

Abstract
RNA is a natural multifunctional polymer, and is an essential component in both complex pathways and structures within the cellular environment. For this reason, artificial self-assembling RNA nanostructures are emerging as a powerful tool with broad applications in drug delivery and metabolic pathway regulation. To date, coordinated delivery of functional molecules via programmable RNA assemblies has been primarily done using nanosize RNA scaffolds. However, larger scaffolds could expand existing capabilities for spatial arrangement of ligands, and enable the controlled delivery of highly concentrated molecular loads. Here, we investigate whether micron-size RNA scaffolds can be assembled and further functionalized with different cargos (e.g. various siRNAs and fluorescent tags) for their synchronized delivery to diseased cells. Since known design approaches to build large RNA scaffolds are still underdeveloped, we apply, for the first time, a tiling method widely used in DNA nanotechnology. DNA tiles have been extensively used to build a variety of scalable and modular structures that are easily decorated with other ligands. Here, we adapt a double crossover (DX) DNA tile motif to design de novo DX RNA tiles that assemble and form lattices via programmed sticky end interactions. Because the assembly protocols used in DNA nanotechnology are not suitable for RNA assembly, we optimize them to guarantee high yield of RNA lattices. The resulting constructs are robust and modular with respect to the presence of distinct siRNAs and fluorophores. RNA tiles and lattices are successfully transfected in either human breast cancer or prostate cancer cells, where they efficiently knockdown the expression of target genes. Blood serum stability assays indicate that RNA lattices are more resilient to nuclease degradation when compared to individual tiles, thus making them better suited for therapeutic purposes. Overall, because of its design simplicity, we anticipate that this approach will be utilized for a wide range of applications in therapeutic RNA nanotechnology.
Introduction
RNA is a key biological regulator with a remarkable natural capacity to form stable, complex assemblies in vivo (the human ribosome, for instance, is a structure containing over 7 thousand nucleotides). Thus, large artificial RNA constructs should be compatible with the cellular environment, providing a programmable platform for delivery and organization of molecular loads. Yet, structural nucleic acid nanotechnology has focused primarily on DNA due to the stability and predictability of its Watson-Crick base-pair interactions1,2,3. Currently, the emerging field of RNA nanotechnology4 offers very few methods that allow programmable multi-strand assembly of RNA constructs with size exceeding a few hundreds of nanometers5,6,7,8,9. We demonstrate that designing principles developed for large-scale DNA self-assembly can be successfully adapted to RNA and support the development of larger programmable RNA scaffolds.
Some of the largest and most robust DNA nanostructures are built starting from nanometer-scale tiles, or monomers, formed by few DNA strands10. The core component of a multi-stranded DNA tile is the crossover junction, where there is a reciprocal exchange of strands between two adjacent helices to create a four-way junction11, which holds the two helices together. Tiles interact through programmable, rationally designed single-stranded regions known as “sticky ends,” which prescribe the controlled assembly patterns1,10,12,13,14,15. The tile-based assembly approach drastically differs from the well-known DNA origami approach2, where a long single-stranded DNA is folded into a desired 2D or 3D patterns by hybridizing with short “staple” strands. One of the most popular classes of DNA tile monomers is characterized by two helices held together by two double crossovers (DX). DX molecules exist in a variety of forms, distinguished by the orientations of their helical axes (parallel or anti-parallel), number of helical half-turns (even or odd) between crossovers, and number of helical half-turns between adjacent tiles (even or odd)10,12. These design parameters can be tuned to obtain lattices or tubular assemblies, and to modulate their melting temperature and polymerization rates. It is well understood how to obtain DNA DX tiles that, a) assemble robustly into structures that can grow up to tens of microns in length; b) can be decorated with a variety of functional domains and ligands12,16,17, and c) can encode information-bearing patterns 18,19. Thus, tile-based assemblies have several advantages as biomolecular scaffolds, including rational design principles, scalable growth, modularity of interactions among distinct tile types, and controllable stoichiometry of functional domains.
Lattices and arrays assembling from RNA tiles, rather than DNA, are promising structures for biomedical applications because they can take advantage of the richer structural motifs, innate functionalities of RNA, and its biocompatibility. Unlike multi-stranded DNA tiles, most existing for self-assembly RNA tiles fold from single stranded molecules (tecto-RNAs), and inter-tile bonds form via conserved tertiary motifs such as for example, loop-receptor 20, 21 or kissing loops6,22,5 interactions. This approach has two main advantages: 1) binding patterns are pre-determined from libraries of known tertiary motifs with controllable geometry, and 2) correctly designed short single strands are less prone to local kinetic traps with respect to multiple, longer strands. However, the size of existing RNA nanostructures is limited to a few hundreds of nanometers; in addition, their sequence design space is restricted to a characterized toolkit of parts, and it is unclear whether they are sufficiently modular and robust to include various functional domains without affecting the correct motif folding4.
