

Abstract
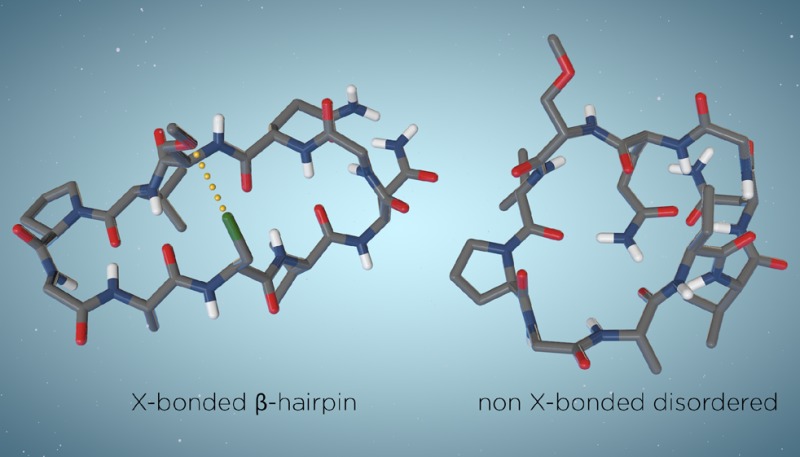
Halogen bonding is a weak chemical force that has so far mostly found applications in crystal engineering. Despite its potential for use in drug discovery, as a new molecular tool in the direction of molecular recognition events, it has rarely been assessed in biopolymers. Motivated by this fact, we have developed a peptide model system that permits the quantitative evaluation of weak forces in a biologically relevant proteinlike environment and have applied it for the assessment of a halogen bond formed between two amino acid side chains. The influence of a single weak force is measured by detection of the extent to which it modulates the conformation of a cooperatively folding system. We have optimized the amino acid sequence of the model peptide on analogues with a hydrogen bond-forming site as a model for the intramolecular halogen bond to be studied, demonstrating the ability of the technique to provide information about any type of weak secondary interaction. A combined solution nuclear magnetic resonance spectroscopic and computational investigation demonstrates that an interstrand halogen bond is capable of conformational stabilization of a β-hairpin foldamer comparable to an analogous hydrogen bond. This is the first report of incorporation of a conformation-stabilizing halogen bond into a peptide/protein system, and the first quantification of a chlorine-centered halogen bond in a biologically relevant system in solution.
The recently rediscovered halogen bond phenomenon,1 i.e., the weak, noncovalent interaction between an electron donor and an electron-poor region of a halogen, has been successfully exploited in crystal engineering,2 supramolecular chemistry,2 organocatalysis,3 functional materials,4 anion recognition,5 and medicinal chemistry,6 for example. It is being developed into a molecular tool that is an alternative to hydrogen bonding7 and is expected to find applications in chemistry and drug discovery. The directionality and strength of halogen bonding can be tuned by changing the halogen atom (F ≪ Cl < Br < I).2 Model studies in solutions have so far typically been performed on the halogen bonds of simplified, perfluorinated, small organic molecules, while studies in biomacromolecules are scarce.8 Halogen bonds have been used in the modulation of binding of a ligand to various receptors,6,9,10 to induce DNA junctions,11 and in rational drug design,12 but they have so far barely been applied in peptide and protein engineering.13,14 Most investigations to date have been performed in silico and in the solid state, whereas solution studies of halogen bonds in a biological environment have lagged behind, most likely as the analysis of week interactions in non-ordered media is highly challenging. In very recent work by Ho and co-workers, the incorporation of a halogen bond donor amino acid analogue into lysozyme T4 resulted in a short iodine–oxygen contact in the solid state yet a simultaneous destabilization of the protein structure in solution.13 Iodine substitution was shown to have a stabilizing effect as compared to another iodinated mutant. This highlights the challenge of developing a biopolymer-based model system in which the isolated effect of a weak halogen bond can be studied, with the additional interactions and geometries remaining unaffected.15 Herein, we demonstrate, for the first time, the utility of halogen bonding in peptide chemistry in solution. We use it for conformational stabilization of a β-hairpin motif, to an extent comparable to that of an analogous interstrand hydrogen bond.
We have designed a peptide model system (Figures 1 and 2) that allows the investigation of a single weak interaction force, here a halogen bond, embedded in a complex, proteinlike environment. To measure its strength, we follow the change in the folding propensity of the peptide upon permitting or preventing the formation of this specific interaction [R3–R8 (Figure 1)], as achieved by the minimal necessary structural alteration. Whereas the overall stability of the folded conformation is established by a series of cooperatively acting weak forces, it is modulated by the alteration of a single, conformation-stabilizing interaction. Studying a weak interaction in an intramolecular model system offers a crucial entropic advantage that permits the characterization of exceptionally weak forces.16 We have previously proven this concept,17,18 and we apply it herein for the exploration of the halogen bond in a biologically relevant model system.
Figure 1.
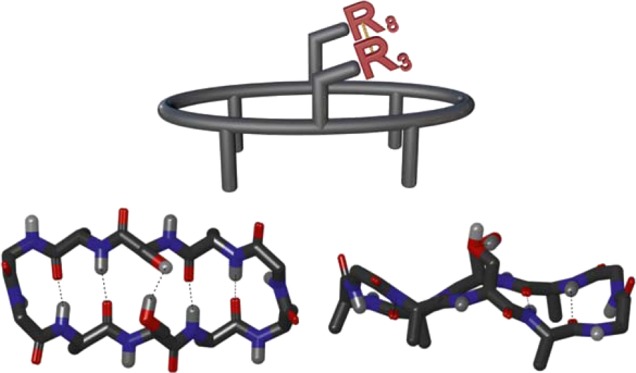
Chemical structure of the studied β-hairpin model system, with the R3–R8 interaction site (top), shown from above (bottom left) and from the side (bottom right).
Figure 2.
