

Abstract
It is now generally accepted that estrogen receptor (ESR1) mutations occur frequently in metastatic breast cancers, however we do not yet know how to best treat these patients. We have modeled the three most frequent hormone binding ESR1 (HBD-ESR1) mutations (Y537N, Y537S, and D538G) using stable lentiviral transduction in human breast cancer cell lines. Effects on growth were examined in response to hormonal and targeted agents, and mutation-specific changes were studied using microarray and western blot analysis. We determined that the HBD-ESR1 mutations alter anti-proliferative effects to tamoxifen (Tam), due to cell-intrinsic changes in activation of the insulin-like growth factor receptor (IGF1R) signaling pathway and levels of PIK3R1/PIK3R3. The selective estrogen receptor degrader, fulvestrant, significantly reduced the anchorage-independent growth of ESR1 mutant-expressing cells, while combination treatments with the mTOR inhibitor everolimus, or an inhibitor blocking IGF1R and the insulin receptor significantly enhanced anti-proliferative responses. Using digital drop (dd) PCR we identified mutations at high frequencies ranging from 12% for Y537N, 5% for Y537S, and 2% for D538G in archived primary breast tumors from women treated with adjuvant mono-tamoxifen therapy. The HBD-ESR1 mutations were not associated with recurrence-free or overall survival in response in this patient cohort, and suggest that knowledge of other cell-intrinsic factors in combination with ESR1 mutation status will be needed determine anti-proliferative responses to Tam.
Keywords: Estrogen Receptor, Mutations, IGF-1, Fulvestrant, Everolimus
INTRODUCTION
Adjuvant hormonal therapy targeting the estrogen receptor (ESR1) in human breast cancer has significantly improved overall survival for patients whose tumors express this receptor1. However, acquired resistance is a major clinical problem in breast cancer treatment. Several mechanisms of hormone resistance have been identified, including loss of estrogen receptor alpha (ERα) expression, altered activity of ER co-activators, and cross-talk with growth factor receptors such as HER2 and IGF1R2. Recently it has been shown that mutations in the hormone binding domain (HBD) of ESR1 occur frequently in metastatic breast tumor tissues3,4,5,6,7, and are selected for during aromatase inhibitor (AI) treatment8.
The first reported HBD-ESR1 mutation, a tyrosine to asparagine missense mutation at amino acid residue 537 (Y537N), was discovered in 1997 by cloning of cDNA from a metastatic lesion, and displayed strong constitutive hormone-independent transcriptional activity9. A second mutation at lysine 303 (K303R) was reported in both atypical ductal hyperplasias and invasive breast cancers, and was hypersensitive to low levels of estrogen10,11. It was therefore proposed that somatic ESR1 mutations might be acquired during resistance to therapy, which could play important roles in the metastatic spread of breast cancer12. However, this hypothesis remained controversial until a large number of metastatic tumors were interrogated using next generation sequencing3,4,5,6,7. Many ESR1 mutations appear to occur at a mutational “hot spot” surrounding residues 536–538 of the HBD, shifting the receptor into a constitutive agonist conformation7 with stabilization of co-activator binding13. Investigators have consistently shown that these ESR1 mutations exhibit elevated estrogen-independent transcriptional activity3,4,5,6,7. Recently it was suggested that the combination of constitutive recruitment of co-activators in the absence of hormone and reduced Tam binding affinity might underlie therapy resistance conferred by the Y537S and D538G ESR1 mutations13. HBD-ESR1 mutations are present at high frequencies in the plasma of metastatic patients using sensitive ddPCR technologies, and indeed may predict reduced sensitivity to subsequent aromatase AI therapy in the metastatic setting8.
The selective estrogen receptor degrader (SERD) fulvestrant is effective in ER-positive patients progressing on hormonal therapy, and is now considered part of a sequential treatment regimen for patients with metastatic disease14. FERGI trial investigators, who examined the addition of a phosphoinositide-3-kinase (PI3K) inhibitor with fulvestrant in AI-resistant metastatic patients, reported that the presence of HBD-ESR1 mutations did not predict resistance to fulvestrant15, thus fulvestrant combined with other targeted therapies may be a viable treatment option in patients with ESR1 mutations. The BOLERO-2 trial demonstrated a 50% reduction in progression when the mTOR inhibitor everolimus was combined with exemestane16, and a subsequent correlative analysis reported that patients with certain of the ESR1 mutations may benefit from this combination as well17.
Although the selective estrogen receptor modulator (SERM) Tam can block estrogen-stimulated ESR1 mutant transcriptional activity3,4,5,6,7, it has not been shown whether Tam will block the growth of ESR1 mutant-expressing cells in patients. Herein, we report a preclinical study showing that sensitivity to the anti-proliferative effects of Tam in some ER-positive cells expressing the HBD hotspot ESR1 mutations is dependent on cross-talk with the IGF1R signaling pathway.
RESULTS
HBD-ESR1 mutations modulate anti-proliferative responses to Tam depending on cellular background
We established stable pools of ER-positive MCF-7 cells expressing either WT ESR1 (as a control for elevated receptor levels), or the most frequent HBD mutants Y537N, Y537S, or D538G. Endogenous ERα (66 kDa), and an m-cherry-tagged exogenous receptor (~95 kDa) were visualized using immunoblot analysis (Fig. 1A), and growth responses were determined using soft agar growth assays. The most noticeable difference was the change in shape and larger size of the ESR1 mutant colonies under control conditions (−). Quantification of colonies greater than 200μm showed that the control (-estrogen) growth of all mutant-expressing colonies was significantly enhanced (Fig. 1B). MCF-7 WT-expressing cells responded to Tam with a 38% and 53% reduction in soft agar growth with 0.1 or 1μM of 4-hydroxy-tamoxifen (designated Tam hereafter) treatment, respectively (Fig. 1C). In contrast, all HBD-ESR1 mutant-expressing cells exhibited reduced anti-proliferative responses to Tam, but with different absolute responses. Tam treatment of mutant cells showed less reduction in colony numbers (ranging from 42 to 13%) compared to WT-expressing cells even at the highest concentration used (Fig. 1C). MCF-7 Y537S ER-expressing cells consistently exhibited the least Tam-responsive phenotype. Similar data was obtained using another MCF-7 subline (designated BK) maintained separately in the laboratory (Figs. S1A and B). These data also suggest that there may be functional biologic differences between the three ESR1 mutations in their anti-proliferative responses to Tam.
