

SUMMARY
Stem cells determine homeostasis and repair of many tissues and are increasingly recognized as functionally heterogeneous. To define the extent of—and molecular basis for—heterogeneity, we overlaid functional, transcriptional, and epigenetic attributes of hematopoietic stem cells (HSCs) at a clonal level using endogenous fluorescent tagging. Endogenous HSC had clone-specific functional attributes in vivo. The intra-clonal behaviors were highly stereotypic, conserved under the stress of transplantation, inflammation, and genotoxic injury, and associated with distinctive transcriptional, DNA methylation, and chromatin accessibility patterns. Further, HSC function corresponded to epigenetic configuration but not always to transcriptional state. Therefore, hematopoiesis under homeostatic and stress conditions represents the integrated action of highly heterogeneous clones of HSC with epigenetically scripted behaviors. This high degree of epigenetically driven cell autonomy among HSCs implies that refinement of the concepts of stem cell plasticity and of the stem cell niche is warranted.
Graphical abstract
INTRODUCTION
Heterogeneity among cells within tissues is increasingly recognized in both normal and malignant conditions (Ding et al., 2012; Lemischka et al., 1986; Notta et al., 2011). Data in the hematopoietic system increasingly point to populations of cells being comprised of subpopulations with divergent properties. These include cells that have distinctive behaviors in terms of cell production and lineage bias (Dykstra et al., 2007; Picelli et al., 2013). Hematopoietic stem cells have been demonstrated to exhibit bias toward myeloid, lymphoid, or megakaryocytic lineage upon transplantation of single cells (Dykstra et al., 2007, 2011; Morita et al., 2010), on ex vivo barcoding and transplantation of populations (Aiuti et al., 2013; Gerrits et al., 2010; Jordan and Lemischka, 1990; Lemischka, 1993; Lemischka et al., 1986; Lu et al., 2011; Mazurier et al., 2004; Shi et al., 2002; Snodgrass and Keller, 1987), or by retrotransposon tagging of endogenous cells (Sun et al., 2014b). Further, single-cell transplant data have been coupled with single-cell gene expression analysis on different cells to resolve subpopulations with corresponding gene expression and repopulation potential (Wilson et al., 2015). Overlaying in vivo functional behavior of endogenous HSC clones with their gene expression and epigenetic characteristics represents a key unresolved challenge. The coupling of function with gene expression and chromatin state at clonal resolution is important for defining what governs stem cells; particularly for defining if HSC function is bounded by cell-autonomous epigenetic constraints. To test whether divergent HSC behaviors could be defined at a clonal level under homeostatic conditions and whether these behaviors were epigenetically determined, we created a multi-fluorescent mouse model that enables both molecular profiling and functional tracking of live cells in vivo.
RESULTS
Generation and Validation of the Multi-color Hue Mouse Model as a Clonal Tracking Tool
We took advantage of the fluorescent tagging system first developed for clonal lineage tracking in the nervous system to generate a transgenic animal bearing fluorescence protein encoding genes that could be recombined to provide a range of distinct colors (Livet et al., 2007). We created a new mouse strain (termed “HUe”) in which the fluorescent tags were driven by a ubiquitously expressed chicken actin promoter with intervening stop sequences flanked by LoxP sites followed by a fluorescent cassette containing GFP, EYFP, tDimer2, and Cerulean intercalated by multiple LoxP pairs (Figure 1A) to enable Cre-induced stochastic recombination and expression. The design is very similar to the independently created “Confetti” mouse (Snippert et al., 2010) with the distinction that the HUe mouse has ~20 tandemly integrated cassettes enabling a wider range (theoretically >103) of possible colors generated by random combinations, in analogy to the color range generated by a television screen using three basic color hues (red, blue, green). We crossed HUe with various promoter-driven Cres to demonstrate marking in mesenchymal or hematopoietic tissue (Figures 1C–1F).
Figure 1. Endogenous Labeling of Individual Cells with Different Colors.
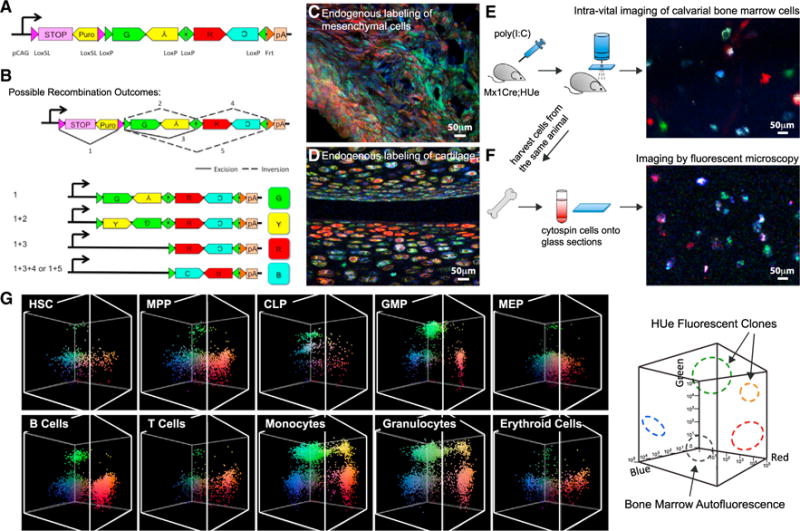
(A) HUe transgene construct contains GFP, EYFP, tDimer2, mCerulean fluorescent cDNAs arranged in tandem invertible segments flanked by four LoxP sites. A LoxP variant floxed STOP sequence was inserted in front of the fluorescent cassette, thereby prohibiting background fluorescence in the absence of Cre recombinase.
(B) Cre-mediated excision of the STOP sequence and random inversion or excision of the fluorescent cassette generates four possible color outcomes. Color complexity is further increased by insertion of multiple copies of transgene into the mouse genome. A HUe founder line with 20 copies of transgene inserted can have 103 color combinations.
(C) Testing the efficiency of expression of fluorescent proteins by crossing the HUe mice with different strains containing a Cre-driving promoter. When the HUe mouse was crossed to the limb mesenchyme-specific Prx1-CreER strain, we observed efficient endogenous labeling of cells in a fracture callus with various colors.
(D) Chondrocytes were labeled with color diversity when the HUe mouse was mated to a collagen-specific Cre driver, Col(II)-CreER.
(E) Hematopoietic cell labeling was assessed by crossing the Mx1-Cre strain with HUe (Mx1-Cre;HUe). When the Mx1-Cre;HUe mouse was given a pulse of pIpC, multi-colored hematopoietic cells within the calvarial cavity could be visualized using an intra-vital fluorescent microscopy system.
(F) Bone marrow cells of the same animal could be extracted and re-visualized on glass sections with fluorescent confocal microscopy.
(G) Clonal quantification of hematopoietic sub-compartments. Flow cytometry can identify and isolate hematopoietic stem cells (HSCs), progenitor cells: multipotent progenitors (MPPs), common lymphoid progenitors (CLPs), granulocyte macrophage progenitors (GMPs), megakaryocyte erythroid progenitors (MEPs), and mature cells of different lineages: B cells, T cells, monocytes, granulocytes, and erythroid cells. Endogenous HUe fluorescence from these populations is shown in the 3D graphs with × axis(tDimer2, red fluorescence),y axis(Cerulean, blue fluorescence), and z axis(EYFP, green fluorescence) representing increasing fluorescent intensities in log scale. The panel shows that hematopoietic cells at all hierarchy can be identified and clones within each compartment can be isolated by flow cytometry.
See also Figures S1 and S2 and Table S1.
To examine the efficiency of HUe in marking hematopoietic cells, we crossed the HUe mouse with the interferon-inducible Mx1-Cre strain (Kühn et al., 1995) (herein Mx1-Cre;HUe). We did not observe background fluorescence in the absence of Cre including in transplantation-mediated stress settings (data not shown). We activated endogenous hematopoietic cell labeling by administering polyinosinic:polycytidylic acid (pIpC) into Mx1-Cre;HUe mice and evaluated mice after an interval (>30 days) when the effects of interferon induction have been long shown to subside (Essers et al., 2009). Intra-vital imaging in live animals showed labeling of cells in the calvarial bone marrow (Figure 1E). These cells can be harvested from the bone marrow, stained with hematopoietic cell surface markers (Table S1), isolated by flow cytometry, and re-visualized by fluorescent microscopy (Figures 1F and 1G). We confirmed HUe fluorescence fidelity upon 12 days of cell division and differentiation by single cell colony assay in vitro (Figure S1A) and transplantation in vivo (Figures S1B and S1C).
Clusters of cells exhibiting distinct color signatures were apparent in the setting of Mx1-Cre-activated HUe mice (Figure 1G). To evaluate whether such clusters represented cell clones, we transplanted single LT-HSCs of distinct colors into 214 lethally irradiated recipients to define the boundaries of clonal populations by flow cytometry (Figure S1C). At 30 weeks post-transplant, the fluorescent positive population in blood and bone marrow was evaluated. We then used similar gates to isolate cells from the bone marrow of activated Mx1-Cre;HUe mice. Single clusters of cells with immunophenotypic signature of granulocyte monocyte progenitor (GMP) were sorted and transplanted into sublethally irradiated mice. Spleens were harvested at day 11 and DNA fingerprinting performed (Figure S2), demonstrating clonal signatures distinctive for each cluster. Clusters of cells with the same color, therefore, likely represent clonal descendants. However, overlapping boundaries can hamper our ability to distinguish individual clones. To minimize that issue, our analyses only involved animals with up to 15 color clones and used statistical treatment that did not require explicit partitioning of clones (Figure S3).
Hematopoiesis Is the Composite Product of Dissimilar Clones with Stereotypical Behaviors
We used Mx1-Cre;HUe mice to examine the clonal dynamics of native hematopoiesis. Sixteen Mx1-Cre;HUe mice were injected with pIpC to induce endogenous labeling of hematopoietic cells. Bone marrow aspirates were collected at 2, 3, 5, and 10 months of age and subjected to flow cytometry to quantify the total number of existing HUe fluorescent clones and the size of each clone (Figure 2; “Statistical Analysis” in the STAR Methods).
