amount of lymphocytes‐B assessed by circulating CD19+ cell count as target‐antigen, and of serum IgG concentrations on rituximab pharmacokinetics in RA patients. In this retrospective single‐centre follow‐up study, a two‐compartment model satisfactorily described rituximab concentrations and pharmacokinetic parameters were reliably estimated, as previously reported 7. Our results show a time‐dependence of rituximab pharmacokinetics in RA patients, which was partly due to the influence of B‐cells, as was previously reported in chronic lymphocytic leukaemia 10. The increase of volume of distribution with body weight and its higher value in males is consistent with previous pharmacokinetics studies of rituximab 7, 19 and other monoclonal antibodies 20, 21.
We observed an increase in rituximab elimination rate constant (k10) correlated with circulating B‐cell counts and also, because B‐cell count decreases with time as an effect of rituximab, a decrease of k10 with time. This is consistent with target‐mediated drug disposition, a mechanism frequently reported for monoclonal antibodies 19, 21, notably in RA for anti‐tumour necrosis factor‐α monoclonal antibodies 21. After binding of rituximab to its CD20 target, antibody‐target antigen complexes are formed, which results in higher rituximab elimination. This target‐mediated elimination is thought to depend not only on antibody concentrations, but also on target‐antigen amount, distribution and turnover 20, 21. Since repeated rituximab injections lead usually to sustained and marked loss of B‐cells, a low B‐cell count should lead to low target‐mediated elimination and therefore a decrease in global rituximab elimination with time in patients with B‐cell depletion induced by rituximab treatment. Therefore, the deeper the B‐cell depletion, the lower the target‐mediated clearance of rituximab and the higher the elimination half‐life. Target‐mediated elimination of rituximab was described in follicular lymphoma 8 and chronic lymphocytic leukaemia 10, but to our knowledge was not described in a noncancer disease. However, since no departure from elimination linearity was observed, target‐mediated drug disposition models could not be assessed. Of note, blood CD19 count is only a fraction of total antigenic burden, since circulating B‐cells represent approximately 3% of total B‐cells. The relationship between CD19 count and rituximab elimination may therefore be an approximation of the actual relationship between antigenic burden and elimination.
In addition, we observed an increase of k10 with IgG serum concentrations. This influence might be explained by the binding to the neonatal Fc receptor (FcRn). FcRn, a MHC‐class I‐like receptor initially described for the transfer of IgG from mother to infant, is involved in recycling and transcytosis process of IgG in humans 23. A high affinity interaction between FcRn molecules and IgG appears at acidific pH rather than neutral pH, leading to the protection of IgG from intracellular catabolism degradation and the long half‐life of 3 weeks of IgG compared to other plasma proteins 23. This receptor was shown to be saturated with high levels of IgG 24. The saturation of FcRn recycling system decreases its recycling efficiency and therefore increases the fractional catabolic rate, and therefore the clearance of all IgG, including therapeutic antibodies as rituximab 24, 25. Unfortunately, pharmacokinetic models that describe FcRn‐mediated recycling kinetics and endogenous elimination of therapeutic antibodies and competition with endogenous IgG 25, 26, 27, 28 were not identifiable in the present study.
The estimation of IOV (interoccasion or intercourse variability) allowed a better description of pharmacokinetic data. Interestingly, a strongly significant trend towards an increase in volume of distribution (Vc) was observed. The increase of Vc in time denotes a gradual decrease of rituximab concentrations in time. Even if the reasons of this phenomenon remain unclear, a possible explanation might be an immunization against rituximab, which was previously described for rituximab in RA patients by Thurlings et al. 13. In their study, patients for whom anti‐rituximab antibodies were detected had significantly lower concentrations than others. However, the presence of such antibodies seemed not to increase rituximab elimination slope, which is consistent with an increase in Vc and not with rituximab elimination 13. Unfortunately, the monitoring research of anti‐rituximab antibodies was not performed in our cohort of patients.
Our model has limitations. First, the parameterization as first‐order transfer and elimination rate constants provided a better characterization of base model than clearance parameterization, which led us to report peripheral volume, and systemic and intercompartment clearances as secondary parameters. Second, neither IIV nor IOV in distribution parameters (k12 and k21) were estimable, which is explained by the low number of samples during the distribution phase 29, 30, 31. Third, models accounting for either target‐mediated kinetics or FcRn‐mediated elimination and competition with endogenous IgG were attempted in the present study, but parameters for either nonlinear elimination or recycling and elimination could not be identified, respectively. Moreover, as IgG subclasses were not available for all the patients and FcRn affinity to IgG differs for different IgG subclasses 32, IgG subclasses were not tested in our model. Fourth, because the total number of B‐cells was not available in clinical routine practice, the actual relationship between rituximab elimination and B‐cell amount cannot be determined. Finally, CD19 counts might have influenced the decision of retreatment in some patients. Even if it may have influenced the IOV, it should not have led to parameter misestimation.