Multi-stranded RNA structures, rationally designed to assemble exclusively via double helical domains, have been demonstrated in vitro23,24,25,26. However, their size is limited to the nanometer-scale as well. Despite of all potential benefits, the thorough characterization of functional RNA tile motifs with the capacity for large-scale, modular assembly is still lacking. The first attempts to build RNA tiles based on the crossover motif were performed to develop a metabolic engineering platform in E. coli, where enzymes could be localized by 0D, 1D or 2D RNA assemblies9,27. Individual tiles were functionalized with aptamer domains that, upon tile assembly, co-localize split fluorescent reporters or catalysts. Although the functional behavior of these assemblies met expectations, confirming the tile interactions, structural features and robustness of assembly are yet to be elucidated. Furthermore, these tiles are not suitable for construction of precisely engineered lattices due to base-pairing mismatches within the tiles, which result in formation of less rigid structures.
RNA is a programmable and scalable nanomaterial that can produce large assemblies with biological functions. Importing DNA nanotechnology design methods to RNA has the potential to yield increasingly complex and scalable nanostructures. Specifically, tile systems used in DNA nanotechnology could yield large RNA nanostructures, which may be useful in biomedical applications as delivery scaffolds for multiple nanoparticles28.
Here we describe and characterize a functional DX RNA tile motif designed de novo, that forms lattices with the capacity to reach the micron-scale, analogously to existing DNA DX tiles. This design is based on a DX tile motif originally designed for synergistic assembly of tiles with hybrid components of both DNA and RNA29. We decorate the DX RNA tile with DSRNA domains, we generate assemblies in vitro, and we demonstrate: 1) the formation of regular lattices that are robust upon the addition of functional domains (as verified via AFM imaging and gel electrophoresis), and in some cases exceed one micron in size; 2) superior stability of lattices in human blood serum compared to individual tiles; 3) the uptake and viability of functional tiles and lattices in human breast cancer and prostate cancer cells; 4) the capacity of functional tiles and lattices to silence target genes in transfected cells, with multivalent siRNA domains. Our results demonstrate that rational design of large, functional RNA nanostructures has the potential to match the complexity and size of scaffolds built with DNA, with improved bio-compatibility for delivery and scaffolding.
Materials and methods
Oligonucleotides
Fluorescently labeled RNA and DNA strands were purchased from Integrated DNA Technologies (Coralville, IA). All other RNA strands were transcribed and gel extracted in-house (see RNA extraction). The full list of DNA sequences is reported in the Supplementary Information (SI) file, Section 1.1, 1.2.
Sa: 5′- GGU GCG ACU AUG CAA CCU GC CUG GCA AGA CCU ACG AUG GAC ACG GUA ACG
Sb: 5′- GUC UUG CCA GGC ACC AUC GUA GGU CUU GCC AGG CAC CAU CGU AG
Sc: 5′- GCA CCC GUU ACC GUG UGG UUG CAU AGU C
Sc-GFP: 5′- GCA CCC GUU ACC GUG UGG UUG CAU AGU CUU CGG UGG UGC AGA UGA ACU UCA GGG UCA
Sc-PLK1: 5′ - GCA CCC GUU ACC GUG UGG UUG CAU AGU CUU CGU CAU UAA GCA GCU CGU UAA UGG UU
GFP sense strand: 5′ - pACC CUG AAG UUC AUC UGC ACC ACC G
PLK1 sense strand: 5′ - pAAC CAU UAA CGA GCU GCU UAA UGA CG
Alexa488 labeled GFP sense strand: 5′ - /AlexF488N/ACC CUG AAG UUC AUC UGC ACC ACC G
Alexa546 labeled PLK1 sense strand: 5′- /AlexF546N/ CCATTAACGAGCTGCTTAATGACG
Dicer Substrate RNA strands are underlined.
RNA extraction
RNA strands were individually transcribed in vitro from the corresponding DNA templates (sequences are reported in the SI file Section 1.1, 1.2) using the AmpliScribe T7 Flash transcription kit Epicenter, Inc.), by running the reaction at 37° C for 4h. RNA strands were gel extracted by either 10% or 15% denaturing PAGE. Next, RNA was eluted using 0.3 M sodium acetate at pH 5.3 and precipitated using ethanol and glycogen and re-suspended in nuclease-free water.
RNA construct purification
After anneal, tiles (blunt-ended) and lattices were electrophoresed through 15% non-denaturing polyacrylamide gels. The desired bands were located and cut out of the gel, and eluted in 1X tris-acetate-EDTA (TAE) and 12.5 mM magnesium chloride (MgCl2) at 4°C for about 48h.
Assembly methods
RNA nanostructures were self-assembled by slowly annealing extracted and purified RNA strands. Each strand was individually transcribed and gel extracted (see RNA extraction). Then, strands Sa, Sb, Sc-AG and/or Sc-AP with sense DNA strands were added in a 2:1:2:2 ratio (required stoichiometry to form a single tile) to a solution of 1X TAE / 12.5 mM MgCl2. This mix was slowly annealed from 70°C to 22°C over 24h.
Atomic force microscopy (AFM)
AFM images were collected in tapping mode using a Digital Instruments Multimode AFM with a Nanoscope (R) III controller, with Bruker SNL-10 silicon tips on a nitride lever with a spring constant of ≈ 0.24 N/m, drive frequency of ≈ 9-10 kHz. Imaging was done using 1X TBE / 12.5 mM MgCl2 buffer.