Figure 2. In Vivo Hematopoietic Dynamics under Homeostatic Conditions.
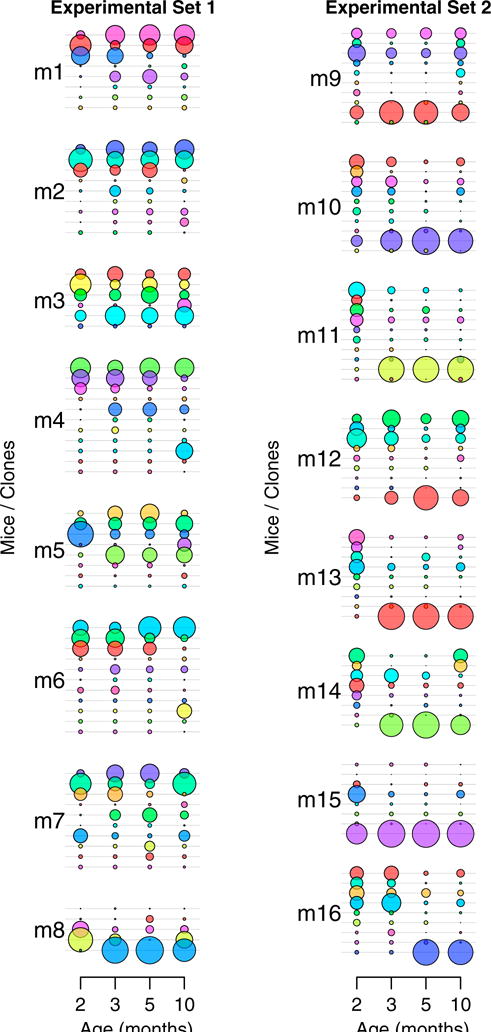
To assess in vivo hematopoietic dynamics in animals under homeostatic conditions, 16 pIpC-induced Mx1-Cre;HUe mice (m1–m16) were subjected to bone marrow aspiration of hematopoietic cells at months 2, 3, 5, and 10. Each uniquely colored circle represents an individual clone in an animal. The area of the circle is proportional to the size of each clone. Bone marrow hematopoietic clonal dynamics under homeostatic conditions were tracked from month 2 to month 10. Clones < 15 cells throughout the tracking period were not scored. See also Figures S3 and S4.
Results revealed that the hematopoietic tissue is composed of both persistent and fluctuating clones. We identified 5–11 clones per mouse with >15 cells throughout the 10-month chase period in a total of 16 animals (Figure 2). The clones had uneven, near-exponential distribution in size, with 1–4 large clones accounting for 80% of all cells in each animal (Figure S4A). Although clonal changes between month 2 and 3 were more pronounced, clonal dynamics were relatively consistent from month 3 to month 10 (Figures 2 and S4B). All animals showed one to four clones that was found at month 2 and persisted until month 10. Among the clones that persisted, some were stable in size, while some others fluctuated over time. Ten out of 16 mice had one to three clones identified at month 2 but disappeared at month 10. Twelve animals showed emergence of one to two new clones during the chase period, and sometimes these new clones became the dominant clones at later time points. Overall, our results show that native murine hematopoiesis is composed of a few major labeled clones that persist and others that expand, disappear or newly emerge.
HSC Behavior Is Highly Cell Autonomous
A major advantage of the HUe model is that we can measure and characterize the behavior of endogenous HSC in vivo, then selectively isolate live HSCs based on fluorescent tagging, transplant them into new hosts, and study their long-term behavior in competition or under varying stress conditions. This cannot be achieved by DNA barcoding or transposon insertion analyses because these methods require the destruction of cells. Transplanting equal aliquots of randomly fluorescent-tagged donor HSCs into 20–40 C57BL/6J recipients resulted in an unanticipated consistency of clonal behavior in recipients (p < 10−16) (Figures 3A, S4C,and S5A–S5C). That is, the individual clones in the recipients behaved after transplant as they had as endogenous HSC in the donor in terms of cell proliferation (defined by clone size) and lineage commitment. Each color-defined clone behaved similarly in different recipients, consistently exhibiting cell activation, proliferation, and lineage differentiation characteristics distinct from the other clones. The individual HUe fluorescent profiles of different cell types (Figure 3A, e.g., B cells from recipient 1) collected from multiple recipients were analyzed using unbiased hierarchical clustering. Clustering of the HUe fluorescent profiles grouped the same cell types together even though they were from different recipients (Figure 3B). This demonstrates that the extent and consistency of clone-specific biases was sufficiently large to distinguish different hematopoietic cell types in recipient mice solely based on their clonal composition, as measured by the fluorescent distribution of each cell type. We termed the group of transplanted recipients a “recipient cohort.” The consistency of behavior in a recipient cohort was striking and suggests cell autonomy governs the in vivo behavior of HSCs.
Figure 3. Hematopoiesis Is Composed of Dissimilar Clones with Cell-Autonomous Behavior.
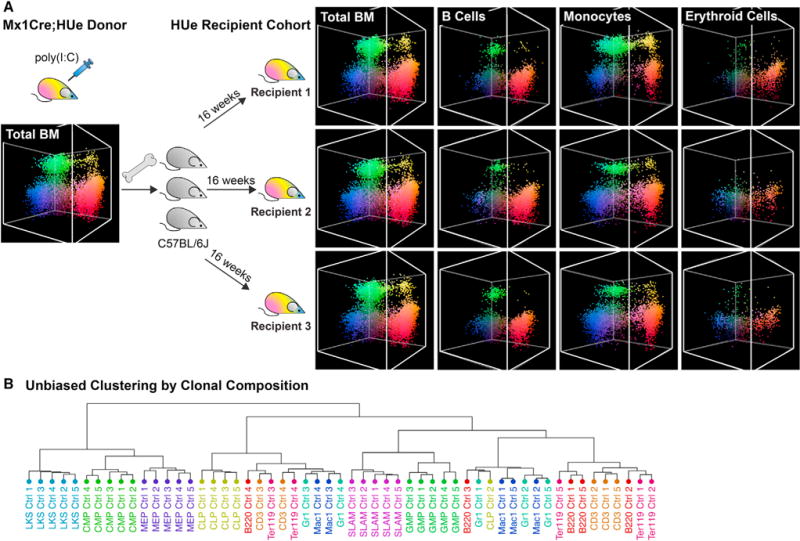
(A) The HUe recipient cohort—a cohort of mice with highly similar clonal fluorescence pattern. To generate a recipient cohort, Mx1-Cre;HUe mice induced with pIpC were used as donors. Randomly labeled fluorescent bone marrow cells from multiple Mx1-Cre;HUe donors were pooled as one mixture and isolated by fluorescence-activated cell sorting (FACS). Fluorescent HSPCs (LineageLoSca+cKit+) mixed with support cells from C57BL/6J were transplanted into each of 20 lethally irradiated C57BL/6J recipients. After 16 weeks of reconstitution, the recipients showed high consistency in clonal output including proliferation, fluorescence, and lineage characteristics in all hierarchy of hematopoietic cell types. HUe clonal fluorescent patterns of B cells, monocytes, and erythroid cells in multiple recipients are shown, illustrating consistency among the recipients and the distinction between different cell compartments.
(B) Unbiased hierarchical clustering of HUe fluorescence profiles. The distribution of HUe fluorescence in each hematopoietic cell type (e.g., B cells in Figure 1G) represents the clonal composition of that cell type in each mouse (Ctrl 1-5). Hierarchical clustering of such HUe profiles is shown. The clustering groups the same cell type samples from different mice together, illustrating that the pronounced pattern of proliferation and lineage bias exhibited by the individual clones is sufficient to consistently distinguish individual cell types from multiple recipients based on their clonal composition.
See also Figures S4 and S5.
HSC Cell Autonomy Is Persistent upon Stress
Using recipient cohorts, we could then test how individual HSC clones respond to a particular stress or perturbation. We divided a recipient cohort into sub-cohorts that received either saline control or inflammatory stress (0.3 mg/kg lipopolysaccharide [LPS], intraperitoneal) (Figure 4A) Similarly, another recipient cohort was divided into sub-cohorts that received either no treatment or genotoxic stress (4.5 Gy total body irradiation) (Figure 4B). Stressed and non-stressed sub-cohorts were harvested and analyzed by flow cytometry. Notably, while individual clones behaved differently (decreased or increased in clone size) in response to stress, consistency of response for a given clone was again observed in all recipients (Figures 4C and 4D). For example, while both HSCs and progenitors responded to LPS, there was a significant reduction in the size of myeloid lineage clones at 12 hr immediately following LPS stress (Figure 4E) but an overall increase in the number of clones (Figure S5D, p < 0.036), consistent across the treated recipient cohort (Figure 4C, p < 0.001). In contrast to LPS, irradiation reduced overall hematopoietic clonal complexity (Figure S5E) and caused expansion of the remaining clones, most notable at day 44 post-radiation when the hematopoietic system had returned to homeostasis (Figure 4F). Again, we observed statistically significant consistency of clonal response at all levels of the hematopoietic hierarchy across all recipient cohorts subjected to irradiation (Figure 4D, p < 0.001).
Figure 4. Hematopoietic Cell Autonomy Is Persistent upon Stress.