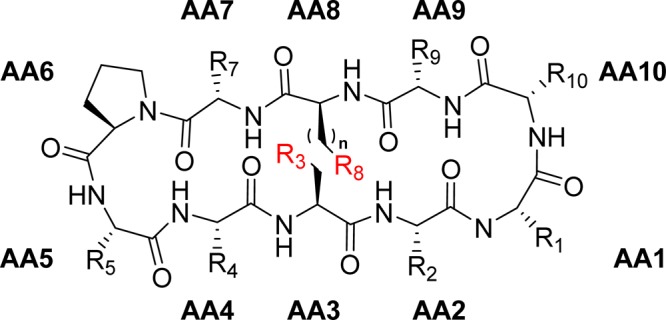
Schematic structure of the investigated cyclic β-hairpin peptides.
Experimental Details
All experimental details are given in the Supporting Information.
Results and Discussion
Design
Our model system (Figures 1 and 2) was designed to be comprised of two three-amino acid strands each, locating the interaction site of interest, R3–R8, on one face of the peptide and the side chains of all other amino acids of the β-strands on the opposite face, thereby avoiding unwanted side chain–side chain interactions. Connecting the strands with strong β-turn inducers19,20 at both ends of the β-strands promotes β-hairpin folding and locates the donor and acceptor sites across from each other, in optimal geometry for interaction. The cyclic nature of the model system provides an entropic advantage and promotes β-hairpin folding,21 which in turn is further supported by the incorporation of β-branched amino acids into the strands.22 Amino acids with nonpolar side chains are also expected to form conformation-stabilizing interstrand hydrophobic interactions,20 which in combination with weak backbone N–H···O=C hydrogen bonds provides an overall stability of the β-hairpin conformation. For evaluation of the β-hairpin content of the model systems, NAMFIS (nuclear magnetic resonance analysis of molecular flexibility in solution) ensemble analysis was used.23,24 This combined nuclear magnetic resonance (NMR) spectroscopic and computational technique deconvolutes time-averaged spectroscopic data (NOE and 3JCαH,NH) to yield the ensemble that describes the solution conformations of flexible systems (vide supra). NAMFIS has previously been successfully utilized to determine the solution conformations of both peptides17,25,26 and small molecules.27,28
Prior to synthesis of a peptide possessing a halogen bond donor site, the system was optimized using an analogous sequence encompassing a hydrogen bond donor side chain functionality. First, peptides encompassing serine or O-methyl-l-serine as a hydrogen bond acceptor at position 8 and serine as a hydrogen bond donor at position 3 [R8 = OH or OMe, and R3 = OH (Figure 2 and Table 1, 1a and 2a)] were synthesized. As references, analogous peptides were used in which the hydrogen bond donor OH of R3 was substituted with a CH3 group (Table 1, 1b and 2b). The CH3 group is of comparable size yet prevents the formation of an attractive R3–R8 interaction. Upon formation of a stabilizing interchain interaction, here a hydrogen (R3 = OH) or a halogen (R3 = Cl/Br) bond, the peptide is expected to show β-hairpin folding propensity higher than that of the corresponding reference (R3 = CH3) that lacks this interaction. The difference in folding propensity of the hydrogen or halogen bond-forming peptide and the references consequently reflects the strength of the R3–R8 interaction.
Table 1. Peptides Evaluated Using NAMFIS Analysis.
| peptide sequenced | R3e | R8 | n | % β-hairpinf | |
|---|---|---|---|---|---|
| 1aa | c(NVSAGD-PVSQG) | OH | OH | 1 | 88 |
| 1ba | c(NVXAGD-PVSQG) | Me | OH | 1 | 50 |
| 2ab | c(NVSAGD-PVS(Me)QG) | OH | OMe | 1 | 58 |
| 2bb | c(NVXAGD-PVS(Me)QG) | Me | OMe | 1 | 29 |
| 3ac | NVSAGD-PVhS(Me)QG-Ac | OH | OMe | 2 | 11 |
| 3bc | NVXAGD-PVhS(Me)QG-Ac | Me | OMe | 2 | 0 |
| 4ac | c(GQSAGD-PVhS(Me)AN) | OH | OMe | 2 | 36 |
| 4bc | c(GQXAGD-PVhS(Me)AN) | Me | OMe | 2 | 31 |
| 5c | c(NVA(Cl)AGD-PVS(Me)QG) | Cl | OMe | 1 | 74 |
| 8c | c(NVA(Br)AGD-PVS(Me)QG) | Br | OMe | 1 | – |
Synthesis and characterization.17
Synthesis and characterization.29
Synthesis and characterization described in the Supporting Information.
The peptide sequence is given according to the numbering in Figure 2. X = ABU (2-aminobutanoic acid). S(Me) = O-methyl-l-serine. hS(Me) = O-methyl-l-homoserine.
OH substitution at position 3 allows a hydrogen cross strand interaction with position 8. When position 3 is substituted with CH3, this interaction is not possible, and the CH3-substituted analogue serves as a reference.
The % β-hairpin in solution as determined by NAMFIS analysis. Peptide 8 could not be evaluated via NAMFIS because of rapid degradation at room temperature.
To explore the geometrical factors influencing the overall β-hairpin stability and the efficiency of the R3–R8 interaction, a series of peptides possessing either a hydrogen bond donor (R3 = OH, 1a–4a) or a non-interacting reference (R3 = CH3, 1b–4b) at position 3 were designed (Figures 1 and 2 and Table 1). The cyclic peptides 1a and 1b have shown a high β-hairpin folding propensity that was efficiently modulated by the presence or absence of the R3–R8 interstrand hydrogen bond [Table 1; 1a, OH···O(H), 88% β-hairpin; 1b, CH3··/··O(H), 50% hairpin].17 Replacement of the R8 OH of 1a and 1b with OCH3, yielding peptides 2a and 2b, respectively, prevents formation of a competing hydrogen bonding interaction in which R8 acts as a Lewis acid, instead of acting only as a Lewis base, which is a necessity for the halogen bond analogue. This change resulted in an ∼30% decrease in the overall level of folding (2a vs 1a). The R3–R8 hydrogen bond-forming peptide 2a showed a level of folding ∼30% higher than that of reference system 2b, which does not permit an attractive interstrand side chain interaction. The 29–58% overall stability of 2a and 2b as compared to that of 1a and 1b (50–88%) puts the model system into the folding region that is most sensitive for detection of small changes, within a two-state equilibrium.