Figure 1. HBD-ESR1 mutations influence Tam response.
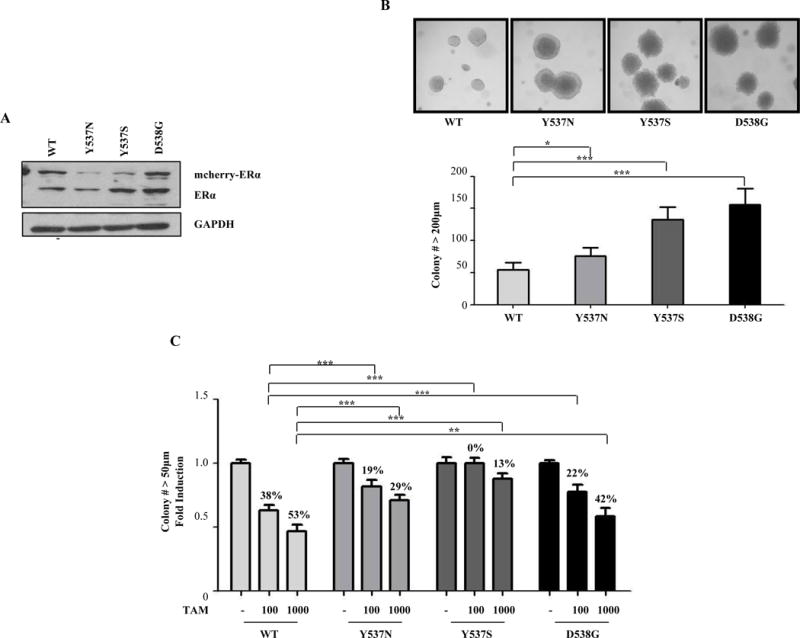
a, immunoblot analysis of detect ER expression in MCF-7 stable clones. GAPDH was used as loading control. b, Representative photographs of three different well plates captured from soft agar assays showing higher number of large colonies (200μm) in mutant-expressing cells. Experiments were performed in triplicate and error bars indicate SD. *P<0.05; ***P<0.001. d, Cells were plated for soft agar assays, and then treated with vehicle or two doses of Tam (100nM or 1000nM). Percentage of growth reduction with Tam treatment is shown. Experiments were performed in triplicate and error bars indicate SD. **P<0.01; ***P<0.001.
We generated ESR1 mutant-expressing pools using two other ER-positive breast cancer cell lines, ZR-75B and T47D (Figs. S1C–F). Both of these models express high mutant levels. ZR-75B and T47D mutant cells responded to Tam treatment equivalent to WT-expressing cells, and colony size was not affected (data not shown). These results indicate that individual anti-proliferative responses to Tam may depend on cellular background, and that other tumor cell-intrinsic factors may be associated with response in cells expressing ESR1 mutants.
Activation of IGF-1 signaling in HBD-ESR1 mutants is associated with Tam response
Since response to Tam requires many cell-specific effectors, we next employed expression microarray profiling, 3-way ANOVA, and canonical pathway analysis using Ingenuity to identify pathways significantly associated with ESR1 mutant expression in MCF-7 compared to ZR-75B cells (Fig. 2A); darker orange bars are pathways most activated in MCF-7 mutant cells, and blue bars are deactivated pathways. The most significantly activated pathway was insulin growth factor-1 (IGF-1) signaling. A known mechanism of hormone resistance is enhanced cross-talk between ER and growth factor receptors, which can feedback and impact ER transcriptional activity. Based on this pathway analysis, we next investigated whether expression of the HBD-ESR1 mutations altered growth factor signaling, but no differences in Tam responses were detected with either epidermal growth factor (EGF) or heregulin co-treatment (data not shown). However higher levels of phosphorylated IGF1Rβ, and phosphorylated insulin receptor substrate-1 (pIRS-1) were seen after IGF-1 treatment of MCF-7 mutant cells (Fig. 2B). Estrogen activation of IGF signaling served as a positive control in this experiment18. In contrast, we did not detect enhanced IGF1Rβ or IRS-1 phosphorylation in ZR-75B Y537S cells (Fig. 2C). Thus, activated IGF-1 signaling was specifically associated with mutant expression in MCF-7 cells exhibiting reduced anti-proliferative effects with Tam, implying that cross-talk with this signaling pathway may be involved. Activated IGF-1 signaling is a known mechanism affecting Tam response, and could be employed by ESR1 mutations as a proliferative escape mechanism in some cellular backgrounds.
Figure 2. IGF-1 signaling pathway activation in HBD-ESR1 mutants.