Denaturing PAGE
A gel pre-mix was prepared by mixing urea, water purified by thermolyne nanopure system (nanopure), and a 40% (v/v) solution of 19:1 acrylamide/bis-acrylamide. To start polymerization, the pre-mix was mixed with tris-borate-EDTA (TBE) buffer, ammonium persulfate (APS), tetramethylethylenediamine (TEMED), and nanopure water in the appropriate amount to obtain the desired polyacrylamide percentage. Gels were cast in 10×10 cm, 1 mm thick mini gel cassettes (Thermo Scientific) and allowed to polymerize for at least 2h before electrophoresis. Gels ran at room temperature at 100V in 1X TBE unless otherwise noted. After electrophoresis the gels were stained in SYBR® Gold Nucleic Acid Gel Stain (Invitrogen) or ethidium bromide; gels were imaged using the Bio-Rad ChemiDoc MP system.
Non-denaturing PAGE
40% (v/v) solution of 19:1 acrylamide/bis-acrylamide, TAE, MgCl2, APS, and TEMED were added together at appropriate concentrations for the desired polyacrylamide percentage, then cast in 10×10 cm, 1 mm thick mini gel cassettes (Thermo Scientific) and allowed to polymerize for at least 2h before use. Gels ran at 4° C at 150 V in 1X TBE buffer unless otherwise noted. After electrophoresis gels were stained in SYBR® Gold Nucleic Acid Gel Stain (Invitrogen) or ethidium bromide, then imaged using the Bio-Rad ChemiDoc MP system.
Dynamic Light Scattering (DLS) experiments
Samples were prepared at 1 μM tile concentration to produce individual tiles or lattices. 100 μL of sample solutions (prepared in 0.2 μm filtered buffer) were measured at 25° C with a Nano-series Zetasizer ZS (Malvern Instruments Ltd) equipped with a red (633 nm) laser. Readings with polydispersity index (PdI) below 0.2 were used.
UV-melting experiments
Experiments were carried out at 260 nm on an Agilent 8453 spectrophotometer coupled with the Agilent 89090 Peltier Temperature Controller. The temperature was gradually increased and the absorbance was recorded every 1°C with 10 seconds equilibration time.
Human blood serum stability assays
Alexa-488 labeled tiles and lattices (1 μM concentration for each strand) were incubated with 10% (v/v) human blood serum at 37° C. Samples were aliquoted over time, quenched on dry ice and visualized on native-PAGE, loaded in reverse order. Fluorescent bands corresponding to the labeled tiles and lattices were quantified and analyzed.
Transfection experiments
Functionalized RNA tiles and lattices were transfected in the human breast cancer cell line MDA-MB-231 (with or without GFP) and the prostate cancer cell line PC-3. Cells were grown in D-MEM medium (Gibco BRL) supplemented with 10% FBS and penicillin-streptomycin (pen-strep) in a 5% CO2 incubator. All transfections were performed using Lipofectamine 2000 (L2K) purchased from Invitrogen. 100X transfection solutions were pre-incubated at room temperature with L2K. Prior to each transfection, the cell media was replaced with OPTI-MEM with added RNA/L2K complexes at a final concentration of 1X. The cells were incubated for 4h followed by the media change (D-MEM, 10%FCS, 1% pen-strep).
Flow cytometry
For flow cytometry experiments, fluorescently labeled assemblies were transfected in MDA-MB-231 cells (with or without GFP) or PC-3 cells grown in 24-well plates. The cells were lifted with cell dissociation buffer 24h later to assess their level of Alexa 488 (Alexa 546) fluorescence or, in the case of MDA-MB-231/GFP cells, 72h later to assess their GFP signal for uptake and silencing experiments, respectively. The measurements were performed through fluorescence activated cell sorting on a FACSCalibur instrument (BD Biosciences, San Jose). At least 20,000 events were collected and analyzed with the CellQuest software to retrieve the geometric mean fluorescence intensity (gMFI) and the standard error of the mean (SEM).
Microscopy
All experiments were performed using an LSM 710 confocal microscope (Carl Zeiss, Oberkochen) and a Plan-Apochromat 63×/1.40 Oil lens. MDA-MB-231 cells plated in glass bottom dishes (Ibidi, Madison) were transfected with fluorescently labeled nanoconstructs. Upon 4h incubation at 37°C, the cells were washed three times with PBS and fixed with 4% paraformaldehyde for 20 minutes at room temperature. For Alexa 488 imaging, the 488 nm line of an argon laser was used as excitation and the emission was collected between 493 and 557 nm. For Alexa 546 imaging, a DPSS 561 laser was used for excitation and emission was collected between 566 and 680 nm. For silencing experiments, MDA-MB-231 eGFP cells were used and visualized 72h post transfection upon fixation. For GFP imaging, the 488 nm line of an argon laser was used as excitation and the emission was collected between 499 and 644 nm.