To evaluate whether an increased overall flexibility could promote a more optimal geometry for formation of an interstrand R3–R8 interaction, expected to provide an even larger difference in folding of the hydrogen bonding and the reference systems, peptides 2a and 2b were converted into their acyclic analogues. In addition, the side chain of amino acid R8 was extended with a methylene group (Table 1, 3a and 3b, n = 2); i.e., a l-serine to l-homoserine substitution was performed at position 8. In this case, a substantial destabilization of the β-hairpin conformation was observed, which is in good agreement with previous reports suggesting cyclization to be an efficient tool in stabilizing the hairpin conformation of small, flexible peptides.21 In addition, the amino acid sequence of 2a and 2b was altered to decrease backbone rigidity, yielding the cyclic analogues 4a and 4b, respectively, with the extended R8 side chain (n = 2). Although the observed β-hairpin content was higher than for acyclic 3a and 3b, the increased flexibility did not facilitate a geometry permitting a stronger R3–R8 hydrogen bond and thus decreased the overall level of folding as well as the difference in folding of the hydrogen bond forming 4a and the reference 4b. On the basis of the collected information, we have synthesized analogues of peptide 2a that possess a chlorine (5) or bromine (8) halogen bond donor functionality in the side chain of R3 and the halogen bond acceptor OCH3 at R8. The impact of an interstrand halogen bond was studied by NMR ensemble analysis in solution, proving the applicability of the halogen bond in peptide and protein engineering and providing a quantitative measure of the relative strength of a halogen bond as compared to an analogous hydrogen bond, 2a, using the non-interacting peptide 2b as a reference.
Synthesis
The synthesis of peptide 5, encompassing β-chloroalanine in position 3, is summarized in Scheme 1. Its linear precursor 6 was prepared by automated solid phase peptide synthesis with a PS3 peptide synthesizer utilizing the Fmoc-t-Bu-Trt protecting strategy30,31 with TBTU as the coupling reagent and diisopropylethylamine as the base on 2-chlorotrityl resin. Following the coupling of the first eight amino acids, the last two were performed manually under conditions carefully optimized to avoid elimination of the chlorine atom (for details, see the Supporting Information) using HATU as a coupling reagent in combination with TMP. Cleavage of the linear decapeptide from the resin was performed with 1% trifluoroacetic acid in dichloromethane, followed by cyclization utilizing a pseudo-high-dilution procedure32 with HATU as the coupling reagent in the presence of TMP. Removal of protecting groups and subsequent purification by RP-HPLC using a C18 column afforded 5 in an overall yield of 18%. The synthesis of peptide 8, encompassing β-bromoalanine at position R3, is summarized in Scheme 2. As the aliphatic carbon–bromine bond is unstable under the conditions of peptide coupling and deprotection, the bromine atom was introduced in the last step of the synthesis by transformation of the S3 hydroxy group of the fully unprotected peptide 2a with the Vilsmeier reagent. This reagent was prepared from phosphorus tribromide and DMF at 0 °C33 and was added to the peptide in the presence of sodium bromide at room temperature in DMF as a solvent.34 It should be noted here that this is the very first use of this synthetic strategy for introduction of bromine functionality into a peptide. This may then be used as a handle for introduction of further functionalities or to study halogen bonding, for example. The method is mild and substitutes an aliphatic hydroxyl group with bromine in a solvent in which many peptides are soluble. Peptide 8 was stored and studied by NMR spectroscopy at temperatures below −10 °C as it was found to rapidly degrade at room temperature in solution. Details of the synthesis of the peptides used for sequence optimization are given in the Supporting Information.
Scheme 1. Outline of the Synthesis of Peptide 5 Encompassing a β-Chloroalanine as a Halogen Bond Donor and O-Methylserine as a Halogen Bond Acceptor.

Scheme 2. Synthesis of Peptide 8, Encompassing an Aliphatic Bromine Halogen Bond Donor Functionality, Starting from Peptide 2a.

NMR Structural Assignment
This process was performed by the TOCSY-NOESY sequential backbone walk strategy.35 Because of the low aqueous solubility of these peptides, similar to numerous other studies of β-hairpins,17,29,36−44 which typically occur in the hydrophobic core of proteins that is much less polar than water,45 DMSO-d6 was chosen as the solvent. For peptide 5, the chemical shifts of the Hαs [δHα (Table 2); for the full assignment see the Supporting Information] and the magnitude of the 3JCαH,NH scalar couplings46,47 were in agreement with the expected β-hairpin conformation.48 The amide temperature coefficients, ΔδNH/ΔT (Table 2), indicate formation of strong intramolecular hydrogen bonds involving the amide protons of A4 and Q9, confirming the two β-turns illustrated with red boxes in Figure 3. In line with the expectations, the intermediate ΔδNH/ΔT values of V2 and V7 indicate that these amide protons are involved in labile intramolecular hydrogen bonds that are in equilibrium with hydrogen bonds in which the solvent acts as an electron donor. The high coefficients of the amides N1, A(Cl)3, S(Me)8, and G10 reflect the fact that these are solvent accessible and do not form intramolecular hydrogen bonds, in agreement with the expected β-hairpin conformation.
Table 2. Chemical Shifts (δHα), Amide Proton Temperature Coefficients (ΔδNH/ΔT, 298–323 K), and Scalar Couplings (3JCαH,NH) for Peptide 5, Obtained in a DMSO-d6 Solution.
| residue position | δHα (ppm) | ΔδNH/ΔT (ppb/K)a | 3JCαH,NH (Hz) |
|---|---|---|---|
| N1 | 4.07 | 6.52 | 6.75 |
| V2 | 4.19 | 3.44 | 8.44 |
| A(Cl)3 | 5.25 | 5.20 | 8.45 |
| A4 | 4.66 | 1.28 | 8.28 |
| G5 | 3.84; 3.39 | 3.40 | – |
| P6 | 4.29 | – | – |
| V7 | 4.30 | 3.48 | 7.98 |
| S(Me)8 | 4.80 | 5.36 | 7.80 |
| Q9 | 4.53 | 1.68 | 8.44 |
| G10 | 3.84; 3.25 | 6.68 | – |
A ΔδNH/ΔT value of <3 indicates a strong intramolecular hydrogen bond. A ΔδNH/ΔT value of 3–5 indicates that the amide proton is in equilibrium between a solvent-exposed and an intramolecular hydrogen bond. A ΔδNH/ΔT value of >5 indicates that the amide proton is solvent-exposed.49
Figure 3.