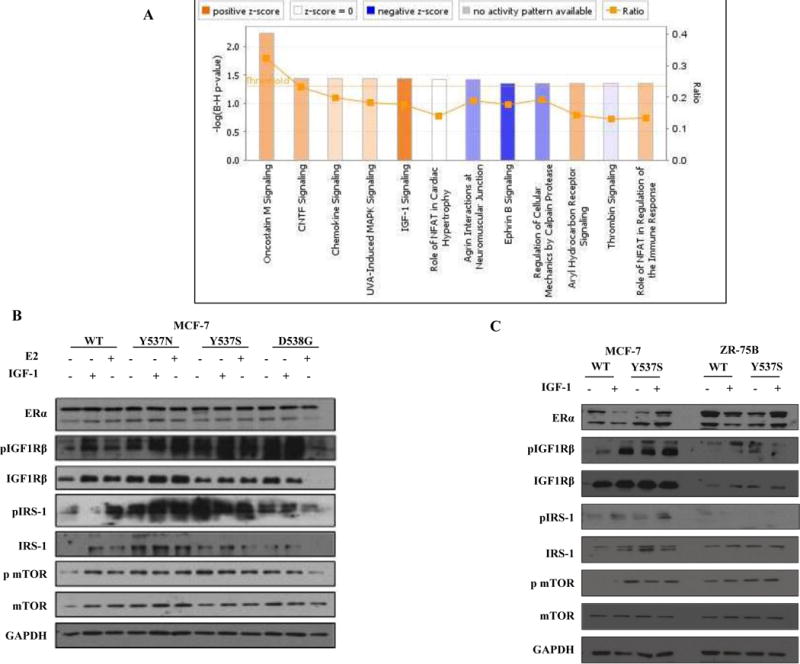
a, Ingenuity Pathway Analysis (IPA) to identify activation of signaling pathways in mutant MCF-7 vs. ZR-75B. b, Total cellular extracts were analyzed for phosphorylation and expression of ER, IGF1Rβ, IRS-1, and mTOR; GAPDH was used as a loading control. Immunoblots show a representative example of three experiments. c, Total cellular extracts were analyzed for phosphorylation and expression of ERα, IGF1Rβ, IRS-1, and mTOR; GAPDH was used as a loading control. Immunoblots show a representative example of three experiments.
We next used an IGF1Rβ inhibitor (GSK1838705A) that also inhibits signaling from the insulin receptor. Mutant ER transcriptional activity was efficiently blocked by Tam treatment, underscoring the limitation of only using transcriptional assays to monitor mutant function (Fig. 3A). The combination of Tam with GSK further enhanced the ability of Tam to block transcriptional activity, demonstrating functional ER cross-talk with IGF1R. We found that GSK treatment enhanced growth sensitivity to Tam in all cells (Fig. 3B). Of note, the growth of Y537S-ESR1 cells was significantly stimulated by Tam treatment in this experiment, and the IGF1Rβ inhibitor was able to block this agonist growth.
Figure 3. Specific blockade of the IGF1R pathway restores anti-proliferative effects of Tam in mutant-expressing cells.
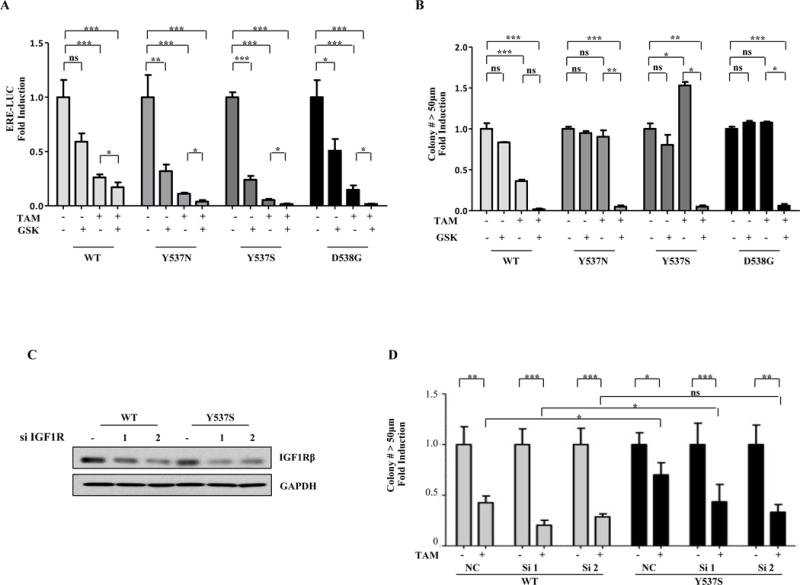
a, ERE-luc assays in MCF-7 cells transiently transfected with WT-, Y537N-, Y537S-, or D538G-ER plasmids. Experiments were performed in triplicate and error bars indicate SD *P<0.05; **P<0.01; ***P<0.001. b, Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; **P<0.01; ***P<0.001. c and d, Cells were transfected for 24 hours with two different IGF1Rβ siRNAs (100nM) and were maintained for 14 days in culture. 10μg of protein were loaded per well and the blot was probed with antibodies detecting IGF1Rβ; GAPDH was used as a loading control (c), Same cells were plated for soft agar assay and then treated with vehicle (-) or with Tam (100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant, *P<0.5.
We performed knockdown of IGF1Rβ levels using two siRNAs to confirm these results, and knockdown enhanced Tam’s anti-proliferative effects (Fig. 3C and D). These results suggest that specific blockade of IGF1R itself or other downstream signaling molecules may be required, along with hormonal therapy, in some mutant-expressing tumors.
Enhanced interactions between IGF1R and the Y537S ESR1 mutant
The MCF-7 Y537S model demonstrated the smallest anti-proliferative effects with Tam treatment, therefore this model was examined for protein interactions between IGF1Rβ and ER using immunoprecipitation coupled with immunoblot analysis. Input extracts were examined and confirmed higher levels of IGF1Rβ protein in MCF-7 mutant, but not in ZR-75B mutant cells; all cells were able to induce pIRS-1 with IGF-1 treatment (Fig. 4A). Higher constitutive binding was observed between the Y537S mutant and IGF1Rβ in MCF-7 (Fig. 4B), but not ZR-75B cells (Fig. 4C). This suggests that although all cells can express pIRS-1, there were diminished anti-proliferative effects in MCF-7 cells with elevated constitutive pIGF1Rβ levels. To further examine for interactions between IGF1R and ER, we also performed proximity ligation assays in MCF-7 and ZR-75B cells (Figs. 4D). Prominent interactions (red speckled staining, arrows) were seen in mutant-expressing cells compared to WT, but only in the MCF-7 cells. We conclude that the role of ESR1 mutations in Tam response may be dependent on a cellular background with elevated pIGF1R.