In conclusion, we report an influence of lymphocytes B as antigenic burden and of baseline IgG serum concentration on the pharmacokinetics of rituximab in RA patients. Increased elimination of rituximab with higher B‐cell counts and higher IgG concentrations might lead to higher chances of underexposure to rituximab in RA patients, and therefore higher chances of decreased clinical response. New studies are needed to investigate the dose–concentration–response of rituximab in rheumatoid arthritis.
Competing Interests
B.L. has given lectures for GSK and has been invited to attend international congresses by Shire, Amgen, Novartis and GSK. S.R.E. received her fellowship from the ABIRISK consortium (www.abirisk.eu). D.M. has received on behalf of his institution consulting fees and speaking fees from Pfizer and MSD (less than $10 000 each). He has participated on behalf of his institution in clinical trials sponsored by Abbott, Roche, Bristol‐Myers Squibb, Pfizer, UCB and MSD. He has been invited to attend international congresses by MSD, Roche, Bristol‐Myers Squibb and Abbott. C.P., C.D., T.B.A., V.G.G., G.T., and J.M. have no competing interests to declare. G.P.'s research team received grants from Novartis, Roche Pharma, Genzyme, MSD, Chugai and Pfizer, outside of the submitted work. D.T. has given lectures for Amgen and Sanofi.
The research leading to these results was conducted as part of the ABIRISK consortium. We would like to acknowledge Anne‐Claire Duveau and Caroline Brochon for their technical assistance in measuring rituximab concentration and to Audrey Dabert, Claudine Prouin and Françoise Vosgien in measuring IgG concentration. We thank Drs Saloua Mammou, Isabelle Griffoul, Emilie Ducourau and Virginie Martaillé for helping with clinical assessment. We are indebted to Nelly Jaccaz‐Vallée, Sergine Gosset, Valérie Angebeau, Laetitia Cornec, Adeline Coutellier, Corinne Depont, Vanessa Fougeray, Valérie Fuseau, Pascale Guibout, Sophie Joncheray, Céline Letot, Isabelle Romier and Elodie Vigneron for blood sampling and their commitment to taking care of patients.
Measurements of adalimumab serum concentrations were carried out within the CePiBAc platform. CePiBAc was cofinanced by the European Union. Europe is committed to the region Centre with the European Regional Development Fund. This work was partly supported by the French Higher Education and Research ministry under the program ‘Investissements d'avenir’ Grant Agreement: LabEx MAbImprove ANR‐10‐LABX‐53‐01.
Contributors
Conceived and designed the experiments: D.T., D.M., J.M. and G.P. Performed the experiments: S.R.E. Analysed the data: D.T., D.M., B.L. and S.R.E. Wrote the paper: B.L., S.R.E., D.T., D.M., T.B.A., C.D., V.G.G., G.P. and CP.
Lioger, B. , Edupuganti, S. R. , Mulleman, D. , Passot, C. , Desvignes, C. , Bejan‐Angoulvant, T. , Thibault, G. , Gouilleux‐Gruart, V. , Mélet, J. , Paintaud, G. , and Ternant, D. (2017) Antigenic burden and serum IgG concentrations influence rituximab pharmacokinetics in rheumatoid arthritis patients. Br J Clin Pharmacol, 83: 1773–1781. doi: 10.1111/bcp.13270.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet 2010; 376: 1094–1108. [DOI] [PubMed] [Google Scholar]
- 4. Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994; 83: 435–445. [PubMed] [Google Scholar]
- 5. Pham T, Fautrel B, Gottenberg JE, Goupille P, Hachulla E, Masson C, et al. Rituximab (MabThera) therapy and safety management. Clinical tool guide. Joint Bone Spine 2008; 75: S1–99. [DOI] [PubMed] [Google Scholar]
- 6. Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2014; 73: 492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti‐CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol 2005; 45: 792–801. [DOI] [PubMed] [Google Scholar]
- 8. Berinstein NL, Grillo‐López AJ, White CA, Bence‐Bruckler I, Maloney D, Czuczman M, et al. Association of serum rituximab (IDEC‐C2B8) concentration and anti‐tumor response in the treatment of recurrent low‐grade or follicular non‐Hodgkin's lymphoma. Ann Oncol 1998; 9: 995–1001. [DOI] [PubMed] [Google Scholar]
- 9. Daydé D, Ternant D, Ohresser M, Lerondel S, Pesnel S, Watier H, et al. Tumor burden influences exposure and response to rituximab: pharmacokinetic‐pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood 2009; 113: 3765–3772. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Zhi J, Wenger M, Valente N, Dmoszynska A, Robak T, et al. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol 2012; 52: 1918–1926. [DOI] [PubMed] [Google Scholar]