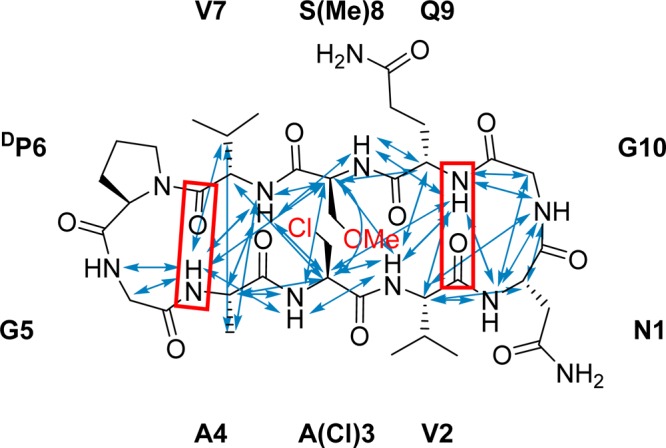
Key NOE correlations (blue arrows) and backbone intramolecular hydrogen bonds (red boxes) observed for 5 in a DMSO-d6 solution. The cross strand NOEs indicate β-hairpin to be the predominant conformer of 5 in solution. See the details in the Supporting Information.
Peptide 8, containing bromine as the halogen bond donor functionality at R3, was assigned following the same strategy as 5, however, at −10 °C in a DMF-d7 solution (for assignments and details, see the Supporting Information). As it was found to rapidly decompose above −10 °C, its amide temperature coefficients could not be reliably determined in DMSO-d6 for comparison to those of 2a, 2b, and 5.
NOESY Buildups
These were acquired with mixing times of 200, 300, 400, 500, 600, and 700 ms without solvent suppression. Interproton distances for 5 (Figure 3) were calculated according to the initial rate approximation from the linear part of the buildups (r2 > 0.98) using the N1β1 and N1β2 geminal protons as an internal distance reference (1.78 Å). In total, 63 NOE correlations were observed (key correlations are shown in Figure 3). Peptide 8 did not show any interstrand NOE correlations, with the spectra obtained at −10 °C in a DMF-d7 solution, suggesting that under these conditions it is unfolded in solution.
Solution Conformations of 5
These were identified like those of 1a–4b were (Table 1), using NAMFIS analysis.23 It should be noted that the conformation of flexible molecular systems cannot be accurately described with a single structure, but rather by deconvolution of their time-averaged spectroscopic data into probability-weighted geometries that are present in solution. Hence, NOESY buildup-based interproton distances and 3JCαH,NH-based dihedral angles were fitted to those back-calculated for the conformers of a computationally generated theoretical ensemble. Interproton distances were derived from NOE buildups as described above, and backbone dihedral angles were derived from 3JCαH,NH scalar couplings using a Karplus equation specifically developed for peptides.47,50 The theoretical ensemble was computed using restraint-free Monte Carlo conformational searches with intermediate torsion sampling and molecular mechanics energy minimization (MCMM) of each generated structure. For 1–4 (Table 1), the conformational search was performed twice, using the OPLS-2005 and AMBER* force fields, as implemented in Macromodel version 9.1. For 5, it was also performed with OPLS-3,51 the first force field also parametrized for halogen bonds of aromatic halogen bond donors. For all computations, geometries within 42 kJ/mol of the global minimum were retained. Output conformations from the calculations using different force fields were combined, and the redundant ones were eliminated to yield a final ensemble that was used as input for the NAMFIS analysis. On the basis of the experimental distance and dihedral data, the probability of the theoretically available individual conformations was estimated (for details about the NAMFIS analysis, see the Supporting Information). The folded β-hairpin conformations of 5 were identified to have 74% probability in solution. This is significantly higher than that observed for the reference peptide 2b (R3 = CH3, 29%). Selected conformers deduced by the NAMFIS analysis are shown in Figure 4, whereas all geometries identified to be present in solution are given in the Supporting Information.
Figure 4.
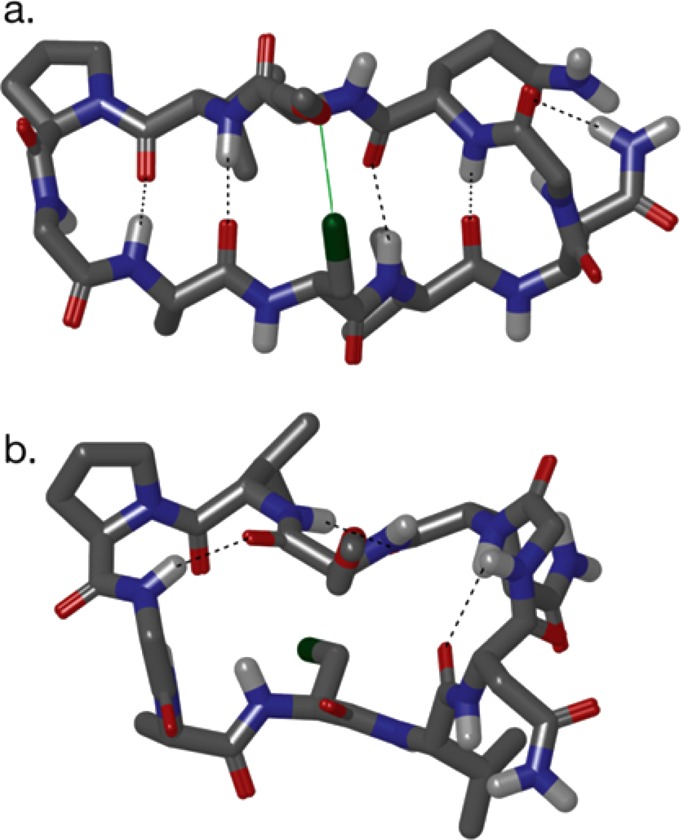
Selected solution conformers of 5, identified by NAMFIS analysis. (a) β-Hairpin conformation with a halogen bond formed at the R3–R8 interaction site (green). The total β-hairpin content was estimated to be 74%. (b) Non-β-hairpin conformer of 5. All conformers deduced by NAMFIS are given in Table S21 and Figure S11. Hydrogen bonds are colored gray; aliphatic protons were omitted for the sake of clarity.