Figure 4. MCF-7 Y537S cells show the lowest anti-proliferative response to Tam through increased interaction between IGF1R/ER.
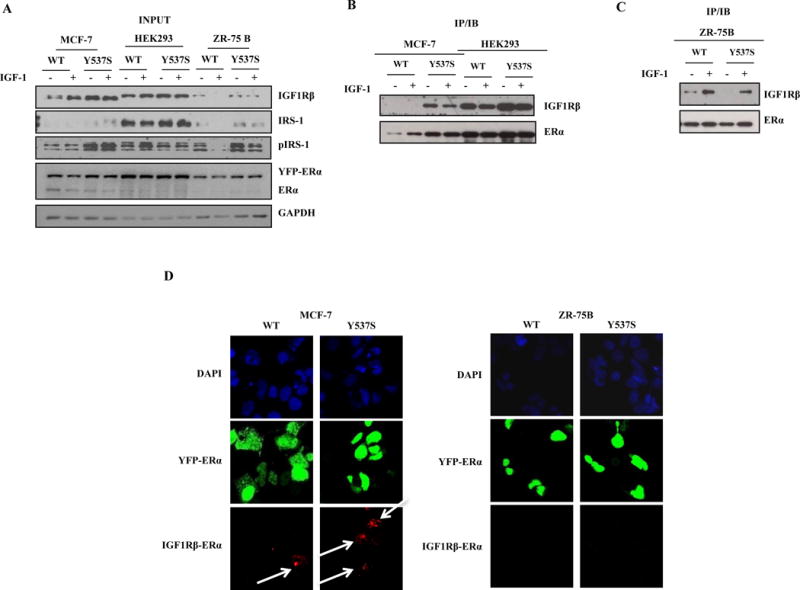
a, b, and c, Immunoblot analysis to detect expression of IGF1Rβ, pIRS-1, IRS-1 and ER in input extracts; GAPDH was used as a loading control (a). Coimmunoprecipitation assays of IGF1R/ER interactions (b and c). Duolink staining (red) demonstrated binding between IGF1R and ER in MCF-7(d) and ZR-75B cells (e). Images are representative of three different experiments.
Everolimus and fulvestrant are effective at reducing growth of stable HBD-ESR1 mutant-expressing cells
Although a number of inhibitors targeting the IGF1R growth pathway have entered clinical trials, they have proven disappointing in randomized clinical trials, possibly because of the lack of patient selection for activation of the IGF1R pathway19. IGF1R and other growth factor receptors activate the PI3K/AKT/mTOR (mammalian target of rapamycin) signaling transduction pathways, which are drivers of acquired hormone resistance in ER-positive patients20. mTOR regulates the transcriptional activity of several proteins involved in cell growth and proliferation,21, and the mTORC1 inhibitor everolimus, in combination with the aromatase inhibitor exemestane, is approved for the treatment of hormone-resistant breast cancer16. Since there is clinical correlative data suggesting that patients with some of the ESR1 mutants can indeed respond to fulvestrant or exemestane17,15, we next tested these combinations in our preclinical MCF-7 mutant models. As expected everolimus treatment reduced downstream pS6 levels, and combination treatments with Tam reduced both mTOR and pS6 phosphorylation in Y537S mutant cells (Fig. 5A). The IGF1R inhibitor GSK in combination with Tam effectively reduced IGF1Rβ and p-mTOR levels, again highlighting IGF1R activation in mutant cells.
Figure 5. Fulvestrant in combination with everolimus or GSK enhances anti-proliferative effects in mutant-expressing cells.
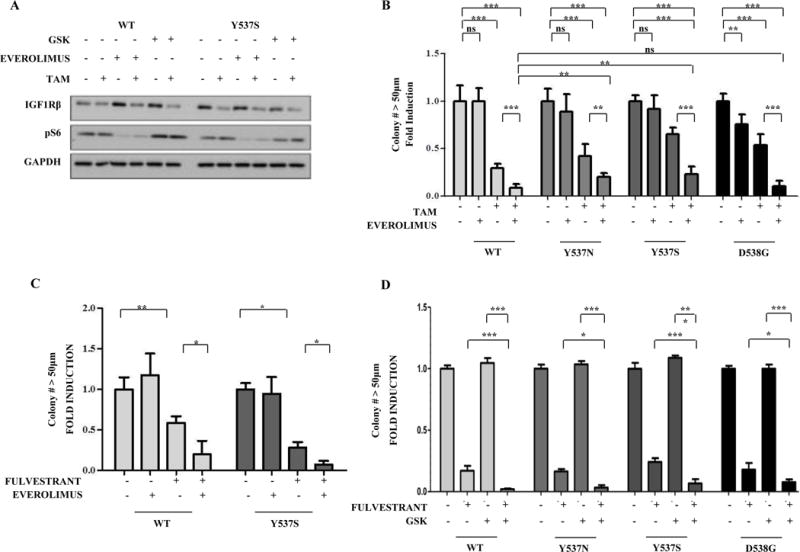
a, Immunoblot analysis to detect IGF1Rβ and pS6 levels; GAPDH was used a loading control. Immunoblots show a representative example of three different experiments. b, Cells were plated in soft agar assay and then treated with vehicle (-), Tam (100nM), or everolimus (1nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; **P<0.01; ***P<0.001. c, Cells were plated in soft agar assay and then treated with vehicle (-) or with fulvestrant (100nM) and everolimus (1nM). Experiments were performed in triplicate and error bars indicate SD. *P<0.05. d. Cells were plated in soft agar assay and treated with vehicle (-), fulvestrant (100nM) and/or GSK (1μM). Experiments were performed in triplicate and error bars indicate SD. *P<0.05, ***P<0.001.