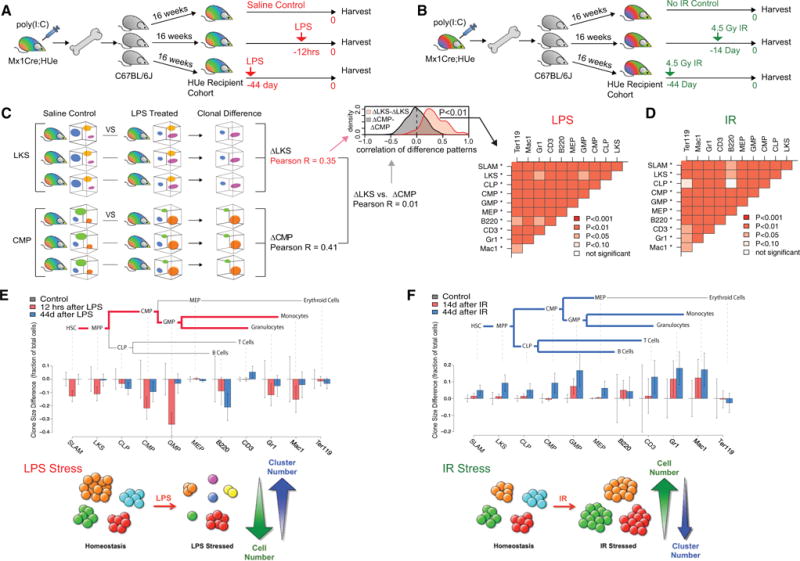
(A) The experimental design to study hematopoietic response upon LPS-mediated inflammatory stress. A HUe recipient cohort was generated by transplanting aliquots of the same mixture of randomly labeled HSPCs into 15 lethally irradiated C57BL/6J recipients. After 16 weeks of reconstitution, the cohort was divided into three sub-cohorts. One sub-cohort received LPS injection 12 hr priorto analysis (12hrs), another sub-cohort at 44 days prior(44 day), and a third sub-cohort received PBS treatment (control). All mice, including the control group, were sacrificed on the same day and assayed by flow cytometry.
(B) The experimental design to study the effect of genotoxic stress on hematopoietic clonal dynamics. An independent HUe recipient cohort was generated as in (A). After16 weeks of hematopoietic reconstitution, the cohort was divided into three sub-cohorts:a control group that received no irradiation, one group received 4.5 Gy irradiation 14 days prior to data collection (14 day), and another group received 4.5 Gy irradiation at 44 days prior to data collection (44 day). All mice, including the control group, were harvested for data collection on the same day.
(C) To test for consistency of clonal response to LPS treatment among cohort recipients, we performed pairwise comparisons of the fluorescent clonal pattern for each hematopoietic compartment (e.g., SLAM, LKS, CLP, etc.) and across each LPS-stressed and control animal. For all pairwise comparisons,the correlation of the LPS-associated clonal changes was significantly higher within a compartment than between compartments. In other words, the clonal response to the LPS insult was distinct among individual hematopoietic cell types, but was highly consistent across multiple HUe recipients (*p < 10−7 significance when comparing a given cell type with all other cell types).
(D) Consistency of the clonal response to irradiation treatment was assessed in the same way as in (C). For the vast majority of pairwise comparisons, the clonal changes within a cell compartment showed significantly higher correlation than between cell compartments. Each cell type was significantly distinct when compared against all other cell types (*p < 10−7) but consistent across multiple recipients.
(E) LPS treatment led to reduction in output of existing clones. Illustration of the hematopoietic clonal response to LPS inflammatory stress at 12 hr and day 44 in comparison to saline-treated controls at the stem, progenitor, and mature stages. To quantify the effect of LPS treatment, we performed pairwise comparison of mice from LPS-treated and control groups, detecting parts of the fluorescent spectra showing statistically significant differences in cell density. The barplot shows average change of cell numbers (measured as a fraction of the total number of cells measured) within such regions (whiskers show 95% confidence interval). The negative values correspond to decrease in the cell counts relative to control group. The analysis shows that at 12 hr following LPS treatment, the majority of existing clones were significantly reduced in size, accompanied by appearance of small new clones (Figure S5D). Such changes preferentially impacted the HSC-MPP-CMP-GMP branch of hematopoiesis, and were largely attenuated at 44 days post LPS treatment.
(F) Irradiation triggered expansion of existing clones. The barplots show the prevalent direction of cell density changes at 14 or 44 days following irradiation treatment. The changes were most pronounced at 44 days and showed widespread increase in the output of individual clones, significantly affecting most of the hematopoietic cell types.
See also Figure S3.
Immunophenotypically Equivalent Stem Cell Subpopulations Have Distinct Functional Attributes that Are Associated with Distinct Transcriptional and Regulatory States
To explore the molecular mechanisms that underpin the remarkably consistent behavior observed in HSC clones, we examined epigenetic and transcriptional states of select clones in parallel with flow cytometric analysis of the functional output of these clones in terms of clonal expansion and lineage outcome. LT-HSCs (LineageLoSca+cKit+CD48−CD150+) belonging to two clones (“Cohort1.Y” and “Cohort1.R”) were picked, and their DNA methylation and transcriptome states were assessed using whole-genome bisulfite sequencing and RNA sequencing (RNA-seq) assays, respectively (Figure 5A). Analysis of clonal contributions to different lineages (Figure 5B) indicated that the exemplar Cohort1.R clone exhibited higher proliferation rates (i.e., contributes higher than expected fraction of cells to the multipotent progenitor [MPP] compartment) (Figure 5C) and biased toward myeloid differentiation (Figure 5D). By contrast, the “Cohort1.Y” clone showed lower proliferation rates and exhibited strong bias toward lymphoid production (i.e., contributed to the common lymphoid progenitor [CLP] but not common myeloid progenitor [CMP] compartment) (Figures 5C and 5D).
Figure 5. Interrogation of the Molecular Signature Associated with Distinct Functions of HSC Clones.
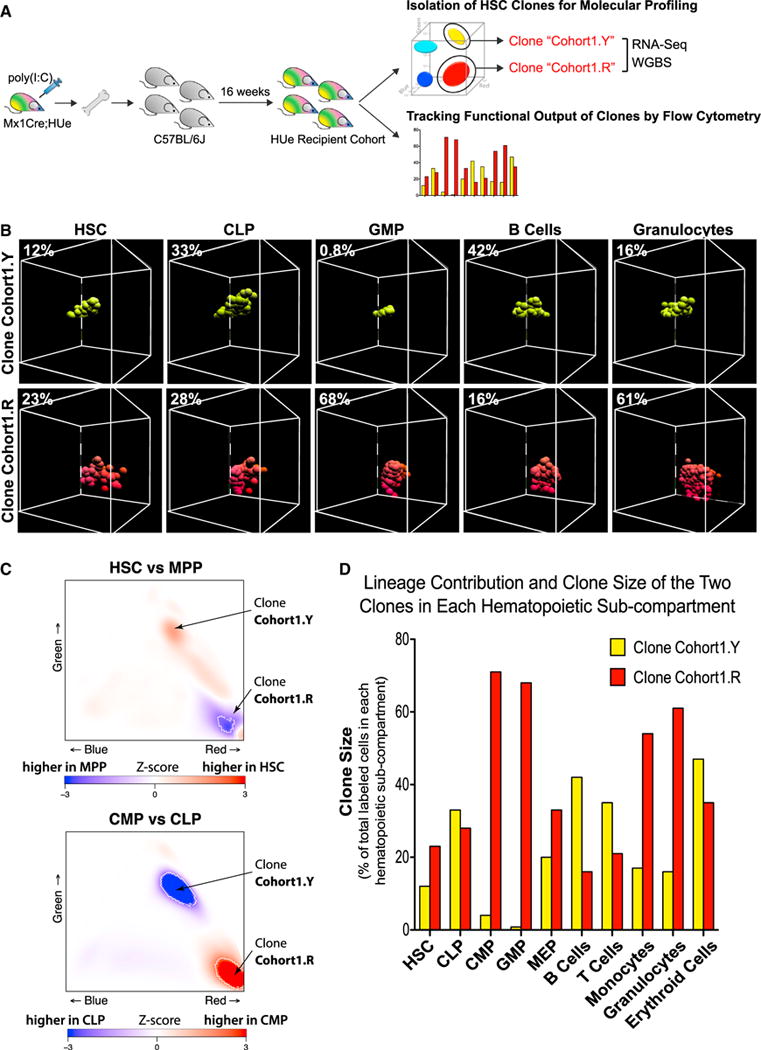
(A) To examine molecular differences associated with phenotypically distinct HSC clones, LT-HSC cells belonging to two selected clones (Cohort1.Y and Co- hort1.R) were harvested from a HUe recipient cohort, subjected to RNA-seq (transcriptome), WGBS (DNA methylation) assays, and flow cytometric measurement of multi-lineage reconstitution.
(B–D) Both long-term lineage contribution and clone size production of the two select LT-HSC (UneageLoSca+cKit+CD48−CD150+) clones (Cohort1.Y and Cohort1.R) to HSC, MPP, CLP, CMP, GMP, MEP, B cells, T cells, monocytes, granulocytes, and erythroid compartments were measured by flow cytometry (B and D) and analyzed as described in Figure S3. The percentage of cells representing either Cohort1.Y or Cohort1.R among all fluorescent cells in each hematopoietic compartment was shown. The Cohort1.R clone exhibited higher proliferation rate as it increased in size (density of cells) from HSC to MPP compartment (C and D) and was present in all hematopoietic compartments particularly toward myelopoiesis. In contrast, the Cohort1.Y clone showed lower proliferation rate (i.e., decreased clone density from HSC to MPP) and a strong presence in the CLP compartment (C), but reduced production in the CMP, GMP, and downstream myeloid compartments.
See also Figures S6 and S7.
Comparing the DNA methylation patterns of the HSCs from the two isolated clones, we found that while both clones were in the epigenetic state of non-differentiated HSCs (Figure 6A), the Cohort1.R clone showed significantly higher DNA methylation at HSC-specific enhancers and promoters and lower transcriptional expression magnitude of such genes (Figure 6B). Consistently, the Cohort1.R clone showed higher expression of genes associated with HSC proliferation (Kittler et al., 2007; Venezia et al., 2004) and G1 phase (Oki et al., 2014) and lower expression of genes characteristic of unmobilized HSCs (Chambers et al., 2007; Forsberg et al., 2010) and G0 phase (Oki et al., 2014) compared to the Cohort1.Y clone (Figure 6C). These consistent patterns of multiple epigenetic and transcriptional regulation highly reflected the enhanced proliferation rate observed for the dominant Cohort1.R clone by flow cytometry.
Figure 6. Immunophenotypically Equivalent HSCs Have Distinct Functional Attributes that Are Associated with Distinct Transcriptional and Epigenetic Regulatory States.