Viability assays
The viability of cells upon transfection was assessed through the CellTiter Blue assay (Promega, Madison) following manufacturer's protocol. Briefly, upon addition of the CellTiter blue reagents to the cells at a 1:6 ratio in DMEM, the fluorescence of the resorufin-forming compound was measured (560 ex/590 em) after 1-2h incubation at 37°C.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation. Informed consent was obtained from all human subjects included in the study. All institutional and national guidelines for the use of human derived materials and biohazard materials were followed. The study was approved by the Institutional Review Board and by the Institutional Biosafety Committee.
Results
Design of functional tiles
We designed DX RNA tiles adapting the motif proposed by Ko et al.29, originally developed to build hybrid DNA-RNA tiles. This motif, qualitatively similar to a DAE DNA tile10, is designed according to the parameters of A-form helical geometry (expected in RNA double helices). The tile consists of five RNA strands, of which only three have a unique sequence and are denoted as Sa, Sb, Sc; these RNA strands interact to generate two helical domains connected by two crossovers (Figure 1A, left). Tiles bind when their sticky-end domains (5 nucleotides long) hybridize, forming a double helix, and upon binding they form an alternating lattice pattern of tiles facing up and down. The intra-tile crossover distance is two full RNA helical turns (22 bp) and the inter-tile crossover distance is about two and a half helical turns (28 bp), as described in previous work30. To functionalize the tile, Dicer Substrate (DS) RNAs31, that provide Dicer-assisted intracellular release of the siRNAs32,33, were introduced. For that, tile strands (Sc) were extended with antisense strands of DS RNAs. As a proof of concept, we tested two DS RNA sequences designed to target 1) green fluorescent protein (GFP), incorporated in Sc strand variant Sc-GFP, and 2) polo-like kinase 1 (PLK1), incorporated in strand Sc-PLK1 (Figure 1A, right). We assembled the tiles that include exclusively either one (Sc-GFP or Sc-PLK1), or both types (Sc-GFP and Sc-PLK1) of DS RNAs. Non-functionalized tiles were used as negative controls. Assemblies including both DS RNAs (50% Sc-GFP and 50% Sc-PLK1) incorporate the domains at random locations in the structure, with an expected uniform distribution. For visualization, sense strands of GFP DS RNAs were labeled with Alexa 488, and the sense strands of PLK1 DS DNAs strand were labeled with Alexa 546. In the case of PLK1, whose downregulation causes an induction of apoptosis34, fluorescently labeled DNAs were used to prevent the dicing and the siRNA release that may cause the undesirable cell death during the uptake experiments.
Figure 1. RNA constructs for delivery of siRNA molecules are prepared in vitro and transfected into cells.
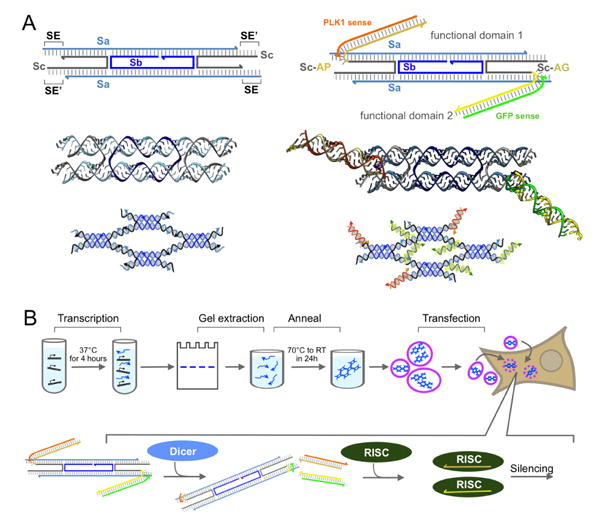
(A) Left: (top) expected secondary structure of DX RNA tile (Ko et al.)29, emphasizing the complementarity pattern of sticky end (SE) domains; (middle) Chimera46 rendering of the RNA DX tile; (bottom) expected lattice assembly. Right: (top) expected secondary structure of the functionalized DX RNA tile, where antisense DS RNA domains are incorporated on strand Sc; to demonstrate functionality we choose to target GFP and PLK1; (middle) Chimera46 rendering of the functional tile; (bottom) expected lattice assembly. (B) Top: Steps of our construct preparation protocol, starting from in vitro transcription to transfection of the constructs in human breast cancer cells; Bottom: summary of the siRNA release pathway.