As all orientations of flexible peptide side chains cannot be expected to be well represented in a Monte Carlo conformational search, to obtain the most accurate results, the NAMFIS analysis was based on the NMR data of the backbone protons only. Importantly, even these analyses identify the conformation in which the R3–R8 interstrand hydrogen bond is formed, as the major one for 1a and 2a.17,29 As none of the currently available force fields are parametrized for halogen bonds of aliphatic halogen bond donors, the molecular mechanics minimization selects conformations for 5 in which the chlorine of R3 avoids approaching the OCH3 of R8. To compensate for this shortcoming of the force field, a conformation optimized with a constrained Cl–OCH3 distance that is 90% of the sum of the van der Waals radii of the interacting atoms (2.94 Å ± 10%) and a restrained C–Cl–O angle of 180° ± 10% was also included in the ensemble for the NAMFIS analysis. This conformer encompasses a halogen bond with a theoretically optimal bond angle and bond length. It was identified by NAMFIS to exist in solution, which confirms that the formation of a halogen bond is compatible with the experimental data. Importantly, the incorporation of this conformer encompassing “optimal halogen bond geometry” into the NAMFIS analysis was not necessary for the evaluation of peptide folding. It was, however, included to compensate for a known weakness of the applied force field and to allow the assessment of whether the halogen-bonded peptide geometry is compatible with the experimental data.
For specific evaluation of the R3 and R8 side chain orientation of 5, to gain an understanding of whether the Cl···O halogen bond is formed, we have performed a NAMFIS analysis using the NMR data of these residues only, along with theoretical data back-calculated for conformations constrained to have all possible low-energy orientations, i.e., gauche+, gauche–, and trans for the involved Cα–Cβ bonds (for details, see the Supporting Information, with the conformations shown in Figure S12 and the experimental and back-calculated distances listed in Table S29). NAMFIS calculations based on experimental NOE data, with permutation of all possible assignments of the β-hydrogens of both amino acid side chains, reliably identified the side chain orientations (92%) that promote formation of a Cl···O halogen bond (Figure 4a) in the dominant conformer. This analysis shows that the orientation of the R3 and R8 side chain permits the formation of a Cl···O halogen bond.
The chemical shift of nuclei in the direct neighborhood of an equilibrium process is expected to show a comparably high temperature dependence, because equilibrium constants describing dynamic processes are temperature-dependent, as described by the van’t Hoff equation. Accordingly, the chemical shift of identical residues of structurally highly similar molecules is a useful reporter tool for identification of nearby equilibrium processes. We have monitored the temperature dependence of the chemical shift of the OCH3 protons of the R8 side chain of 2a, 2b, and 5 in the temperature interval of 298–373 K. The obtained NMR data corroborate the conclusion that 2b and 5 form interstrand hydrogen and halogen bonds, respectively, between their R3 and R8 side chains as the temperature dependence of their OCH3 protons is significantly higher (Δδ/ΔT values for 2a of 0.19 ppm/K and for 5 of 0.32 ppm/K) than that of the OCH3 of 2b (Δδ/ΔT value for 2b of 0.07 ppm/K), with the latter peptide possessing a non-interacting CH3 at position 3.
We have also performed variable-temperature NMR measurements to detect the melting curve describing the thermodynamic stability of the β-hairpin of peptides 2a, 2b, and 5. To avoid extensive signal overlaps and to detect comparably large chemical shift changes, we have incorporated a 13C isotope at the β-carbon of alanine 7, permitting the selective detection of its chemical shift within minutes. Analysis of these data has, however, revealed that the chemical shift of a single nucleus in a peptide does not reliably reflect the overall folding of a peptide but may instead be dominantly reporting on local geometrical changes.29 Therefore, these data were not used to characterize the relative thermodynamic stability of the folded conformation of the studied peptides.
Quantum Chemical Computations
These were performed to assess the possibility of Cl···O interaction in 5. The computational cost for a comprehensive investigation of the low-energy conformations of 5 with density functional theory (DFT) would have been prohibitive; therefore, the bonding situation was analyzed on the basis of a simplified model (see the Supporting Information for details). The calculated Cl···O bond length of 3.291 Å and C–Cl···O bond angle of 173.1° for the Me2O···ClCH3 dimer indicate that the analogous S(Me)8 and A(Cl)3 side chains of 5 should allow for some halogen bonding, which was further corroborated by the results from a previous high-level ab initio calculation for H2CO···ClCH3 (3.26 Å, 166.8°).52 The computed molecular electrostatic potential surface indicates the existence of a slightly positive region, i.e., a σ-hole, on the chlorine (Figure 5).
Figure 5.
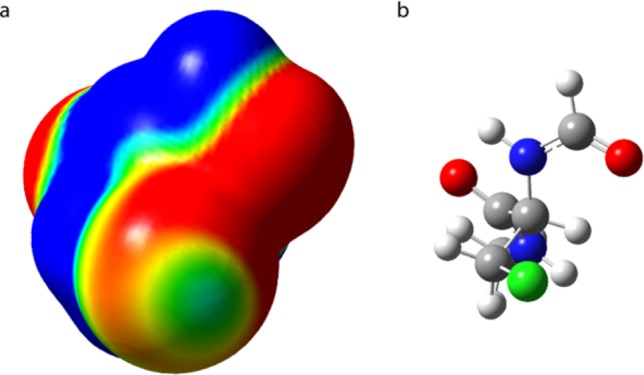
(a) Computed surface electrostatic potential of (b) β-chloroalanine indicating the presence of a σ-hole, i.e., a slightly positive region on the chlorine atom, promoting the formation of a halogen bond. The surface was computed on the 0.004 au contour of the electronic density. Color ranges are as follows: red, less than −50 kJ mol–1; yellow, between −50 and −3 kJ mol–1; green, between −30 and −20 kJ mol–1; light blue, between −20 and 0 kJ mol–1; and blue, greater than 0 kJ mol–1.