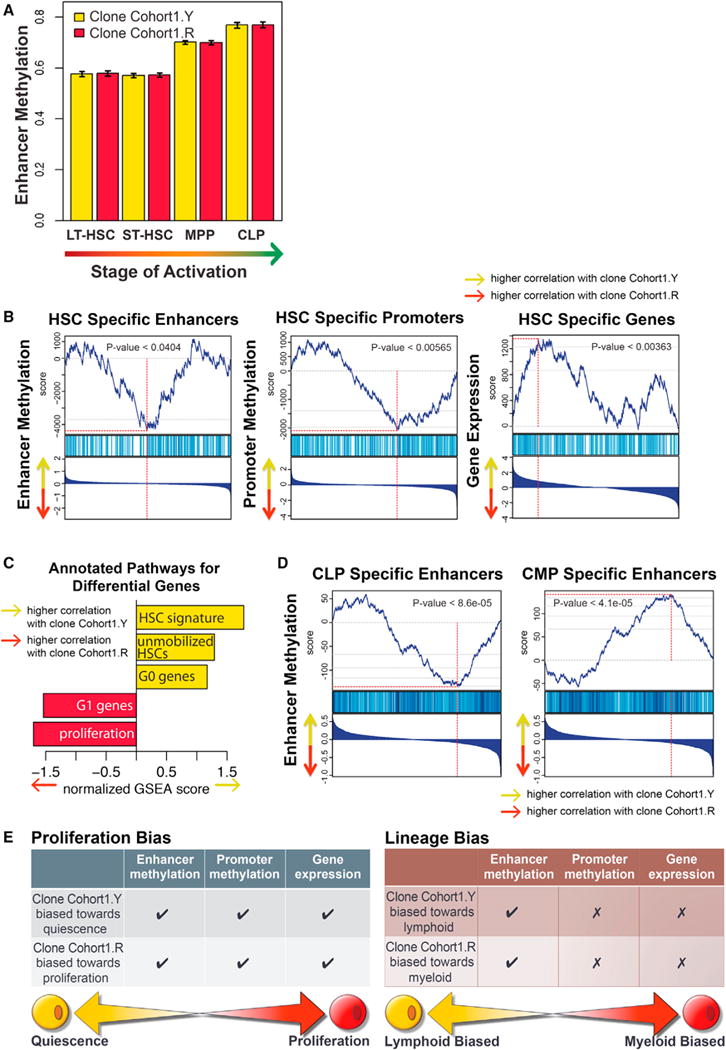
(A) The epigenetic state of both Cohort1.Y and Cohort1.R clones matched that expected of the HSCs. The DNA methylation state of enhancers activated at different stages of hematopoiesis was examined in the two clones. Both clones showed equally low methylation levels at the enhancers active at the HSC stage, with higher methylation observed at the enhancer regions activated at later MPP and CLP stages. Whiskers represent 95% confidence interval.
(B) Higher proliferative bias of the Cohort1.R clone was apparent from its epigenetic state. Gene set enrichment analysis (GSEA) analysis showed higher DNA methylation of HSC-specific enhancers and lower methylation of MPP-specific enhancers in the Cohort1.R clone relative to the Cohort1.Y clone. Similarly, Cohort1.R clone showed higher DNA methylation at HSC-specific and lower at MPP-specific promoter regions. Combined with the correspondingly higher expression of MPP- and lower expression of HSC-specific genes in the Cohort1.R clone, all three types of molecular signatures reflect higher proliferative bias of the Cohort1.R clone. In each GSEA plot, the genes (enhancer/promoters) are ranked according to their relative expression (DNA methylation level) ration between Cohort1.Y and Cohort1.R, with the highest Y/R ratios positioned on the left. The top plot shows rank sum statistics with the point of maximum deviation from 0 considered to be the enrichment score of that set (red vertical line). The middle plot marks the positions of the genes (promoters/enhancers) that belong to the set. The bottom plots show log2 fold ratio of expression (DNA methylation) magnitudes between Cohort1.Y and Cohort1.R.
(C) GSEA analysis showed higher expression of proliferation-associated genes and genes associated with G1 phase in the Cohort1.R clone compared to the Cohort1.Y clone, consistent with higher relative contribution of the Cohort1.R clone to the MPP compartment observed in fluorescence data. Higher relative expression of genes associated with unmobilized HSC and G0 phase signature was seen in the Cohort1.Y clone.
(D) Enhancer state reflected lymphoid-specific bias of the Cohort1.Y clone. Consistent with the pronounced lymphoid bias observed for the Cohort1.R clone in fluorescence data, Cohort1.Y clone showed lower DNA methylation at CLP-specific enhancer elements and higher methylation at CMP-specific enhancers relative to the Cohort1.R clone.
(E) Despite both Cohort1.R and Cohort1.Y clones having been immunophenotypically defined as HSCs, molecular profiling of their epigenetic and transcriptional landscape revealed distinctive signatures reflective of their differential functional behavior. Consistent with its larger clone size, the Cohort1.R clone had distinctive DNA methylation pattern at enhancer and promoter regions, as well as transcription of genes indicative of a proliferative cell state. In comparison, the Cohort1.Y clone showed a pronounced lymphoid output and such lineage preference was manifested by lower DNA methylation of lymphoid-specific enhancer regions, while no discernable pattern was detected in terms of promoter methylation or gene transcription.
See also Figures S6 and S7.
The strong lymphoid bias observed for the Cohort1.Y clone was reflected in the epigenetic state of regulatory regions with significantly lower DNA methylation levels in CLP-specific enhancer regions (Figure 6D). However, no significant differences were observed in the expression magnitude of the CLP- or CMP-specific genes or the DNA methylation state of their promoters (not shown). These results suggest that physiological differences between clones, such as lineage bias, can arise due to distinct epigenetic configuration of the regulatory regions at the level of HSCs (Figure 6E). Furthermore, these differences may not manifest themselves in transcriptional differences until later stages of differentiation. Therefore, a “poised” equipotent state may be evident in the transcriptome, but the epigenome provides lineage-constraining boundaries within which lineage bias will eventually be resolved.
Given that the enhancer DNA methylation state was particularly informative about lineage bias, we also examined whether other aspects of epigenetic state, such as chromatin accessibility, are also informative about inter-clonal variation. We have isolated an independent set of HSC clones (“Cohort2.G” and “Cohort2.P”) and in addition to measuring gene expression and DNA methylation, applied assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Lara-Astiaso et al., 2014) to assess genome-wide chromatin accessibility profile for each clone, in parallel with flow cytometric analysis of their functional output (Figure 7A). Analysis of clone size and clonal contribution to different lineages showed that while the Cohort2.G HSC clone was larger and contributed to both myeloid and lymphoid production, the Cohort2.P HSC clone was smaller and mostly contributed to lymphoid production (Figures 7B and 7C). Analysis of both chromatin accessibility and DNA methylation states of known enhancer regions confirmed that the regulatory state of the collected cells was more similar to that of HSCs than progenitor or effector cells (Figures 7D and 7E). The Cohort2.G clone exhibited epigenetic signatures of CMP-specific enhancers (Figure 7F), associated with a strong myeloid output as measured by flow cytometry (Figures 7B and 7C). The ATAC-seq differences were particularly prominent, revealing significantly higher chromatin accessibility of the CMP-specific enhancers in the Cohort2.G clone when compared between the two clones or to a set of CLP-specific enhancers within the Cohort2.G clone (Figure 7F). At the same time, analysis of RNA-seq data did not show significant differences in CMP/CLP transcriptional bias between the clones (data not shown).
Figure 7. Enhancer Methylation State Reflects Functional Differences between HSC Clones.
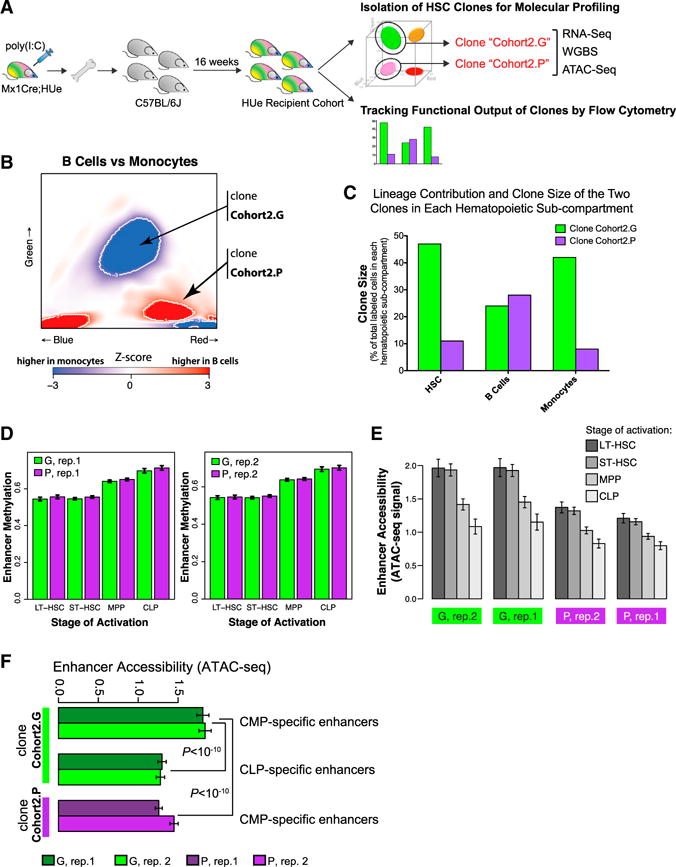
(A) In a second independent experiment, LT-HSC cells belonging to two independently selected clones (Cohort2.G and Cohort2.P) were harvested from an independent HUe recipient cohort, subjected to RNA-seq (transcriptome), WGBS (DNA methylation), and ATAC-seq (chromatin accessibility) assays, as well as flow cytometric measurement of clone size and multi-lineage reconstitution.