Tile and lattice assembly and characterization
All constructs were obtained by slowly annealing RNA strands mixed in the appropriate stoichiometry; each RNA strand was separately transcribed and gel extracted (Figure 1B). Lattices were assembled from RNA strands including sticky ends, which are required for polymerization; individual tiles were assembled by removing the sticky end domains. Assembly of tiles and structures in vitro was characterized via AFM imaging and non-denaturing PAGE. AFM images in Figure 2A show that lattices assemble in all variants of our functional tiles (either a single type of DS RNA domain, or two distinct DS RNA domains). The DS RNA domains sticking out of the lattices are not visible because they are connected to the lattice by a loose linker (UU). Tiles yield lattices with similar nanoscale features, but different morphologies (flat lattices and tubular structures). In some cases assemblies appear to have a fixed width, which suggests that tubular structures may form in solution and open upon landing on the mica (Figure S8 in the SI file). Some of the lattices reach the micron size (Figure S8 in the SI file). The presence of DS RNAs in the lattices was verified via gel fluorescence analysis, by using fluorescently labeled sense DS RNA strands. As shown in Figure 2B, fluorescent bands co-localized with bands corresponding to lattices and tiles indicate successful incorporation of DS RNAs (additional images are in SI Figure S2). To further confirm the relative size difference between assembled lattices and individual tiles, we performed Dynamic Light Scattering (DLS) experiments. The hydrodynamic diameters of tiles functionalized with GFP and PLK1 DS RNAs are estimated to be 39 ± 1.3 nm and 42 ± 0.37 nm, respectively (Figure S4 in the SI file). The DLS results for assembled lattices show a significant increase in size distributions with the substantial increases in polydispersity index. This can be attributed to the formation of lattices with non-uniform size. Additionally, we estimated the relative resilience of tiles and lattices to nuclease degradation via human blood serum stability assays. The results of these assays (SI file Figure S5) show higher stability for lattices (t1/2=52 ± 16.7 min) compared to tiles (t1/2=6.6 ± 0.53 min). The melting temperature (Tm) of functionalized tiles was measured to be 64.4 ± 0.6°C with a melting profile represented by a single step curve. The functionalized lattices exhibited a two-step melting curve with the inflection points at 33.7 ± 0.2°C and 64.6 ± 1.1°C for each step, respectively (supporting Figure S6). The thermodynamic stability of lattices can be potentially fine-tuned by simply changing the lengths and composition of sticky-ends required for their assembly.
Figure 2. Functional DX RNA tiles assemble into lattices.
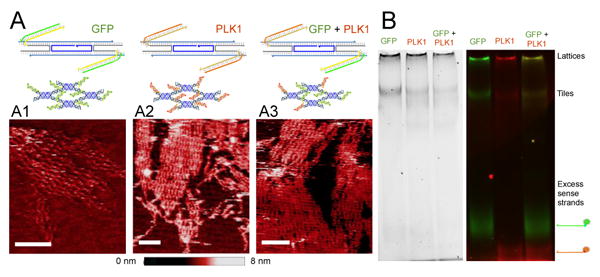
(A) Construct schematics and AFM images of lattices assembling from tiles functionalized with GFP DS RNA domains (A1), PLK1 DS RNA domains (A2) and both GFP and PLK1 domains (A3). Scale bar: 50 nm. (B) Gel electrophoresis of constructs including fluorescently labeled sense RNA molecules. Fluorescent bands indicate that DS RNA sense strands are incorporated in complexes that do not enter the gel (presumed to be lattices/polymerized tiles) and in individual tiles without sticky ends; the same pattern is observed in mono- and multi-valent tiles (with both PLK1 and GFP domains).
Transfection of tiles and lattices
Assembled functional tiles and lattices were transfected in human cells, and we assessed their capacity for gene silencing of either GFP, or PKL1, or both (Figure 3). All functional constructs were gel purified prior to transfection, as detailed in the Methods section. Control experiments indicate that association with Lipofectamine 2000 (L2K) does not disrupt the assembly of individual tiles (SI file Figure S3). In all transfection experiments, equivalent concentrations of free DS RNAs were used as control. To assess the relative uptake efficiencies, human breast cancer cells were transfected with fluorescently labeled functional tiles and lattices (Figure 3A-B). The results demonstrate dose dependent response with relatively higher transfection efficiencies for tiles (blunt-ended), which are the smallest tested constructs. Interestingly, the uptake efficiencies at lower concentrations (5 nM or lower) are very comparable between the constructs and free DS RNAs, while at 20 nM ∼3-fold increase in relative uptake is observed to free DS RNAs compared to functionalized assemblies. This difference can be explained by the saturation of the transfection agent (L2K) with RNA in the case of larger assemblies. The viability of cells was not affected by transfections with different assemblies (Figure 3C). To confirm that the assemblies stay intact in cells after the transfection, fluorescence co-localization experiments were carried out (Figure 3D-E and SI file Figure S7). In these experiments, tiles and lattices carrying in their composition both fluorescently labeled strands (Alexa 488 and Alexa 546) were visualized after the transfection. While we observe some separation of the dyes from the constructs (e.g. upon degradation), multiple co-localized signals were still measured in both transfected tiles and lattices. These results suggest that at least some of the transfected constructs remain structurally intact.
Figure 3. Cellular uptake of RNA tiles and lattices.
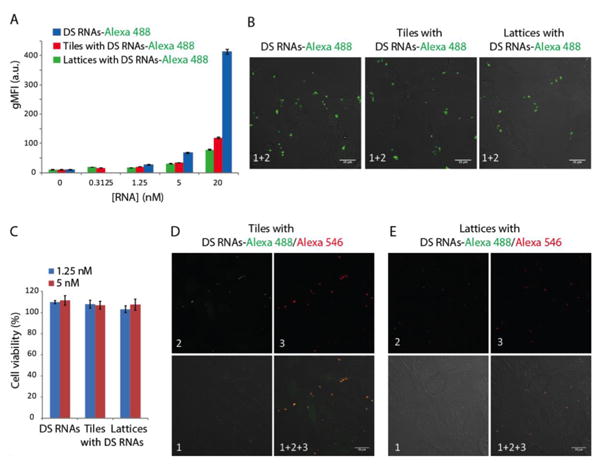
Human breast cancer cells (MDA-MB-231) were transfected with fluorescently labeled DS RNAs and gel purified lattices and tiles using Lipofectamine 2000. On the next day, the relative uptake was assessed by flow cytometry (A) and fluorescent microscopy (B). The viability of transfected cells was assessed at different construct concentration (C). For co-localization experiments, gel purified tiles (D) and lattices (E) labeled with two different dyes (Alexa 488 and Alexa 546) were transfected and analyzed by microscopy. Image numbers in D and E correspond to: differential interference contrast (DIC) images (1), Alexa 488 emission (2), and Alexa 546 emission (3). Images (1+2+3) are superposition of three different images. Error bars in A and C denote ± S.E.M.