Fulvestrant, alone or in combination with a steroidal AI, is mostly used after treatment has failed with a nonsteroidal aromatase inhibitor in postmenopausal women22. Everolimus in combination with fulvestrant has shown significant efficacy in Phase II studies in heavily pretreated metastatic ER-positive breast cancer patients23. Everolimus treatment alone exhibited little effects on cellular proliferation, but in combination with Tam significantly enhanced anti-proliferative effects in mutant-expressing cells (Fig. 5B). Fulvestrant treatment alone also significantly blocked growth, and in combination with everolimus further inhibited growth of the Y537S ESR1 mutant (Fig. 5C). Fulvestrant plus GSK was particularly effective at reducing the anchorage-independent growth of ESR1 mutations (Fig. 5D). Similar results were seen in the other MCF-7 BK line (Fig. S2). Y537S and D538G-expressing cells also demonstrated enhanced response to IGF treatment in wound-healing scratch motility assays, and GSK and effectively blocked IGF-induced motility of WT and Y537S-expressing cells (Fig. S3). Collectively these preclinical data demonstrate that everolimus/fulvestrant is an effective combination for the Y537S mutant, and also suggest that fulvestrant plus strategies to completely block IGF1R signaling24 should be explored in the metastatic setting in ESR1 mutant patients.
HBD-ESR1 mutations in primary tumors do not significantly impact outcomes in Tam-treated patients
Based on the very low frequency of ESR1 mutations in either the TCGA primary breast tumor database or primary tumors from the BOLERO-2 trial using next generation sequencing7, it was concluded that these mutations must develop as a consequence of treatment selection during the evolution of metastasis, perhaps via emergence from an undetectable subclonal mutant population, and recent reports strongly suggest this to be true8. Using sensitive ddPCR assays, Takeshita et al. reported that 2.5% of primary breast cancer specimens contained ESR1 mutations, with the Y537S mutation being the most frequent25. To address whether ESR1 mutations might predict response to Tam in primary breast cancers, we used ddPCR sequencing of archived DNAs from 203 primary tumors treated with Tam monotherapy11. Positive ESR1 mutation status was associated with smaller tumor size and progesterone receptor (PR) negativity (Supplementary Table 1). Mutations were found at relatively high frequencies using a sensitive cut-off in this cohort, ranging from 12% for Y537N, 5% for Y537S, and 2% for D538G (Table 1). However allele frequencies were low in primary tumors, and are shown as the percent of mutant compared to WT ESR1 (Fig 6A), suggesting that these three mutations indeed represent a minor subpopulation within the primary tumor. We assessed the clinical impact of ESR1 mutations on outcomes in this cohort, and for patients with any (Y537N/S or D538G) mutation, patients with an ESR1 mutant exhibited slightly improved recurrence-free survival ([RFS], Fig 6B, p=0.0118), but no difference was seen in overall survival (OS, Fig. 6C). Effects of individual mutations on RFS or OS are shown in Supplementary Fig. S4. Thus the HBD-ESR1 mutations were not significantly associated with resistance to Tam as predicted by many preclinical studies. These results suggest that measurement of ESR1 mutation status as a single biomarker may not provide significant information in primary breast cancers treated with Tam.
Table 1.
Mutation frequency in invasive breast tumors treated with Tam
| Mutation | Frequency (%) | Number of tumors |
|---|---|---|
| Y537N | 12 | 74/195 |
| Y537S | 5 | 29/199 |
| D538G | 2 | 4/195 |
Figure 6. ESR1 mutation do not predict outcomes in patient treated with adjuvant Tam.
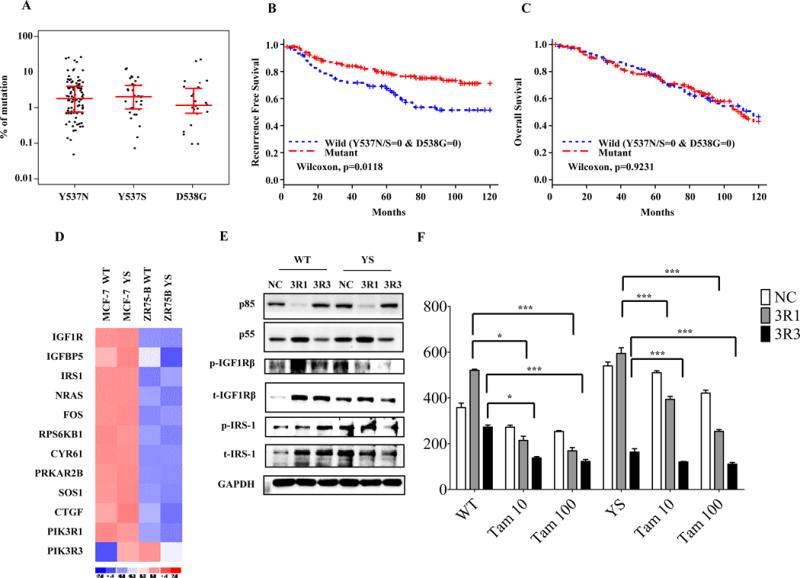
a, ddPCR analysis showing allele frequencies of three HBD-ESR1 mutations in primary breast cancers. b and c, Kaplan–Meier analysis of RFS (b), and OS (c). (d) Heatmaps of mRNA levels of genes involved in IGF-1 signaling pathway comparing MCF-7 and ZR-75B stable. Immunoblot analysis to detect phosphorylation and total protein expression of IGF1Rβ, pIRS-1, p85 and p55; GAPDH was used as a loading control (e). Immunoblot of cells transduced with siRNAs for p55 and p85. (f). Cells were also plated in soft agar assays and treated with vehicle (-) or with Tam (10 and 100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; ***P<0.001.