(B and C) We assessed both long-term lineage contribution and clone size production of the Cohort2.G and Cohort2.P clones toward myeloid (Mac+) and lymphoid (B220+) lineages by flow cytometry. The Cohort2.G clone had increased clone size (density of cells) at the HSC stage (C). While it contributed moderately to lymphoid cells, it had a strong myeloid output (C), consistent with the Z score heatmap indicating statistically significant (p < 10−3) bias of the Cohort2.G clone toward the myeloid lineage (B). In comparison, the Cohort2.P HSC clone was smaller, contributed moderately to lymphoid cells and had reduced production in myeloid cells.
(D) Both clones exhibited chromatin methylation pattern representative of LT-HSC and ST-HSC but not progenitors at the lineage-specific enhancer regions.
(E) Average chromatin accessibility, as measured by the ATAC-seq assay, at the lineage-specific enhancer regions in the Cohort2.G and Cohort2.P clones (two replicate measurements are shown for each clone). Consistent with the DNA methylation results shown in D, ATAC-seq assay indicated highest average accessibility at enhancers associated with LT- and ST-HSC states.
(F) Analogous to (E), Cohort2.G clone showed higher accessibility of the CMP-specific enhancers (relative to CLP-specific enhancers, and relative to the CMP-specific enhancers in the Cohort2.P clone), consistent with the strong myeloid bias observed for the Cohort2.G clone in the flow cytometric measurements.
(D)–(F) Whiskers give 95% confidence interval.
See also Figures S3, S6, and S7 and Table S2.
The analysis of molecular signatures underlying clonal biases can be limited by intra-clonal variability. To evaluate how the degree of transcriptional variability within each clone relates to the variability between clones, we performed single-cell RNA-seq analysis on HSCs associated with different clones in an independent cohort (see the STAR Methods). We found that consistent with the bulk measurements, different clones showed statistically significant bias in their distribution within the transcriptional space. However, the extent of intra-clonal variation varied from one clone to another (Figure S6). For instance, while one of the clones was preferentially found outside of the transcriptional state with a mitotic signature indicating overall lower cell-cycle frequency, there were cells from that clone that were also found within the mitotic state. Presence of high-coverage whole-genome bisulfite sequencing (WGBS) data also allowed us to check whether the clones isolated from each cohort showed notable genomic differences that could potentially impact their phenotype (see the STAR Methods). Despite good sensitivity of the approach, genomic copy number variation (CNV) analysis showed no difference between clones that belong to the same cohort (Figure S7), suggesting that the different HSC phenotypes we observed in these clones is a true biological phenomenon and not due to transgene-induced aberrant chromosome rearrangement. Together, these data show that HSC clones exhibit inter-clonal variation in behavior that is mirrored by the differences in their epigenetic state.
DISCUSSION
Our data demonstrate that endogenous clonal behavior can be quantitatively monitored in vivo under varying conditions and the clonal cells can be contemporaneously assessed for transcriptional and epigenetic characteristics. The results indicate that endogenous hematopoiesis is a composite of highly heterogeneous clones with very different cell kinetics, roughly balancing multipotent clones that are transient (generating cells for short intervals) with clones that provide persistent cell output. These data are consistent with the recent findings of Busch et al. (2015) where pulsed labeling of the pool of HSC was used to model cell kinetics. HSC were found to infrequently (~1/110 HSC per day) generate downstream progeny producing blood cells while maintaining the HSC pool. Our data are also consistent with recent data in mouse (Verovskaya et al., 2014) and human (Biasco et al., 2016), that the majority of the hematopoietic population is sustained by a few major HSC clones despite the existence of smaller clones. This is in contrast to the report by Sun et al. (2014b), which suggests that murine hematopoiesis is maintained by thousands of progenitor clones rather than HSCs. This discrepancy in clone number is likely due to the limited sensitivity of our method and the inability to measure clone size in the transposon-based tagging method. We found that a few labeled clones support the production of progeny for up to ~1 year, but we cannot estimate what proportion of the active HSC were labeled by our method. Of note, we did not find labeled progenitors without a corresponding labeled stem cell, but again our method does not have the ability to comprehensively scan for cell clones. Rather, we can define the attributes of a limited number of clones over time. Inferring from them, our data support a model where hematopoiesis is the composite of multipotent stem cells where some HSC persist for long intervals while others are transient. The advantages of such a composite model where clones turn cell production “on” and “off” or maintain cell production “on” seem self-evident as a means of sustaining hematopoiesis in the context of widely varying and at times hostile, physiologic challenges.
In addition, our data points to an unanticipated stereotypical behavior of clones upon transplantation. While it has been previously reported that single isolated cells can be serially transplant with retained behavior (Dykstra et al., 2007; Picelli et al., 2013), it is not clear that this behavior reflects the behavior of the endogenous cells. The system reported here enables comparison of endogenous and transplanted HSC. While the transient induction of interferon with pIpC used to activate the Mx-1 promoter may not be considered an unperturbed state, it is modest compared with transplantation, and it has been shown that HSC functions revert to baseline shortly after pIpC exposure (Essers et al., 2009). Overall, our data are consistent with behavioral features of individual HSC clones being established in development prior to young adulthood and persistently manifest under varying conditions.
Lineage bias and proliferative potency has been demonstrated previously (Dykstra et al., 2007; Morita et al., 2010; Muller-Sieburg et al., 2004; Picelli et al., 2013), including at the clonal level (Sun et al., 2014b). The data here indicate that multiple other characteristics including sensitivity to inflammation or radiation are also clone-specific features. The consistency of these characteristics despite clones residing in different hosts again points to rather remarkable cell autonomy. While stem/progenitors are generally thought of as relatively plastic cells with the capacity to respond variably to their specific environment, a wide range of behaviors appear to be highly constrained by cell intrinsic features. The functional characteristics of cell pools are therefore likely to reflect an ensemble phenotype of individual clones with much more bounded behaviors. These stereotyped behaviors have distinct molecular features at least as we defined by using the fluorescence system for isolation and analysis of clones with specific functions. While we assessed a limited number of cells with particular functional attributes, we envision that these data will encourage a more comprehensive analysis of clones with different lineage biases. proliferation, and response to inflammation or tolerance of genotoxicity as we defined here, but include a far greater range of activities. From the data we have and the consistency of behaviors in multiple hosts, we anticipate that each of the functional clonal behaviors will have molecular signatures. Defining these signatures may provide both new insights into how in vivo HSC functions are governed and identify points where molecular manipulation can change specific in vivo outcomes.
Accomplishing a link of functional features with gene expression and epigenetic characterization is a challenging dimension of stem cell biology as noted and explored by others (Wilson et al., 2015). Single cell studies of function and gene expression have been conducted (Tsang et al., 2015; Wilson et al., 2015), however, they are generally performed on different cells within an immunophenotypically, not functionally or clonally isolated subset. Similarly, detailed epigenetic characteristics have been assessed in populations of cells isolated by immunophenotype (Bock et al., 2012; Sun et al., 2014a). We have attempted to accomplish these analyses at a clonal level with clonal functional features defined in vivo. The result is that HSCs appear to have epigenetic features dictating how they will behave. The transcriptome was not consistently correlated with behavior. Rather, DNA methylation and chromatin accessibility data provide a clearer window into the ultimate function of specific clones. HSCs therefore appear to establish and retain a memory imposed on them prior to the testing we conducted in the young adult stage of organism development. This epigenetic memory is persistent and guides their function even with stress at times when they are presumably exposed to highly different exogenous conditions.