Functional tiles and lattices successfully silence target genes
To validate the intracellular release of functional siRNAs through dicing, we carried out specific gene silencing experiments. First, as a proof of concept, GFP expressing breast cancer cells (MDA-MB-231/GFP) were transfected with constructs carrying anti-GFP DS RNAs (Figure 2 A1) and free DS RNAs. The results in Fig. 4B and 4C show concentration dependent GFP silencing with comparable efficiencies (of ∼90%) for assemblies and DS RNA at nanomolar concentrations. However, at lower picomolar range of concentrations, DS RNA was shown to be the most efficient silencer followed by tiles with the lattices being the least efficient (Figure 4C). To attribute the silencing of GFP to the presence of functionalized RNA constructs, non-functionalized tiles and lattices were tested as negative controls and showed no silencing (Figure 4B). The GFP silencing was observed for 10 days (SI file Figure S8) with a quicker fluorescence recovery for cells treated with functionalized lattices. To show the generality of the approach and the therapeutic potential of novel RNA nanoassemblies, human prostate cancer cell line (PC3) was transfected with constructs functionalized with anti-PLK1 DS RNAs (Figure 2 A2). PLK1 is a well-validated molecular target34, whose down-regulation causes an induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells; thus, PLK1 silencing studies have implications for the treatment of prostate cancer. Cell viability experiments (Figure 4D) clearly demonstrated dose-response with the results consistent with what observed in GFP silencing – at higher concentrations, constructs and DS RNAs cause comparable extent of cell deaths while at lower concentrations, tiles outperform lattices.
Figure 4. Specific gene silencing triggered by functionalized RNA tiles and lattices.
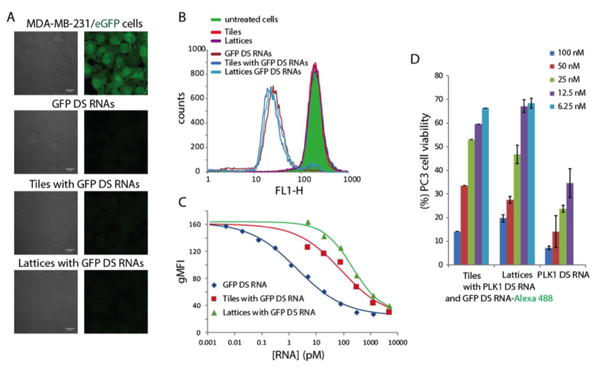
Down regulation of GFP expressed by human breast cancer cells was assessed by A: fluorescent microscopy and B: flow cytometry. C: Different concentration of constructs tested for relative silencing efficiencies. D: As an alternative, PLK1 silencing in human prostate cancer cells was assessed through cell viability assays. Cell viabilities are normalized to the non-treated cells and error bars denote ± SD, N=3.
Discussion
We demonstrated that the DX tile motif, well known in DNA nanotechnology, can be used to build functional RNA tiles assembling into large lattices. Tiles were functionalized by incorporating two distinct DS RNA domains for GFP and PLK1, which can be released intracellularly via dicing. Lattices and tiles were transfected into human breast cancer cells, where they were stable, viable, and performed the desired inhibition of gene expression.
Constructs robustly assembled in the presence of the DS RNA double stranded domains. However, we observed that lattice growth is affected by the presence of the DS RNAs. In particular we found that, depending on the sequence of the DS RNA domain, constructs may either form lattices or tubular assemblies. When no functional domains were present, we observed tubular structures that typically have a left-handed chirality (the source of chirality is yet to be elucidated)30. Tubular structures also formed when both DS RNAs domains are targeted against GFP (SI file Figure S9). However, lattices were observed when both DS RNAs attached were targeted against either solely PLK1 or against both GFP and PLK1 simultaneously. We speculate that, upon further investigation, this feature may be rationally exploited to obtain structures with switchable 3D organization which may be enzymatically controlled (for instance, by dicing the pre-programmed DS RNAs).
DNA and RNA nanostructures have been successfully used to deliver several nanoparticles, especially in targeting the stimulation of cellular immune response35 or for gene silencing36. For instance, DNA structures were used to deliver gold nanoparticles or antibodies3, CpG37 and doxorubicin38; 10 nm-sized DNA tetrahedra were also used to deliver siRNA molecules39. Yet, nm-sized DNA nanotubes of functionalized with siRNA40 were not stable in cells, and no siRNA activity was observed. Delivery of siRNA has been demonstrated with several RNA nano-scale structures such as pRNA-based assemblies41, nanocubes42 and kissing loop motifs used to build the RNA nanorings 43,44. To the best of our knowledge, our RNA lattices are the largest programmable multi-stranded RNA assemblies that are successfully used for siRNA delivery purposes (Figure S9 in the SI file).