To estimate the stabilization energy provided by the Cl···O interaction, we reoptimized one of the (OPLS-351) equilibrium geometries of 5 using a two-layer ONIOM53 procedure, treating the residues involved in the interaction by DFT and the remaining residues on the PM6 level.54 The pair of residues S(Me)8 and A(Cl)3 were then isolated from the DFT geometry, and the energies of the S(Me)8···A(Cl)3 dimer and of the residues S(Me)8 and A(Cl)3 were estimated separately, with all geometries kept frozen. This procedure gave a bond energy of 41.2 kJ mol–1 for the S(Me)8 and A(Cl)3 interaction. This value is, however, dominated by the two hydrogen bonds between the residues. To isolate the contribution of the Cl···O bond, the chlorine atom in A(Cl)3 was replaced with a hydrogen atom, whose position was reoptimized while the rest of the system remained frozen. The bond energy between the two residues was recalculated, yielding a value of 35.1 kJ mol–1. Thus, the pure Cl···O bond energy contribution is estimated to be 41.2 – 35.1 = 6.1 kJ mol–1. The actual halogen bond energy contribution is expected to be slightly smaller as there is probably some dispersion interaction between the Cl in A(Cl)3 and one of the methyl H atoms in S(Me)8. Thus, this value is in good agreement with the literature (4.4 kJ/mol)52 and with the experimental finding that there is a weak conformation stabilizing Cl···O interaction in 5.
Conclusion
Folded peptides are mini-proteins that are common model systems for assessing the fundamental determinants of protein folding and stability.55 Using a 10-amino acid model system, the chlorine-centered halogen bond was demonstrated to provide conformational stabilization comparable to that of an analogous hydrogen bond, proving the applicability of the halogen bond in protein engineering. This is the first peptide/protein system in which a halogen bond was rationally incorporated and shown to improve the stability of the folded conformation in solution. The sensitivity of the applied technique for detecting weak interactions is reflected by its ability to observe the influence of a weak, chlorine-centered halogen bond. For peptide 5, the Cl···O halogen bond increases the molar fraction of the folded β-hairpin conformation by 44%, as compared to that of peptide 2b, in which no interstrand R3–R8 interaction is promoted. Because of the poor NMR properties of chlorine, the halogen bond interaction could not be directly detected but was deduced to exist on the basis of NOE distance analysis (NAMFIS) and variable-temperature measurements of the OCH3 protons on the R8 halogen bond acceptor. The formation of a halogen bond is the most plausible explanation for the observed thermodynamic stability of 5 being higher than that of 2b. The computed Cl···O bond energy is in good agreement with the experimental findings. Chlorine may be involved in van der Waals interactions, however, not significantly stronger than the analogous methyl functionality of 2b at its R3. We have not observed formation of a bromine-centered halogen bond, which is most likely attributable to the larger size of bromine. Hence, formation of a bromine-centered, linear halogen bond in 8 may require an altered side chain orientation as compared to 5 that may be available for this peptide only with a significant energetic cost, or in a conformation that does not permit formation of interstrand N–H···O=C or hydrophobic side chain–side chain interactions. Synthesis of an analogue encompassing an iodine halogen bond donor was not attempted as the aliphatic C–I bond was expected to have a chemical stability even lower than that of the C–Br bond of 8. The comparable conformation stabilizing ability of a chlorine-centered halogen bond to a hydrogen bond is remarkable. This may also be due to a R3–R8 side chain distance and an orientation that provides better geometry for formation of a weak halogen (5) than a weak hydrogen (2a) bond. The applicability of halogen bonding in peptide/protein engineering needs to be explored further to gain a detailed understanding of all steric and electronic factors that are of essential importance for the rational use of this so far unexplored molecular force in structural biology and drug discovery. Our experimental data will also be helpful for the parametrization of computational force fields to describe halogen bonding.
Halogen bonds are expected to be widely applicable in medicinal chemistry and protein engineering far beyond the modulation of conformational stability. They may, for example, be utilized in rational drug discovery to modulate the affinity and selectivity of molecular recognition events. Moreover, halogen bonding will likely be useful for the modulation of protein–protein interactions for which model studies of peptide and protein model systems in solution are of greatest significance.
We have developed a distinct model system that allows investigation of a specific weak interaction force in a complex molecular system. It has been carefully optimized to facilitate the investigation of any type of weak chemical force in a biologically relevant environment in solution, with its true potential being demonstrated here for the first time, through a comparative study of the halogen and the hydrogen bonds.
Glossary
Abbreviations
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- RP-HPLC
reversed phase high-pressure liquid chromatography
- TBTU
N′-tetramethyluronium tetrafluoroborate
- TMP
2,4,6-trimethylpyridine.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00429.
Details of the synthesis, spectroscopic data for compound identification, and details of the NMR and computational investigations (PDF)
European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) ERC Grant Agreement n° 259638 and Swedish Research Council Grant 2016-03602.
The authors declare no competing financial interest.