On their surface, the results of this study argue against the concept of the HSC as a plastic cell capable of different functions in response to particular organismal needs. Rather, our data indicate that HSC have clone-specific stereotyped behavior that is epigenetically constrained. Varied responses by the HSC pool to particular stresses may therefore reflect differential activation of clones of cells that each have their own predefined function, rather like a set of chess pieces. Achieving a nuanced, condition-specific response may then reflect differential activation of particular clones or particular combinations of clones. The data also argue against at least some aspects of the niche hypothesis: that cells are dependent upon a specific microenvironment for their regulated self-renewal and differentiation. HSC behaviors such as lineage and proliferation outcomes were preserved even after transplantation into an independent host and thereby, a presumably independent niche. It may be that the niche governs only fundamental aspects of HSC behavior like survival or aspects of cell state other than the ones we measured. However, the data are also consistent with a facultative model where the niche is generic and responds to stem/progenitor cells to provide the specific support functions dictated by the particular stem/progenitor cells it serves. The largely cell autonomous HSC might “condition” its own niche. An alternative model is that specific HSC clones find and localize to very specific niches that match their needs. There may be a heterogeneity among niches comparable in complexity to the HSC pool and transplantation succeeds when specific functional niches pair with specific functional HSC partners. Distinguishing between these alternatives will help define the relative importance of niche components to hematopoietic function and guide efforts to control cell production.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B220-biotin | Biolegend | Cat#:103204; RRID: AB_312989 |
| CD3e-biotin | Biolegend | Cat#:100244; RRID: AB_2563947 |
| CD4-biotin | Biolegend | Cat#:100508; RRID: AB_312711 |
| CD8a-biotin | Biolegend | Cat#:100704; RRID: AB_312743 |
| CD19-biotin | Biolegend | Cat#:115504; RRID: AB_313639 |
| CD11b-biotin | Biolegend | Cat#:101204; RRID: AB_312787 |
| Gr1-biotin | Biolegend | Cat#:108404; RRID: AB_313369 |
| Ter119-biotin | Biolegend | Cat#:116204; RRID: AB_313705 |
| CD11c-biotin | Biolegend | Cat#:117304; RRID: AB_313773 |
| NK1.1-biotin | Biolegend | Cat#:108704; RRID: AB_313391 |
| Streptavidin-Pacific Orange | Invitrogen | Cat#:S32365 |
| cKit-APC-Cy7 | Biolegend | Cat#:105826; RRID: AB_1626278 |
| Sca-PE-Cy7 | Biolegend | Cat#:108114; RRID: AB_493596 |
| Sca-PE-Cy5 | Biolegend | Cat#:108110; RRID: AB_313347 |
| CD48-APC | Biolegend | Cat#:103412; RRID: AB_57199 |
| CD150-PE-Cy5 | Biolegend | Cat#:115912; RRID: AB_493598 |
| CD127-PE-Cy7 | eBioscience | Cat#:25-1271-82; RRID: AB_469649 |
| CD16/32-PE-Cy7 | eBioscience | Cat#:25-0161-82; RRID: AB_469598 |
| CD34-efluor660 | eBioscience | Cat#:50-0341-82; RRID: AB_10596826 |
| B220-APC | eBioscience | Cat#:17-0452-83; RRID: AB_469396 |
| CD3-APC | Biolegend | Cat#:100236; RRID: AB_2561456 |
| Mac1-APC | Biolegend | Cat#:101212 |
| Gr1-APC | Biolegend | Cat#:108412; RRID: AB_313377 |
| Ter119-APC | Biolegend | Cat#:116212; RRID: AB_313713 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polyinosinic:polycytidylic acid | Amersham | Cat#:24939-03-5 |
| Isoflurane | Sigma-Aldrich | Cat#:792632-250MG |
| Lipopolysaccharide from E. coli 055:B5 | Sigma-Aldrich | Cat#:L2880-10MG |
| Medium 199 | ThermoFisher Scientific | Cat#:11043023 |
| Fetal bovine serum | ThermoFisher Scientific | Cat#:10082139 |
| RNase Out | ThermoFisher Scientific | Cat#:10777019 |
| Calcein AM | ThermoFisher Scientific | Cat#:C3099 |
| Propidium iodide | ThermoFisher Scientific | Cat#:P3566 |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat#:M0541L |
| Invitrogen SYBR Green I Dye | Invitrogen | Cat#:S7563 |
| 4OH-tamoxifen | Sigma | Cat#:T-5648 |
| Progesterone | Sigma | Cat#:P-3972 |
| Critical Commercial Assays | ||
| Smart-seq2 protocol | Picelli et al., 2013 | N/A |
| Nextera XT | Illumina | Cat#:FC-131-1024 |
| NuGen’s Ovation® Ultralow Methyl-Seq Library Systems | NuGen | Cat#:0335 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| QIAGEN Epitect Bisulfite kit | QIAGEN | Cat#:59104 |
| Illumina Nextera DNA Preparation Kit | Illumina | Cat#:FC-121-1030 |
| QIAGEN MinElute PCR purification Kit | QIAGEN | Cat#:28004 |
| Deposited Data | ||
| HUe RNA-Seq data | GEO datasets http://www.ncbi.nlm.nih.gov/gds | GEO: GSE60101 |
| 129P2 WGBS data | GEO datasets http://www.ncbi.nlm.nih.gov/gds | GEO: GSE56986 |
| BALBcJ WGBS data | GEO datasets http://www.ncbi.nlm.nih.gov/gds | GEO: GSE60485 |
| 129P2 sequence structure annotation files | http://www.sanger.ac.uk/science/programmes/mouse-and-zebrafish-genetics | 129P2 |
| BALBcJ sequence structure annotation files | http://www.sanger.ac.uk/science/programmes/mouse-and-zebrafish-genetics | BALBcJ |
| Referenced RNA-Seq data of hematopoietic subtypes | Lara-Astiaso et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| HUe mouse | This paper | N/A |
| C57BL6/J | Jackson Laboratories | 000664 |
| Mx1-Cre | Jackson Laboratories | 003556 |
| Prx1-CreER | This paper | N/A |
| Col(II)-CreER | Jackson Laboratories | 006774 |
| Recombinant DNA | ||
| HUe fluorescent cassette | Livet et al., 2007 | #6 - CMV-Brainbow-2.1 “R” [GYRC] |
| pCAG promoter | www.addgene.org | Plasmid #13777 |
| LoxP-STOP-LoxP-TOPO sequence | www.addgene.org | Plasmid #11584 |
| LoxSL: ATAACTTCGTATA GTTCAACT TATACGAAGTTAT | This paper | N/A |
| Sequence-Based Reagents | ||
| Custom primers used for ATAC-Seq | This paper | Table S2 |
| Software and Algorithms | ||
| FlowJo software | TreeStar Inc. | N/A |
| PAGODA package V1.99.3 | Fan et al., 2016 | http://pklab.med.harvard.edu/scde |
| MASS R package | N/A | https://cran.r-project.org/ |
| MClust R package | N/A | https://cran.r-project.org/ |
| BSMap 2.7 | N/A | N/A |
| BIC-seq2 | Xi et al., 2016 | http://www.math.pku.edu.cn/teachers/xirb/downloads/software/BICseq2/BICseq2.html |
| bowtie2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| tophat2 | Johns Hopkins University | https://ccb.jhu.edu/software/tophat/index.shtml |
| DESeq | Anders and Huber, 2010 | https://www.bioconductor.org/ |
| Homer package | Heinz et al., 2010 | http://homer.salk.edu/homer/ |
| SPP package 1.13 | Kharchenko et al., 2008 | http://compbio.med.harvard.edu/Supplements/ChIP-seq/ |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Other | ||
| BD FACSAria II Cell Sorter | BD Biosciences | N/A |
| Cesium 137 irradiator | N/A | N/A |
| Illumina Nexseq 500 instrument | Illumina | N/A |
CONTACT FOR REAGENTS AND RESOURCE SHARING
Requests should be addressed to and will be fulfilled by Lead Contact David T. Scadden (david_scadden@harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of the HUe Mouse Model
Development of the HUe transgenic construct utilized a fluorescent cassette that was originally adapted for the neural system (Livet et al., 2007), with some modifications. In brief, a STOP sequence flanked by a pair of LoxP variants was inserted in front of a fluorescent cassette containing GFP, EYFP, tDimer2, and Cerulean cDNA sequence interspersed by multiple LoxP sites such that no background fluorescence was expressed in the absence of Cre recombinase (Figure S1). The whole construct was placed under a ubiquitous chicken beta actin promoter and the linearized construct was microinjected into C57BL6/J embryos to generate transgenic mouse lines. Six founder lines were established and fluorescence was observed in multiple founders. Experiments performed in this study used founder six, which has approximately 20 copies of transgene insertion.
Mouse Models
HUe, Prx1-CreER, Mx1-Cre, Col(II)-CreER, and C57BL6/J strains were used and cross-bred as needed in this study. Mouse strains HUe and Prx1-CreER were made in-house, while B6.Cg-Tg(Mx1-cre)1Cgn/J (Mx1-Cre), FVB-Tg(Col2a1-cre/ERT)KA3Smac/J (Col(II)-CreER), and C57BL6/J were obtained from Jackson Laboratory. To image labeled cells of limb bud mesenchyme, Prx1-CreER was crossed with HUe to create Prx1-CreER;HUe. 2mg of 4OH-tamoxifen and 1mg of progesterone was injected into < 20 g pregnant females at E18.5. Mice were sacrificed for imaging at post-natal day 1 to 1 month of age. To image cells of labeled cartilage, Col(II)-CreER was crossed HUe to create Col(II)-CreER;HUe. Col(II)-CreER;HUe mice at 2 months of age was injected with 2mg of 4OH-tamoxifen and sacrificed for imaging 2–4 weeks post-injection. To study hematopoiesis, Mx1-Cre was crossed with HUe to create Mx1-Cre;HUe strain. To induce hematopoietic cell labeling, Mx1-Cre;HUe mice were injected with 12.5ug pIpC/g BW at two weeks of age. For most transplantation studies, 6–8 months old Mx1-Cre;HUe and C57BL6/J mice were used. To track endogenous hematopoiesis, bone marrow aspirates were obtained from Mx1-Cre;HUe mice at 2, 3, 5, and 10 months old. For all studies, age matched littermates were used as experimental controls. All animal housing, usage, and procedures performed were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
METHOD DETAILS
Flow Cytometry
For each mouse, tibiae, femurs, iliac crests, and spines were collected for bone marrow cells. Isolation and enumeration of different hematopoietic cell types was performed by flow cytometry. Bone marrow cells harvested from each animal were ACK lysed before antibody staining. We routinely stain 5×107 cells per sample for the stem population, and 1×107 cells per sample for each progenitor and mature population. Lineage cocktail consists of biotinylated B220, CD3e, CD4, CD8a, CD19, CD11b, Gr1, Ter119, CD11c, and NK1.1 antibodies. Fluorescence conjugated to streptavidin was used to recognize lineage cocktail. Using the following antibody combinations, we were able to identify hematopoietic subpopulations at the stem-cell level: hematopoietic stem cells (HSCs) (Lineage-Pacific Orange, cKit-APC-Cy7, Sca-PE-Cy7, CD48-APC, CD150-PE-Cy5), at the stem/progenitor level: multipotent progenitor cells (MPPs) (Lineage-Pacific Orange, cKit-APC-Cy7, Sca-PE-Cy5), common lymphoid progenitors (CLPs) (Lineage-Pacific Orange, cKit-APC-Cy7, Sca-PE-Cy5, CD127-PE-Cy7), common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs), megakaryocyte erythroid progenitors (MEPs) (all three with Lineage-Pacific Orange, cKit-APC-Cy7, Sca-PE-Cy5, CD16/32-PE-Cy7, CD34-efluor660), as well as mature lineages: B cells (B220-APC), T cells (CD3-APC), monocytes (Mac1-APC), granulocytes (Gr1-APC), and erythroid cells (Ter119-APC) using a BD FACSAria II Cell Sorter equipped with ultraviolet, violet, blue, yellow/green, red lasers.
Bone Marrow Aspiration
Bone marrow aspiration was performed under full body anesthesia using 3% isoflurane and 2 L/min O2. Fur was removed from the knee joint to expose intact skin. A PBS-wetted 27-gauge needle coupled with a 1mL syringe was inserted from the femur-tibial joint longitudinally into the bone marrow cavity of the tibia with negative pressure applied to extract 10ul of bone marrow. Mice after surgical procedure were placed under the heat lamp for 3–5 min to aid recovery from anesthesia and were monitored daily for any signs of discomfort following the Pain Assessment Protocol published by the National Research Council (US) Committee. All animal usage and procedures performed were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
Generation of the HUe Recipient Cohort
PIpC induced Mx1-Cre;HUe mice at 6–8 weeks old were used as donors. Bone marrow cells from 8–12 donors were pulled, lineage-depleted and flow sorted for LineageL°Sca+cKit+ cells before transplantation. One hundred thousand flow sorted fluorescent LineageLoSca+cKit+ cells mixed with 500,000 Sca− C57BL/6J support cells were transplanted into each of 20 lethally irradiated C57BL/6J recipients. Sixteen weeks were allowed for hematopoietic reconstitution. Experiment was repeated 6–8 times.