Thermal annealing is required to form regular lattices with our DX tiles; this finding is in disagreement with previous reports27, but largely in agreement with the literature on DNA DX nanostructures12. Attempts to fold lattices isothermally only yielded formation of loose filaments30. Thus, the current design of tiles is not suited for in vivo expression and co-transcriptional assembly. Addition of hairpins to hierarchically expose sticky-ends only upon formation of tile crossovers appears to have enabled isothermal assembly in an RNA DX variant expressed in vivo9.
In conclusion, we presented the DX tile motif that self-assembles and yields RNA lattices of unprecedented size, which are viable in human cells and more stable than individual tiles in blood serum. These constructs are therefore potentially useful for in vivo delivery applications and for stoichiometric co-localization of a large number of components on a planar or tubular surface. This feature might be important in bio-catalysis or metabolic pathway engineering. One disadvantage our approach is the inability to precisely control size of the lattices. However, this limitation can be overcome because the architecture we presented is modular: in principle it is possible to engineer several sequence-distinct tile-types with programmable (and even algorithmic) interactions45,17,18 to form assemblies of controllable, monodisperse size. The most notable advantage of the tiles we presented is their relative designing simplicity and robustness to addition of functional domains; we expect that, in addition to siRNA functions, it will be possible to decorate tiles with aptamers for localization of a variety of ligands.
Supplementary Material
Acknowledgments
This work has been funded in part by the U.S. National Science Foundation under CAREER grant DMR-1450747 to EF and by the U.S. Department of Energy under grant SC0010595 to EF. This work has been funded in part with the start-up funds provided by UNC Charlotte to KAA. This work has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under Contract No. HHSN261200800001E. The authors thank Dr. Jeanette M. Bennett (UNCC) for sharing reagents.
References
- 1.Winfree E, Liu F, Wenzler LA, Seeman NC. Nature. 1998;394(6693):539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 2.Rothemund PW. Nature. 2006;440(7082):297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 3.Douglas SM, Bachelet I, Church GM. Science. 2012;335(6070):831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 4.Guo P. Nature nanotechnology. 2010;5(12):833–842. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geary C, Rothemund PW, Andersen ES. Science. 2014;345(6198):799–804. doi: 10.1126/science.1253920. [DOI] [PubMed] [Google Scholar]
- 6.Chworos A, Severcan I, Koyfman AY, Weinkam P, Oroudjev E, Hansma HG, Jaeger L. Science. 2004;306(5704):2068–2072. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 7.Lee JB, Hong J, Bonner DK, Poon Z, Hammond PT. Nature materials. 2012;11(4):316–322. doi: 10.1038/nmat3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koyfman AY, Braun G, Magonov S, Chworos A, Reich NO, Jaeger L. Journal of the American Chemical Society. 2005;127(34):11886–11887. doi: 10.1021/ja051144m. [DOI] [PubMed] [Google Scholar]
- 9.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Science. 2011;333(6041):470–474. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 10.Fu TJ, Seeman NC. Biochemistry. 1993;32(13):3211–3220. doi: 10.1021/bi00064a003. [DOI] [PubMed] [Google Scholar]
- 11.Seeman NC. Annual review of biophysics and biomolecular structure. 1998;27(1):225–248. doi: 10.1146/annurev.biophys.27.1.225. [DOI] [PubMed] [Google Scholar]
- 12.Rothemund PW, Ekani-Nkodo A, Papadakis N, Kumar A, Fygenson DK, Winfree E. Journal of the American Chemical Society. 2004;126(50):16344–16352. doi: 10.1021/ja044319l. [DOI] [PubMed] [Google Scholar]
- 13.Wei B, Dai M, Yin P. Nature. 2012;485(7400):623–626. doi: 10.1038/nature11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke Y, Ong LL, Shih WM, Yin P. science. 2012;338(6111):1177–1183. doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Chen Y, He Y, Ribbe AE, Mao C. Angewandte Chemie International Edition. 2006;45(12):1942–1945. doi: 10.1002/anie.200504022. [DOI] [PubMed] [Google Scholar]
- 16.O’Neill PR, Young K, Schiffels D, Fygenson DK. Nano letters. 2012;12(11):5464–5469. doi: 10.1021/nl3017797. [DOI] [PubMed] [Google Scholar]
- 17.Sharma J, Chhabra R, Cheng A, Brownell J, Liu Y, Yan H. Science. 2009;323(5910):112–116. doi: 10.1126/science.1165831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothemund PW, Papadakis N, Winfree E. PLoS Biol. 2004;2(12):e424. doi: 10.1371/journal.pbio.0020424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujibayashi K, Hariadi R, Park SH, Winfree E, Murata S. Nano Letters. 2007;8(7):1791–1797. doi: 10.1021/nl0722830. [DOI] [PubMed] [Google Scholar]