Supplementary Material
References
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. (2013) Definition of the halogen bond. Pure Appl. Chem. 85, 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. (2016) The halogen bond. Chem. Rev. 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfield D.; Huber S. M. (2016) Halogen bonding in organic synthesis and organocatalysis. Chem. - Eur. J. 22, 14434–14450. 10.1002/chem.201601844. [DOI] [PubMed] [Google Scholar]
- Priimagi A.; Cavallo G.; Metrangolo P.; Resnati G. (2013) The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 46, 2686–2695. 10.1021/ar400103r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.; Beer P. D. (2016) Halogen bonding anion recognition. Chem. Commun. 52, 8645–8658. 10.1039/C6CC03638D. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. (2013) Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 56, 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Metrangolo P.; Neukirch H.; Pilati T.; Resnati G. (2005) Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 38, 386–395. 10.1021/ar0400995. [DOI] [PubMed] [Google Scholar]
- Erdélyi M. (2012) Halogen bonding in solution. Chem. Soc. Rev. 41, 3547–3557. 10.1039/c2cs15292d. [DOI] [PubMed] [Google Scholar]
- Scholfield M. R.; Vander Zanden C. M.; Carter M.; Ho P. S. (2013) Halogen bonding (X-bonding): A biological perspective. Protein Sci. 22, 139–152. 10.1002/pro.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P. S. (2014) Biomolecular halogen bonds. Top. Curr. Chem. 358, 241–276. 10.1007/128_2014_551. [DOI] [PubMed] [Google Scholar]
- Voth A. R.; Hays F. A.; Ho P. S. (2007) Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. U. S. A. 104, 6188–6193. 10.1073/pnas.0610531104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford M. C.; Ho P. S. (2016) Computational tools to model halogen bonds in medicinal chemistry. J. Med. Chem. 59, 1655–1670. 10.1021/acs.jmedchem.5b00997. [DOI] [PubMed] [Google Scholar]
- Scholfield M. R.; Ford M. C.; Carlsson A.-C. C.; Butta H.; Mehl R. A.; Ho P. S. (2017) Structure-energy relationships of halogen bonds in proteins. Biochemistry 56, 2794. 10.1021/acs.biochem.7b00022. [DOI] [PubMed] [Google Scholar]
- Bertolani A.; Pirrie L.; Stefan L.; Houbenov N.; Haataja J. S.; Catalano L.; Terraneo G.; Giancane G.; Valli L.; Milani R.; Ikkala O.; Resnati G.; Metrangolo P. (2015) Supramolecular amplification of amyloid self-assembly by iodination. Nat. Commun. 6, 7574. 10.1038/ncomms8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdelyi M. (2017) Application of the halogen bond in protein systems. Biochemistry 56, 2759. 10.1021/acs.biochem.7b00371. [DOI] [PubMed] [Google Scholar]
- Thorson R. A.; Woller G. R.; Driscoll Z. L.; Geiger B. E.; Moss C. A.; Schlapper A. L.; Speetzen E. D.; Bosch E.; Erdélyi M.; Bowling N. P. (2015) Intramolecular halogen bonding in solution: 15N, 13C, and 19F NMR studies of temperature and solvent effects. Eur. J. Org. Chem. 2015, 1685–1695. 10.1002/ejoc.201403671. [DOI] [Google Scholar]
- Danelius E.; Brath U.; Erdélyi M. (2013) Insight into β-hairpin stability: Interstrand hydrogen bonding. Synlett 24, 2407–2410. 10.1055/s-0033-1339670. [DOI] [Google Scholar]
- Niebling S.; Danelius E.; Brath U.; Westenhoff S.; Erdélyi M. (2015) The impact of interchain hydrogen bonding on β-hairpin stability is readily predicted by molecular dynamics simulation. Biopolymers 104, 703–706. 10.1002/bip.22671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs A. C.; Bjorndahl T. C.; Hodges R. S.; Wishart D. S. (2002) Probing the structural determinants of type II′ β-turn formation in peptides and proteins. J. Am. Chem. Soc. 124, 1203–1213. 10.1021/ja011005e. [DOI] [PubMed] [Google Scholar]
- Lewandowska A.; Ołdziej S.; Liwo A.; Scheraga H. A. (2010) β-Hairpin-forming peptides; models of early stages of protein folding. Biophys. Chem. 151, 1–9. 10.1016/j.bpc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdélyi M.; Karlén A.; Gogoll A. (2006) A new tool in peptide engineering: A photoswitchable stilbene-type β-hairpin mimetic. Chem. - Eur. J. 12, 403–412. 10.1002/chem.200500648. [DOI] [PubMed] [Google Scholar]
- Minor D. L.; Kim P. S. (1994) Measurement of the β-sheet-forming propensities of amino acids. Nature 367, 660–663. 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- Cicero D. O.; Barbato G.; Bazzo R. (1995) NMR analysis of molecular flexibility in solution: A new method for the study of complex distributions of rapidly exchanging conformations. Application to a 13-residue peptide with an 8-residue loop. J. Am. Chem. Soc. 117, 1027–1033. 10.1021/ja00108a019. [DOI] [Google Scholar]
- Nevins N.; Cicero D.; Snyder J. P. (1999) A test of the single-conformation hypothesis in the analysis of NMR data for small polar molecules: A force field comparison. J. Org. Chem. 64, 3979–3986. 10.1021/jo9824450. [DOI] [Google Scholar]
- Koivisto J. J.; Kumpulainen E. T. T.; Koskinen A. M. P. (2010) Conformational ensembles of flexible β-turn mimetics in DMSO-d6. Org. Biomol. Chem. 8, 2103–2116. 10.1039/b921794k. [DOI] [PubMed] [Google Scholar]
- Andersson H.; Demaegdt H.; Vauquelin G.; Lindeberg G.; Karlén A.; Hallberg M.; Erdélyi M. t.; Hallberg A. (2010) Disulfide cyclized tripeptide analogues of angiotensin iv as potent and selective inhibitors of insulin-regulated aminopeptidase (IRAP). J. Med. Chem. 53, 8059–8071. 10.1021/jm100793t. [DOI] [PubMed] [Google Scholar]
- Grimmer C.; Moore T. W.; Padwa A.; Prussia A.; Wells G.; Wu S.; Sun A.; Snyder J. P. (2014) Antiviral atropisomers: Conformational energy surfaces by NMR for host-directed myxovirus blockers. J. Chem. Inf. Model. 54, 2214–2223. 10.1021/ci500204j. [DOI] [PubMed] [Google Scholar]
- Fridén-Saxin M.; Seifert T.; Hansen L. K.; Grøtli M.; Erdelyi M.; Luthman K. (2012) Proline-mediated formation of novel chroman-4-one tetrahydropyrimidines. Tetrahedron 68, 7035–7040. 10.1016/j.tet.2012.06.077. [DOI] [Google Scholar]
- Andersson H.; Danelius E.; Jarvoll P.; Niebling S.; Hughes A. J.; Westenhoff S.; Brath U.; Erdelyi M. (2017) Assessing the ability of spectroscopic methods to determine the difference in the folding propensities of highly similar β-hairpins. ACS Omega 2, 508–516. 10.1021/acsomega.6b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidro-Llobet A.; Álvarez M.; Albericio F. (2009) Amino acid-protecting groups. Chem. Rev. 109, 2455–2504. 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- Góngora-Benítez M.; Tulla-Puche J.; Albericio F. (2013) Handles for Fmoc solid-phase synthesis of protected peptides. ACS Comb. Sci. 15, 217–228. 10.1021/co300153c. [DOI] [PubMed] [Google Scholar]
- Malesevic M.; Strijowski U.; Bächle D.; Sewald N. (2004) An improved method for the solution cyclization of peptides under pseudo-high dilution conditions. J. Biotechnol. 112, 73–77. 10.1016/j.jbiotec.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xin X.; Liang Y.; Lin Y.; Zhang R.; Dong D. (2009) A facile and efficient one-pot synthesis of substituted quinolines from α-arylamino ketones under Vilsmeier conditions. Eur. J. Org. Chem. 2009, 4165–4169. 10.1002/ejoc.200900430. [DOI] [Google Scholar]
- Fmoc-l-serine methyl ester and 3-phenyl-1-propanol were used as model substrates for the reaction and were both successfully transformed to the desired brominated products, as confirmed from NMR spectroscopic and liquid chromatography–mass spectrometry analysis data.