LPS Stress Experiment
A HUe recipient cohort (15 mice) was divided into three sub-cohorts: saline control (5 mice), treatment at 12 hr prior to tissue harvest (5 mice), and 44 days prior (5 mice). Mice received an intraperitoneal injection of either PBS or 0.3mg/kg BW LPS at the respective time points and all three groups of mice were euthanized for bone marrow harvest on the same day. Bone marrow cells were stained with antibodies summarized in Table S1 to identify HSCs, CLPs, GMP, MEPs, B cells, T cells, monocytes, granulocytes, and erythroid cells by flow cytometry, n = 5 for each data point. Experiment was independently performed twice with different recipient cohorts.
Irradiation Stress Experiment
A HUe recipient cohort (15 mice) was divided into three sub-cohorts including no treatment control (5 mice), 4.5 Gy irradiation at 14 days prior to tissue harvest (5 mice), and 4.5 Gy irradiation 44 days prior (5 mice). All mice were euthanized for bone marrow harvest on the same day. Bone marrow cells were stained with antibodies to identify HSCs, CLPs, GMP, MEPs, B cells, T cells, monocytes, granulocytes, and erythroid cells by flow cytometry (n = 5 for each data point). Experiment was independently performed twice with different recipient cohorts.
Whole Genome Bisulfite DNA Sequencing (WGBS) and Bulk RNA-Seq
A HUe recipient cohort of 36 mice was divided into two sub-cohorts: one group for monitoring lineage output using flow cytometry, one group for isolation of LT-HSC clones. Cells from a red and a yellow LT-HSC (LineageLoSca+cKit+CD48−CD150+) clone were flow sorted from multiple recipient cohort mice by flow cytometry and subjected to whole genome bisulfite DNA sequencing (WGBS) and RNA-seq. For WGBS, purified genomic DNA was fragmented to a size range of 100–400bp, 20–50ng of DNA fragments from each sample were end-repaired and ligated with indexed adapters using the NuGen’s Ovation Ultralow Methyl-Seq Library Systems. Adaptor-equipped DNA fragments were subjected to bisulfite treatments using the QIAGEN Epitect Bisulfite kit followed manufacture’s recommendation with modifications. Bisulfite converted library DNA was PCR-amplified and sequenced at the Broad Institute Genomics Platform. For RNA-seq, RNA was extracted from sample, reverse transcribed to cDNA, and was subjected to whole genome sequencing at Partners Healthcare Center for Personalized Genetic Medicine. Each data point represents triplicate of samples. The differential expression analysis was performed using CuffDiff 2.0, and the GSEA analysis was conducted on the resulting log-fold change values.
Single-cell RNA-Seq
HUe mice were induced by pIpC two weeks before single-cell sort of LKS SLAM cells. Whole bone marrow was isolated from femurs and tibiae by softly crushing bones, filtered by 40 μm cell strainer, and resuspended in Media 199 (ThermoFisher Scientific) supplemented with 2% fetal bovine serum (ThermoFisher Scientific) and RNase Out (ThermoFisher Scientific). Cells were stained for LKS SLAM markers, Calcein AM (ThermoFisher Scientific) and propidium iodide for cell viability detection. Single-cells were sorted using a BD FACSAria II (BD Biosciences) into PCR 384 well plates (ThermoFisher Scientific) containing standard lysis buffer. Whole transcriptome amplification was performed using the Smart-seq2 protocol (Picelli et al., 2013), and libraries prepared by Nextera XT (Illumina). Samples were pooled and sequenced on an Illumina Nexseq 500 instrument using a 50 bp paired-end-reads. The analysis was carried out using PAGODA package v1.99.3(Fan et al., 2016).
ATAC-Seq
We used an independent cohort of HUe recipient mice to select new clones to for a second set of experiment that integrated ATAC-, DNA- and RNA-seq analysis in correlation with HSC behavior. A new HUe recipient cohort of 32 mice was divided into two sub-cohorts: one group for monitoring lineage output using flow cytometry, one group for isolation of LT-HSC clones. Two new LT-HSC (LineageLoSca+cKit+CD48−CD150+) clones (Red2 and Yellow2) were isolated from multiple recipient cohort mice by flow cytometry and subjected to ATAC-Seq, WGBS, and RNA-seq. For ATAC-Seq, 10,000 cells of each clone were lysed in 50 μL of cold lysis buffer (10 mM Tris-HCl pH 7.4,10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) and immediately subjected to a transposition reaction at 37°C for 30 min with 2.5 μL transposase enzyme (Illumina Nextera DNA Preparation Kit). Transposed DNA was purified using QIAGEN MinElute PCR purification Kit and subjected to library amplification using NEBNext High-Fidelity 2X PCR Master Mix, Invitrogen SYBR Green I Dye, and primers (Table S2). Prior to sequencing, the ATAC-Seq library was assayed for quality using TapeStation and BioAnalyzer instruments, and qPCR. WGBS and RNA-seq libraries were prepared as described previously. All libraries of this second set of experiment were sequenced at the Bauer Core Facility of Harvard FAS Center for Systems Biology.
QUANTIFICATION AND STATISTICAL ANALYSIS
Identifying Significant Differences between Fluorescence Patterns
The fluorescent patterns between any two samples were compared based on the density of cells on a unit sphere in the space of color composition. The uncertainty of the color composition measured for each cell was determined based on the intensity of each color, using power law dependency between intensity and the variance. The power coefficients were fit based on technical replicates of homeostatic condition mixtures, and were in the 0.5–0.75 range. To determine regions of statistical significance, 1000 sampling rounds were performed, resampling the exact positions of every cell on the color composition surface using the appropriate variance for each cell. Smoothed cell density was calculated on the surface, and compared using Student’s t test with degrees of freedom corresponding to n-2, where n is the minimum number of cells in either sample. The color composition regions showing significant differences (Z-score < 3) were identified as regions of significant differences between the samples. The change in the fractional cell density between the samples was measured as a fraction of the total number of cells recorded in a given experiment.
Enumerating Individual Clones
Because of the naturally occurring intensity differences, the clones can have highly non-spherical shape, making identification and separation of individual clones particularly challenging. To provide the initial definition of the clones, all pairwise cell-to-cell distances were calculated in terms of the number of standard deviations (based on the intensity-variance model for each color). The distance structure was then projected into a 2D space using non-metric multi-dimensional scaling (as implemented in MASS R package). The clones were identified using elliptical clustering method implemented by the MClust R package. Despite such approach, some of the very similar clones occurring in different samples were split or merged. In such cases, the count was corrected manually.
Whole-Genome Bisulfite Sequencing (WGBS) Processing
To calculate DNA methylation estimates for individual CpG positions (beta values), whole genome-bisulfite sequencing libraries were aligned to the mouse genome mm9/NCBI Build 37 using BSMap 2.7 with the following parameters: -v 10 -f 40 -q 5 -S 1. Subsequently, CpG methylation calls were made, excluding duplicate reads as well as the first four bases of each read, only taking into account CpGs with quality scores ≥ 20 as well as requiring that surrounding bases exhibit quality scores ≥ 10. Here, CpG methylation calling is defined as the computation of the number of reads overlapping a particular CpG harboring a C or a T at the cytosine coordinate of the CpG. Let m be the number of C’s and u be the number of T’s. The value beta = m/(m + u) then gives the methylation ratio of each CpG that was used as a basis of subsequent analysis.
CNV Analysis of WGBS Data
To determine whether there are significant genomic alterations that may distinguish clones within the same mouse/cohort, we used BIC-seq2 (Xi et al., 2016), a read-depth-based CNV calling algorithm to detect copy number variation (CNVs) from the bisulfite WGBS data of the mouse clones. Briefly, BIC-seq2 divides genomic regions into disjoint bins and counts uniquely aligned reads in each bin. Then, it combines neighboring bins into genomic segments with similar copy numbers iteratively based on Bayesian Information Criteria (BIC), a statistical criterion measuring both the fitness and complexity of a statistical model. BIC-seq2 provides two different CNV calling methods: 1) paired-sample CNV calling that takes a pair of samples as input and detects genomic regions with different copy numbers between the two samples, and 2) control-free CNV calling that takes only one sample as input and calls CNVs in the sample. We used a bin size of ~1000 bp and a lambda of 50 (a smoothing parameter for CNV segmentation). We called segments as copy gain or loss when their log2 copy ratios were larger than 0.2 or smaller than −0.2, respectively.
To evaluate the sensitivity of our CNV calling method for bisulfite whole genome sequencing data, we analyzed published bisulfite whole genome sequencing data for two different mouse strains: 129P2 (GSE56986) (Lu et al., 2014) and BALBcJ (GSE60485) (Wu et al., 2015). The CNVs for the three mouse strains were annotated by Sanger mouse genome project. We downloaded the FASTQ files from the NCBI GEO database and mapped the reads using the same procedure as described in the previous section. We then applied BIC-seq2 with the same parameters (bin size and lambda) as we analyzed our own data. We also downloaded annotation files for structural variation of the three mouse strains from the Sanger mouse genome project web site (http://www.sanger.ac.uk/science/programmes/mouse-and-zebrafish-genetics). Using annotated deletions larger than 10 kbp as a gold standard set, we calculated the sensitivity as the fraction of the gold standard CNVs that were detected by BIC-seq2. The estimated CNV sensitivity was 58.6% and 44.7% for 129P2 and BALBcJ.