- 20.Jaeger L, Westhof E, Leontis NB. Nucleic acids research. 2001;29(2):455–463. doi: 10.1093/nar/29.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaeger L, Leontis NB. Angewandte Chemie International Edition. 2000;39(14):2521–2524. doi: 10.1002/1521-3773(20000717)39:14<2521::aid-anie2521>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Afonin KA, Lindsay B, Shapiro BA. DNA and RNA Nanotechnology. 2013;1(1) doi: 10.2478/rnan-2013-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Afonin KA, Bindewald E, Yaghoubian AJ, Voss N, Jacovetty E, Shapiro BA, Jaeger L. Nature nanotechnology. 2010;5(9):676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afonin KA, Kireeva M, Grabow WW, Kashlev M, Jaeger L, Shapiro BA. Nano letters. 2012;12(10):5192–5195. doi: 10.1021/nl302302e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu J, Liu Z, Jiang W, Wang G, Mao C. Nature communications. 2015;6 doi: 10.1038/ncomms6724. [DOI] [PubMed] [Google Scholar]
- 26.Bindewald E, Afonin K, Jaeger L, Shapiro BA. ACS nano. 2011;5(12):9542–9551. doi: 10.1021/nn202666w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sachdeva G, Garg A, Godding D, Way JC, Silver PA. Nucleic acids research. 2014;42(14):9493–9503. doi: 10.1093/nar/gku617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart JM, Franco E. DNA and RNA Nanotechnology. 2015;2(1) doi: 10.1515/rnan-2015-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko SH, Su M, Zhang C, Ribbe AE, Jiang W, Mao C. Nature chemistry. 2010;2(12):1050–1055. doi: 10.1038/nchem.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart JM, Subramanian HK, Franco E. 2016 Submitted Manuscript can be downloaded here: http://www.engr.ucr.edu/∼efranco/Stewartetal.html.
- 31.Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, Rossi JJ, Behlke MA. Nucleic acids research. 2005;33(13):4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bindewald E, Afonin KA, Viard M, Zakrevsky P, Kim T, Shapiro BA. Nano letters. 2016;16(3):1726–1735. doi: 10.1021/acs.nanolett.5b04651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afonin KA, Viard M, Tedbury P, Bindewald E, Parlea L, Howington M, Valdman M, Johns-Boehme A, Brainerd C, Freed EO. Nano letters. 2016;16(3):1746–1753. doi: 10.1021/acs.nanolett.5b04676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reagan-Shaw S, Ahmad N. The FASEB journal. 2005;19(6):611–613. doi: 10.1096/fj.04-2910fje. [DOI] [PubMed] [Google Scholar]
- 35.Khisamutdinov EF, Li H, Jasinski DL, Chen J, Fu J, Guo P. Nucleic acids research. 2014:gku516. doi: 10.1093/nar/gku516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okholm AH, Schaffert D, Kjems J. Chemical Biology of Nucleic Acids. Springer; 2014. Towards Defined DNA and RNA Delivery Vehicles Using Nucleic Acid Nanotechnology; pp. 325–345. [Google Scholar]
- 37.Schüller VJ, Heidegger S, Sandholzer N, Nickels PC, Suhartha NA, Endres S, Bourquin C, Liedl T. ACS nano. 2011;5(12):9696–9702. doi: 10.1021/nn203161y. [DOI] [PubMed] [Google Scholar]
- 38.Jiang Q, Song C, Nangreave J, Liu X, Lin L, Qiu D, Wang ZG, Zou G, Liang X, Yan H, Ding B. J Am Chem Soc. 2012;134(32):13396–13403. doi: 10.1021/ja304263n. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Lytton-Jean AK, Chen Y, Love KT, Park AI, Karagiannis ED, Sehgal A, Querbes W, Zurenko CS, Jayaraman M. Nature nanotechnology. 2012;7(6):389–393. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kocabey S, Meinl H, MacPherson IS, Cassinelli V, Manetto A, Rothenfusser S, Liedl T, Lichtenegger FS. Nanomaterials. 2014;5(1):47–60. doi: 10.3390/nano5010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shu D, Shu Y, Haque F, Abdelmawla S, Guo P. Nature nanotechnology. 2011;6(10):658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Afonin KA, Viard M, Kagiampakis I, Case CL, Dobrovolskaia MA, Hofmann J, Vrzak A, Kireeva M, Kasprzak WK, KewalRamani VN. ACS nano. 2014;9(1):251–259. doi: 10.1021/nn504508s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grabow WW, Zakrevsky P, Afonin KA, Chworos A, Shapiro BA, Jaeger L. Nano letters. 2011;11(2):878–887. doi: 10.1021/nl104271s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Afonin KA, Viard M, Koyfman AY, Martins AN, Kasprzak WK, Panigaj M, Desai R, Santhanam A, Grabow WW, Jaeger L. Nano letters. 2014;14(10):5662–5671. doi: 10.1021/nl502385k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan H, Park SH, Finkelstein G, Reif JH, LaBean TH. Science. 2003;301(5641):1882–1884. doi: 10.1126/science.1089389. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Journal of computational chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.