Knockdown of PI3K regulatory units PIK3R1 and PIK3R3 enhances anti-proliferative effects of Tam
Undoubtedly, primary breast cancers are heterogeneous, and a combination of molecular alterations will define the functional consequences determining therapeutic sensitivity to hormone therapy. To explore other cell-intrinsic effectors of Tam response in mutant cells, we further examined the biology of two of the differentially-expressed IGF-1 pathway genes identified using comparative microarray analysis of WT and the Y537S mutant in MCF-7 vs. ZR-75B cells (Fig. 6D). Up-regulated IGF-1 pathway members the PI3K regulatory subunits 1 and 3 (PIK3R1 and PIK3R3). In addition to their roles in PI3K signaling, PIK3R1 and 3 can serve as adaptors for cellular signaling. PIK3R3 can physically interact with IGF1R and impact signaling26, and naturally-occurring mutations in PIK3R1 activate PI3K/MAPK signaling and dictate sensitivity to MAPK pathway inhibitors27. Since RNA levels of both PIK3R1 and 3 were elevated in MCF-7 but not ZR-75B Y537S mutant-expressing cells (Fig. 6D), we used siRNAs for specific knockdown of the 3R1 (p85) and 3R3 (p55) subunits, and examined effects on signaling and Tam response. Knockdown of PIK3R3 exhibited the greatest effect on reducing pIGF1Rβ levels in mutant cells, and affected proliferation of both WT and mutant cells (Fig. 6E and F). Knockdown of both regulatory subunits potentiated the anti-proliferative effects of Tam, especially in mutant-expressing cells. Thus, altered PI3K regulatory subunit expression may be cell-intrinsic factors associated with Tam response in breast cancer, especially in those patients expressing ESR1 mutants. These observations will require validation in clinical material from ESR1 mutant-positive patients.
DISCUSSION
The “rediscovery” of ESR1 mutations in metastatic breast cancer has reopened a number of important clinical questions about their role in acquired resistance to hormonal agents, and the metastatic spread of breast cancer28. The most important clinical question of course is whether the mutations are targetable? To date, it is well-demonstrated that ER transcriptional activity associated with mutations in the HBD is hormone-independent, and that the mutations display different transcriptional responses to Tam and fulvestrant4,5,6,7. Molecular modeling of the Y537S and D538G mutations show that they shift the receptor into an agonist conformation, which may account for their elevated basal transcriptional activity7, and recently it has been shown these two mutations stabilize binding to co-activators and reduce binding affinity for Tam13. Using stable expression of three hot spot ESR1 mutations, we demonstrate that although the mutations reduce the absolute anti-proliferative effects of Tam depending on the cellular background, Tam is an effective agent to reduce anchorage-independent growth of cells expressing ESR1 mutations.
We employed microarray analysis to identify cell-intrinsic mediators associated with reduced Tam effects in MCF-7 compared to ZR-75B cells, and activation of the IGF-1 pathway was the most significant differentially-expressed pathway in Ingenuity analyses. Our data suggests that activation of IGF1Rβ was a key determinant of Tam response and was driven by cross-talk with the HBD-ESR1 mutations. Enhanced binding between the Y537S ESR1 mutant and IGF1Rβ was detected. Similarly, increased binding and cross-talk between IGF1Rβ/HER2 and the K303R ESR1 mutation has previously been reported29,30. Enhanced cross-talk between WT ER and these growth factor receptors is a well-studied mechanism of hormone resistance in breast cancer31,32. In K303R ESR1 mutant-expressing cells, components both upstream and downstream of the IGF1R pathway were altered with mutant expression29. In ZR-75B cells expressing the HBD-ESR1 mutations Y537N, Y537S and D538G where differences in anti-proliferative effects to Tam were not observed, IGF-1 signaling was similarly not engaged. Thus, the ESR1 mutations may employ a common mechanism via cross-talk with the IGF1R pathway to escape hormone therapy in some cellular backgrounds.
Although previous preclinical studies have suggested that HBD-ESR1 mutants may be relatively resistant to the SERD fulvestrant4,5,33, in this study, fulvestrant was very effective alone, or in combination with either an IGF1R or mTOR-targeting agent. It has been shown that the oral SERD AZD9496 inhibited the growth of a patient-derived xenograft (PDX) harboring the D538G mutation34, and the SERM/SERD hybrid pipendoxifen inhibited the growth of a PDX with the Y537S mutation35. An early report from the FERGI trial suggests that ESR1 mutations are not associated with differential PFS in the fulvestrant control arm, thus the mutations were not associated with resistance to fulvestrant34. Thus this collective data strongly support the use of fulvestrant in metastatic patients harboring ESR1 mutations.
Another important clinical question is which component of the activated signaling cascade observed in ESR1 mutant tumors might be the most optimal therapeutic target for combination therapy with fulvestrant? Fulvestrant or exemestane combined with IGF1R inhibition was unsuccessful in a phase II trial of advanced breast cancer, which effectively halted the use of hormonal therapies combined with an IGF1R inhibitor in breast cancer36. Our results showed that the combination of fulvestrant with the GSK inhibitor was very effective in ESR1 mutant cells with activated IGF-1 signaling. The assessment of the IGF-1 molecular signature and alternate IGF1R modulators, such as PIK3R3 also mediating IGF1R escape, within the context of a clinical trial, might improve patient selection for combined hormonal/IGF1R strategies in ESR1 mutant metastatic patients.
The PI3K/AKT/mTOR pathway is commonly altered in luminal tumors, and preclinical studies have shown that inhibition of these pathways can restore hormone sensitivity37,38,39. We show that this pathway is highly activated in ESR1 mutant-expressing cells, and everolimus in combination with either fulvestrant or Tam exhibited significant anti-proliferative effects in cells expressing the Y537N/S and D538G ESR1 mutations. Preliminary results from the BOLERO-2 trial suggest that metastatic patients harboring the D538G, but not the Y537S mutation respond to exemestane plus everolimus17. This is the first report in patients that selective ESR1 mutations might respond differentially to targeted therapies, and this significant finding awaits confirmation from other patient cohorts. A small phase II study of fulvestrant and everolimus in metastatic patients after AI treatment failure showed some efficacy in delaying fulvestrant resistance, and another phase II study of everolimus in combination with Tam also showed clinical benefit in AI-resistant metastatic patients40, warranting further study of these combination in tumors with ESR1 mutations. It is interesting to note that elevated expression of PIK3R3, as was seen in MCF-7 YS cells, was reported to be a biomarker predicting for response to everolimus41. An important challenge of mTOR inhibition however is signaling feedback via elevated expression and activity of growth factor receptors, providing alternative survival pathways to evade therapy42. One could speculate that this could become particularly problematic in the treatment of ESR1 mutant tumors with constitutively activated pIGF1R.