Computational Validation of the HSC States of the Selected Clones
To validate that the Cohort1.Y, Cohort1.R, Cohort2.G, Cohort2.P clones are at the HSC states, we compared DNA methylation levels of enhancers activated at different hematopoietic stages. The enhancer positions and their epigenetic states within hematopoiesis were taken from Lara-Astiaso et al. (2014). Following the described methods, we tallied H3K4me1 and H3K27ac ChIP-seq read counts for 48415 enhancers across 16 hematopoietic cell types. The H3K4me1 read counts were used to categorize the states of enhancers, ‘on’, ‘off’ or ‘intermediate’, and H3K27ac read counts were used to further determine whether enhancers with ‘on’ states are ‘active’ or ‘poised’, as described previously (Lara-Astiaso et al., 2014). We focused on early lymphopoiesis involving the ‘LT-HSC’ (long-term HSC), ‘ST-HSC’ (short-term HSC), ‘MPP’ (multipotent progenitors) and ‘CLP’ (lymphoid progenitors) stages. For each stage, we selected a set of enhancers that were ‘on’ in that cell type as well as all downstream cell types, but ‘off’ in all upstream cell types. For instance, for MPP enhancers we selected those that are ‘on’ in MPP and CLP, but are off in LT-HSC and ST-HSC. Average DNA methylation (beta values) were calculated within each enhancer region and compared between the selected clones, respectively (e.g., Figures 6A and 7D).
For the ATAC-seq data, the signal for each enhancer was quantified as a number of the ATAC-seq fragment centers that fall within the ± 1kb region around the enhancer center, normalized by the library size (measured in million of reads). The read counts were based on the paired-end alignment (using bowtie2) to the mm9 genome assembly, removing duplicate reads.
Gene Set Enrichment Analysis for Testing Cell Type Bias of Clones
Gene set enrichment analysis (GSEA) was performed to investigate whether gene expression profiles of the selected clones show bias toward a specific hematopoietic cell type. In the GSEA, the genes were ranked by the Z score corresponding to the p value of the expression differences between the clones (using tophat2 (Kim et al., 2013) with default parameters, and DESeq (Anders and Huber, 2010) with local fit option). GSEA was used to test gene sets obtained based on differential expression analysis of RNA-seq data across 16 different hematopoietic cell types from Lara-Astiaso et al. (2014). Following the same steps as described by Lara-Astiaso, the RNA-seq data (GEO: GSE60101) were aligned to the mm9 mouse genome assembly using bowtie2 (Langmead and Salzberg, 2012) with default parameters. Read counts of genes were calculated using ‘analyzeRNA.pl’ from the Homer package (Heinz et al., 2010). Differential gene expression analysis was performed based on DESeq between a given pair of cell types (i.e. HSC and MPP). Genes significantly higher expression in a given cell type (FDR-corrected p value < 0.05) were selected as a set for GSEA analysis (Figure 6B).
GSEA was also applied to test whether the selected clones also show significant bias in DNA methylation at promoters (within 2 kb upstream of transcription start sites) of the identified cell-type-specific genes (Figure 6B). In the GSEA, promoters were ranked based on the maximum likelihood estimates of log2 fold ratio of promoter methylation of the clones, calculated based on the SPP package (Kharchenko et al., 2008).
Finally, GSEA was also employed to study potential cell type bias of the two clones in DNA methylation of enhancers (Figures 6B and 6D). For the GSEA, enhancers were ranked by the maximum likelihood estimates of log2 fold ratio of DNA methylation of the ‘Yellow’ and ‘Red’ clones. For a given pair of cell types (HSC versus MPP in Figure 6B, or CLP versus CMP in Figure 6D), we selected top 500 enhancers that are most ‘active’ (as determined by the H3K27ac read counts) in one cell type and are not ‘on’ (determined by H3K4me1 counts) in the paired cell type, and vice versa, for the GSEA analysis.
DATA AND SOFTWARE AVAILABILITY
HUe RNA-seq, WGBS and ATAC-seq data accessible through GEO datasets (GEO: GSE87527).
Supplementary Material
Highlights.
Clonal tracking demonstrates clone-specific functional heterogeneity in vivo
Stereotypical functions of HSCs are preserved under stress or tissue injury
HSC clonal behavior is associated with a distinct epigenetic pattern
HSC is epigenetically constrained with limited plasticity in response to cues
Acknowledgments
We are grateful to Drs. Jeff W. Lichtman, Jean Livet, and Joshua R. Sanes for their kind gifts of the Brainbow fluorescent vectors. We also thank Laura Prickett, Kathryn E. Folz-Donahue, and Meredith Weglarz at the Flow Cytometry Core Facility of the Harvard Stem Cell Institute for their technical assistance. We are grateful for support from Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, and the National Genomics Infrastructure, funded by the Swedish Research Council, for assistance with single-cell RNA-seq measurements. This work was supported by NIH grants 1R21HL126070-01A1 to V.W.C.Y., DK103074 and CA193461 and the Gerald and Darlene Jordan Chair | to D.T.S, and the Ellison Medical Foundation AG-NS-0965-12 and NIA 5K25AG037596 to P.V.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.10.045.
AUTHOR CONTRIBUTIONS
V.W.C.Y. created the mouse model, designed concept, performed experiments, interpreted data, and wrote the manuscript. R.Z.R. contributed flow cytometric expertise and participated in flow experiments. T.O. performed gene set enrichment analysis. J.W. provided biocomputational contribution. B.S. and C.C. participated in multiple experiments in this project. N.B. participated in single-cell isolation. H.G. prepared DNA libraries for sequencing. X.W. and M.Z. contributed to DNA sequence analysis. P.V.K. designed the approach for statistical analysis of flow cytometric data, performed DNA and RNA sequence analysis, and was involved in manuscript writing. C.L. provided expertise on optical physics and intra-vital imaging. E.L. carried out CNV analysis of WGBS data. A.M. provided expertise on epigenetic experiments. D.T.S. supervised the project and was involved in concept design, data interpretation, and manuscript writing.
References
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, Scaramuzza S, Baricordi C, Ferrua F, Cicalese MP, et al. In vivo tracking of human hematopoiesis reveals patterns of clonal dynamics during early and steady-state reconstitution phases. Cell Stem Cell. 2016;19:107–119. doi: 10.1016/j.stem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012;47:633–647. doi: 10.1016/j.molcel.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, Rodewald HR. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Boles NC, Lin KY, Tierney MP, Bowman TV, Bradfute SB, Chen AJ, Merchant AA, Sirin O, Weksberg DC, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee SJ, Brinkman R, Eaves C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1:218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208:2691–2703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J, Kharchenko PV. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016;13:241–244. doi: 10.1038/nmeth.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Passegué E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, Weissman IL. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS ONE. 2010;5:e8785. doi: 10.1371/journal.pone.0008785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits A, Dykstra B, Kalmykowa OJ, Klauke K, Verovskaya E, Broe-khuis MJ, de Haan G, Bystrykh LV. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood. 2010;115:2610–2618. doi: 10.1182/blood-2009-06-229757. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT, Lemischka IR. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev. 1990;4:220–232. doi: 10.1101/gad.4.2.220. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Pelletier L, Heninger AK, Slabicki M, Theis M, Miroslaw L, Poser I, Lawo S, Grabner H, Kozak K, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol. 2007;9:1401–1412. doi: 10.1038/ncb1659. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. Immunogenetics. Chromatin state dynamics during blood formation Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemischka IR. Retroviral lineage studies: some principals and applications. Curr Opin Genet Dev. 1993;3:115–118. doi: 10.1016/s0959-437x(05)80351-x. [DOI] [PubMed] [Google Scholar]
- Lemischka IR, Raulet DH, Mulligan RC. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 1986;45:917–927. doi: 10.1016/0092-8674(86)90566-0. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Lu R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol. 2011;29:928–933. doi: 10.1038/nbt.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28:2103–2119. doi: 10.1101/gad.248005.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurier F, Gan OI, McKenzie JL, Doedens M, Dick JE. Lentivector-mediated clonal tracking reveals intrinsic heterogeneity in the human hematopoietic stem cell compartment and culture-induced stem cell impairment. Blood. 2004;103:545–552. doi: 10.1182/blood-2003-05-1558. [DOI] [PubMed] [Google Scholar]
- Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med. 2010;207:1173–1182. doi: 10.1084/jem.20091318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Sieburg CE, Cho RH, Karlsson L, Huang JF, Sieburg HB. Myeloid-biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood. 2004;103:4111–4118. doi: 10.1182/blood-2003-10-3448. [DOI] [PubMed] [Google Scholar]
- Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–367. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, Sakaue-Sawano A, Niida A, Miyano S, Aburatani H, et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0–G1 transition. Sci Rep. 2014;4:4012. doi: 10.1038/srep04012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Shi PA, Hematti P, von Kalle C, Dunbar CE. Genetic marking as an approach to studying in vivo hematopoiesis: progress in the non-human primate model. Oncogene. 2002;21:3274–3283. doi: 10.1038/sj.onc.1205320. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Snodgrass R, Keller G. Clonal fluctuation within the haematopoietic system of mice reconstituted with retrovirus-infected stem cells. EMBO J. 1987;6:3955–3960. doi: 10.1002/j.1460-2075.1987.tb02737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014a;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD. Clonal dynamics of native haemato-poiesis. Nature. 2014b;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang JC, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P. Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol. 2015;16:178. doi: 10.1186/s13059-015-0739-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, Goodell MA. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2:e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verovskaya E, Broekhuis MJ, Zwart E, Weersing E, Ritsema M, Bosman LJ, van Poele T, de Haan G, Bystrykh LV. Asymmetry in skeletal distribution of mouse hematopoietic stem cell clones and their equilibration by mobilizing cytokines. J Exp Med. 2014;211:487–497. doi: 10.1084/jem.20131804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sánchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, et al. Combined single-cell functional and gene expression analysis resolves heterogeneitywithin stem cell populations. Cell Stem Cell. 2015;16:712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhou H, Fan X, Zhang Y, Zhang M, Wang Y, Xie Z, Bai M, Yin Q, Liang D, et al. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. 2015;25:67–79. doi: 10.1038/cr.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Lee S, Xia Y, Kim TM, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 2016;44:6274–6286. doi: 10.1093/nar/gkw491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.