- Jacobsen N. E. (2007) NMR spectroscopy explained: Simplified theory, applications and examples for organic chemistry and structural biology, 1st ed., John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- Das C.; Raghothama S.; Balaram P. (1998) A designed three stranded beta-sheet peptide as a multiple beta-hairpin model. J. Am. Chem. Soc. 120, 5812–5813. 10.1021/ja973739q. [DOI] [Google Scholar]
- Shankaramma S. C.; Singh S. K.; Sathyamurthy A.; Balaram P. (1999) Insertion of methylene units into the turn segment of designed beta-hairpin peptides. J. Am. Chem. Soc. 121, 5360–5363. 10.1021/ja990412b. [DOI] [Google Scholar]
- Haque T. S.; Little J. C.; Gellman S. H. (1996) Stereochemical requirements for beta-hairpin formation: Model studies with four-residue peptides and depsipeptides. J. Am. Chem. Soc. 118, 6975–6985. 10.1021/ja960429j. [DOI] [Google Scholar]
- Chung Y. J.; Huck B. R.; Christianson L. A.; Stanger H. E.; Krauthauser S.; Powell D. R.; Gellman S. H. (2000) Stereochemical control of hairpin formation in beta-peptides containing dinipecotic acid reverse turn segments. J. Am. Chem. Soc. 122, 3995–4004. 10.1021/ja993416p. [DOI] [Google Scholar]
- Nowick J. S.; Insaf S. (1997) The propensities of amino acids to form parallel beta-sheets. J. Am. Chem. Soc. 119, 10903–10908. 10.1021/ja972074p. [DOI] [Google Scholar]
- Nowick J. S. (1999) Chemical models of protein beta-sheets. Acc. Chem. Res. 32, 287–296. 10.1021/ar970204t. [DOI] [Google Scholar]
- Haque T. S.; Little J. C.; Gellman S. H. (1994) Mirror-image reverse turns promote beta-hairpin formation. J. Am. Chem. Soc. 116, 4105–4106. 10.1021/ja00088a067. [DOI] [Google Scholar]
- Varedian M.; Erdelyi M.; Persson A.; Gogoll A. (2009) Interplaying factors for the formation of photoswitchable beta-hairpins: The advantage of a flexible switch. J. Pept. Sci. 15, 107–113. 10.1002/psc.1103. [DOI] [PubMed] [Google Scholar]
- Erdelyi M.; Karlen A.; Gogoll A. (2006) A new tool in peptide engineering: A photoswitchable stilbene-type beta-hairpin mimetic. Chem. - Eur. J. 12, 403–412. 10.1002/chem.200500648. [DOI] [PubMed] [Google Scholar]
- GarciaMoreno B.; Dwyer J. J.; Gittis A. G.; Lattman E. E.; Spencer D. S.; Stites W. E. (1997) Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophys. Chem. 64, 211–224. 10.1016/S0301-4622(96)02238-7. [DOI] [PubMed] [Google Scholar]
- Kessler H. (1982) Conformation and biological activity of cyclic peptides. Angew. Chem., Int. Ed. Engl. 21, 512–523. 10.1002/anie.198205121. [DOI] [Google Scholar]
- Schmidt J. M. (2007) A versatile component-coupling model to account for substituent effects: Application to polypeptide ϕ and χ1 torsion related 3J data. J. Magn. Reson. 186, 34–50. 10.1016/j.jmr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333. 10.1016/0022-2836(91)90214-Q. [DOI] [PubMed] [Google Scholar]
- Erdélyi M. t.; Langer V.; Karlen A.; Gogoll A. (2002) Insight into β-hairpin stability: A structural and thermodynamic study of diastereomeric β-hairpin mimetics. New J. Chem. 26, 834–843. 10.1039/b111241d. [DOI] [Google Scholar]
- Kessler H.; Griesinger C.; Lautz J.; Mueller A.; Van Gunsteren W. F.; Berendsen H. J. C. (1988) Conformational dynamics detected by nuclear magnetic resonance NOE values and J coupling constants. J. Am. Chem. Soc. 110, 3393–3396. 10.1021/ja00219a008. [DOI] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. (2016) OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Riley K.; Hobza P. (2008) Investigations into the nature of hlogen bonding including symmetry adapted preturbation theory analyses. J. Chem. Theory Comput. 4, 232–242. 10.1021/ct700216w. [DOI] [PubMed] [Google Scholar]
- Dapprich S.; Komáromi I.; Byun K. S.; Morokuma K.; Frisch M. J. (1999) A new ONIOM implementation in gaussian98. Part i. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct.: THEOCHEM 461–462, 1–21. 10.1016/S0166-1280(98)00475-8. [DOI] [Google Scholar]
- Stewart J. J. P. (2007) Optimization of parameters for semiempirical methods v: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polticelli F.; Raybaudi-Massilia G.; Ascenzi P. (2001) Structural determinants of mini-protein stability. Biochem. Mol. Biol. Educ. 29, 16–20. 10.1111/j.1539-3429.2001.tb00057.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.