The TCGA breast cancer next generation sequencing study did not detect ESR1 mutations in primary ER-positive tumors18; however, a recent report from Takeshita et al. using ddPCR reported a frequency of 2.5% in primary breast cancer43. Emerging data using cell free circulating tumor DNA have reported ESR1 mutant frequencies at almost 40% in selected metastatic patients17. We also used a sensitive ddPCR technique to detect the three HBD-ESR1 mutations, and report a high frequency of these mutations in primary tumors. The low allele frequency of the ESR1 mutations in our cohort suggests that the mutations exist in only a minor subpopulation of the primary tumor. Using single cell sequencing it has been demonstrated that mutations in breast cancer occur at low frequencies and evolve gradually over time44. In a small series of patients monitored for circulating tumor DNA, it has been shown that ESR1 mutations are rarely selected during adjuvant AI therapy, but are commonly selected for during AI therapy for metastatic disease. Since AI therapy might provide a selective advantage to the HBD-ESR1 mutations, our data suggest that the use of targeted ESR1 mutation sequencing might be warranted earlier in patients with advanced breast cancer. Since the ESR1 mutations might also confer enhanced metastatic potential and/or aggressive biological attributes, testing of this possibility in archived material from adjuvant studies is warranted.
MATERIALS AND METHODS
Reagents, Hormones, and Antibodies
4-OH-tamoxifen (Tam), 17β-estradiol, insulin growth factor-1 (IGF-1) and puromycin were purchased from Sigma (St. Louis, MO, USA). GSK1838705A (GSK) and everolimus were obtained from Selleck Chemicals (Houston, TX, USA). MEM, RPMI 1640, DMEM, L-glutamine, penicillin/streptomycin, MEM non-essential amino acids and SeaPlaque™ Agarose were from Lonza (Walkersville, MD, USA). Fetal bovine serum was obtained from Gemini Bio Products (West Sacramento, CA, USA). SuperScript III reverse Transcriptase, qPCR probes (IGF1Rβ and GAPDH), and lipofectamine LTX was provided by Life Technologies (Grand Island, NY, USA). Antibodies were: ERα (Vector Laboratories, Burlingame, CA, USA); total IGF1R, IRS-1, mTOR, phosphorylated IGF1R(Tyr1131), mTOR(Ser2448), pS6(Ser240, 244), p85 and p55 (Cell signaling Technology, Beverly, MA, USA); IRS-1(Tyr612) (Invitrogen, Carlsbad, CA, USA); and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Goat anti-mouse and anti-rabbit secondary antibodies were from Amersham Bioscences (Piscataway, NJ, USA). The Renilla Luciferase assay kit was from Promega (Madison, WI, USA). GFP-nAB beads were from Allele Biotechnology (San Diego, CA, USA).
Plasmids
Y537N, Y537S, and D538G-ERα constructs were generated using QuickChange Site-Directed Mutagenesis (Stratagene, La Jolla, CA, USA) to make the indicated point mutations in LV111-mCherry-ERα (purchased from Genecopoeia, Rockville, MD, USA). The primer sequences for construction of the point mutations were as follows: Y537N, 5′-cgtggtgcccctcaatgacctgctgctggag-3′; Y537S, 5′-cgtggtgcccctctctgacctgctgctggag-3′; D538G, 5′-cgtggtgcccctctatggcctgctgctggag-3′ with the mutated nucleotides italicized and underlined. The entire nucleotide sequence of the construct then was verified using Sanger Sequencing.
Empty vector (EV), IGF1R SiRNAs, PIK3R1 and PIK3R3 SiRNAs, were provided by Addgene (Cambridge, MA. USA).
Cell Culture
MCF-7 and MCF-7 BK cells were grown in MEM; T47D and ZR-75B cells (generously obtained from Dr. Marc Lippman) in RPMI-1640, HEK293 in DMEM without sodium pyruvate were grown supplemented with 10% FBS, 100 I.U./ml Penicillin, 100μg Streptomycin, and 0.1mM non-essential amino acids. All stable clones were maintained in puromycin (1μg/μl).
Stable Clones
Transduced cells were generated as previously described45. A growth disadvantage was observed during selection, especially with the Y537N and Y537S clones,.
Transfection Assays
Transfection for ERE-luciferase assays, siRNA, immunoblot, coimmunoprecipitation and PLA assays was performed using the lipofectamine reagent as recommended by the manufacturer.
Proximity Ligation Assays
Proximity ligation assays was performed using Duolink detection reagent kits (Sigma-Aldrich, St. Louis, MO, USA). Fluorescence was detected using a Leica confocal microscope TCS SP5 (Leica Microsystems, Buffalo Grove, IL, USA) and Leica Application Suite Software (LAS, Wetzlar, Germany, EU).
Cell Extraction, Immunoblot and Coimmunoprecipitation Analysis
Cells were cultured in regular media or were starved in 5% charcoal stripped-serum media for 24h, and were treated with IGF-1 50ng/ml for 15′ and E2 100nM for 2h, GSK 1μM, everolimus 100nM and Tam 100nM for 5 days (immunoblot analysis), with IGF-1 50ng/ml for 2h (coimmunoprecipitation analysis) before lysis. Immunoblot analyses were performed as previously described45. Coimmunoprecipitation analyses were performed using the GFP-nAb beads.