- 11. Couderc M, Mathieu S, Pereira B, Glace B, Soubrier M. Predictive factors of rituximab response in rheumatoid arthritis: results from a French university hospital. Arthritis Care Res 2013; 65: 648–652. [DOI] [PubMed] [Google Scholar]
- 12. Sellam J, Hendel‐Chavez H, Rouanet S, Abbed K, Combe B, Le Loët X, et al. B cell activation biomarkers as predictive factors for the response to rituximab in rheumatoid arthritis: a six‐month, national, multicenter, open‐label study. Arthritis Rheum 2011; 63: 933–938. [DOI] [PubMed] [Google Scholar]
- 13. Thurlings RM, Teng O, Vos K, Gerlag DM, Aarden L, Stapel SO, et al. Clinical response, pharmacokinetics, development of human anti‐chimaeric antibodies, and synovial tissue response to rituximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis 2010; 69: 409–412. [DOI] [PubMed] [Google Scholar]
- 14. Mélet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab‐induced T cell depletion in patients with rheumatoid arthritis: association with clinical response. Arthritis Rheum 2013; 65: 2783–2790. [DOI] [PubMed] [Google Scholar]
- 15. Lavielle M, Mulleman D, Goupille P, Bahuaud C, Sung HC, Watier H, et al. Repeated decrease of CD4+ T‐cell counts in patients with rheumatoid arthritis over multiple cycles of rituximab treatment. Arthritis Res Ther 2016; 18: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blasco H, Lalmanach G, Godat E, Maurel MC, Canepa S, Belghazi M, et al. Evaluation of a peptide ELISA for the detection of rituximab in serum. J Immunol Methods 2007; 325: 127–139. [DOI] [PubMed] [Google Scholar]
- 17. Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J 2009; 11: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brendel K, Comets E, Laffont C, Mentré F. Evaluation of different tests based on observations for external model evaluation of population analyses. J Pharmacokinet Pharmacodyn 2010; 37: 49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Müller C, Murawski N, Wiesen MHJ, Held G, Poeschel V, Zeynalova S, et al. The role of sex and weight on rituximab clearance and serum elimination half‐life in elderly patients with DLBCL. Blood 2012; 119: 3276–3284. [DOI] [PubMed] [Google Scholar]
- 20. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49: 633–659. [DOI] [PubMed] [Google Scholar]
- 21. Ternant D, Bejan‐Angoulvant T, Passot C, Mulleman D, Paintaud G. Clinical pharmacokinetics and pharmacodynamics of monoclonal antibodies approved to treat rheumatoid arthritis. Clin Pharmacokinet 2015; 54: 1107–1123. [DOI] [PubMed] [Google Scholar]
- 22. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic–pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol 2009; 5: 803–812. [DOI] [PubMed] [Google Scholar]
- 23. Roopenian DC, Akilesh S. FcRn: the neonatal fc receptor comes of age. Nat Rev Immunol 2007; 7: 715–725. [DOI] [PubMed] [Google Scholar]
- 24. Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2‐microglobulin‐containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A 1996; 93: 5512–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brambell FW, Hemmings WA, Morris IG. A theoretical model of gamma‐globulin catabolism. Nature 1964; 203: 1352–1354. [DOI] [PubMed] [Google Scholar]
- 26. Ferl GZ, Wu AM, DiStefano JJ. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal fc receptor (FcRn). Ann Biomed Eng 2005; 33: 1640–1652. [DOI] [PubMed] [Google Scholar]
- 27. Garg A, Balthasar JP. Physiologically‐based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild‐type and FcRn‐knockout mice. J Pharmacokinet Pharmacodyn 2007; 34: 687–709. [DOI] [PubMed] [Google Scholar]
- 28. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin Immunol 2007; 122: 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol 2009; 65: 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ternant D, Aubourg A, Magdelaine‐Beuzelin C, Degenne D, Watier H, Picon L, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit 2008; 30: 523–529. [DOI] [PubMed] [Google Scholar]
- 31. Xu Z, Seitz K, Fasanmade A, Ford J, Williamson P, Xu W, et al. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol 2008; 48: 681–695. [DOI] [PubMed] [Google Scholar]
- 32. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014; 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]