Cell Proliferation Assays
Cell proliferation was measured using a soft agar anchorage-independent assay as previously decribed45.
Expression Microarray Analysis
Cells were plated in regular media for 24 hours and then treated for another 24 hours with Tam 100nM. RNA was extracted using the RNeasy micro kit (Qiagen. Valencia, CA, USA). Labeled cRNA was hybridized onto Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix Inc. Santa Clara, CA, USA) with three chips each. Expression values were estimated using RMA method with Partek software (http://partek.com/). Three-way ANOVA with contrasts were run using Partek to find differentially-expressed genes. Differentially-expressed genes with FDR (Fold Discovery Rate) = 0.01 and Fold=3 was then used for Ingenuity Pathway Analysis (http://www.ingenuity.com/).
Wound-Healing Scratch Assays
Cell monolayers were scraped and wound closuremonitored over 12–24h, and cells fixed and stained with Coomassie brilliant blue. Pictures were taken at 10X magnification using phase-contrast microscopy. The rate of wound healing was quantified from the images using Scion Image Program.
Mutation Dection using ddPCR
DNA was isolated from archived, formalin fixed paraffin-embedded patient samples and amplified using a QX100 ddPCR system. In this study we used invasive breast cancer obtained from women in the United Staes, and maintained in an archived tumor bank at Baylor College of Medicine. Patients were diagnosed between 1973 and 1993 and treated with mastectomy or lumpectomy plus axillary dissection, with or without postoperatvie radiation therapy. All patients had been treated with adjuvant Tam monotherapy11. The fraction of positive droplets determined the concentration of the target molecules in the sample; any drop was considered positive. Samples were analyzed by fractional abundance (molecules of mutation/molecules of WT). Specific assays for ESR1 Y537N, Y537S or D538G were designed and optimized using MCF-7 stable expression cell lines. FAM was used for Y537S, Y537N and D538G mutations, and HEX for wide type.
Sequences for amplicons generated: Y537N WT: TACAGCATGAAGTGCAAGAACGTGGTGCCCCTCTATGACCTGCTGCTGGAGATGCTGGACG; Y537N Mutant:GTACAGCATGAAGTGCAAGAACGTGGTGCCCCTCAATGACCTGCTGCTGGAGATGCTGGACG; Y537S WT: GTACAGCATGAAGTGCAAGAACGTGGTGCCCCTCTATGACCTGCTGCTGGAGATGCTGGACGCCC; Y537S Mutant: TACAGCATGAAGTGCAAGAACGTGGTGCCCCTCTCTGACCTGCTGCTGGAGATGCTGGACGCCC; D538G WT: GTACAGCATGAAGTGCAAGAACGTGGTGCCCCTCTATGACCTGCTGCTGGAGATGCTGGACGCCC; D538G Mutant: GTACAGCATGAAGTGCAAGAACGTGGTGCCCCTCTATGGCCTGCTGCTGGAGATGCTGGACGCCC.
Statistical Analyses
Data were analyzed by student’s t-test using GraphPAD Prism5 software (GraphPad Software, Inc. San Diego, CA, USA). Standard deviations (SD) are shown.
Supplementary Material
Figure S1. The effects of ESR1-HBD mutations in modulating the anti-proliferative Tam response in different breast cancer cell lines. a, c, e, immunoblot analysis to detect ERα in MCF-7BK, ZR-75B and T47D stable clones. GAPDH was used as a loading control. b, d, f cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM). Cells were allowed to grow for 14 days, and the number of colonies greater than 50μm were counted graphed. Experiments were performed in triplicate and error bars indicate SD. ns, not significant, *P<0.05.
Figure S2. HBD-mutant growth effects in MCF-7BK. a, Y537N, Y537S, and D538G-ERα showed activation of IGF-1 signaling pathway. Immunoblot analysis to detect phosphorylation and total protein expression of IGF1Rβ and ERα. GAPDH was used as a loading control. b, Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and GSK (1μM). Cells were allowed to grow for 14 days, and the number of colonies greater than 50μm were counted. Experiments were performed in triplicate and error bars indicate SD. *P<0.5. c, Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and everolimus (100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant; **P<0.01 d, fulvestrant in combination with everolimus displayed a greater cell growth reduction in mutant expressing cells. Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and fulvestrant (100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; **P<0.01; ***P<0.001.
Figure S3. Effects of HBD-ESR1 mutations on cell motility. Wound-healing assays in cells treated as indicated. Fields were photographed immediately after wounding (inset, time 0) and 24 hours later for WT and Y537S-MCF-7 expressing cells, respectively. Representative images from each condition are shown (a and c). The histograms represent the percentage of wound closure calculated by image analysis using Scion Image software (b and d). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; ***P<0.001.
Figure S4. Effects of individual mutations on recurrence free survival or overall survival. a, b, c, d, e and f, Kaplan–Meier analysis for recurrence free survival (a,b,c) and overall survival of each HBD-ESR1 mutations (d, e, f).
Supplementary Table 1. Clinical characteristerics of breast cancer cases and by Y537N/S and D538G mutation status
Acknowledgments
This work was supported by NIH/NCI R01-CA72038, CPRIT RP120732, and the Breast Cancer Research Foundation to SAWF, and FIRC-AIRC GRANT n16487 to LG.
Footnotes
Disclosure of Potential Conflicts of Interest
Authors have nothing to disclose
References
- 1.Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 2.Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:1979–1987. doi: 10.1158/1078-0432.CCR-09-1823. [DOI] [PubMed] [Google Scholar]
- 3.Fuqua SA, Gu G, Rechoum Y. Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast cancer research and treatment. 2014;144:11–19. doi: 10.1007/s10549-014-2847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer research. 2013;73:6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 6.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nature genetics. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Science translational medicine. 2015;7:313ra182. doi: 10.1126/scitranslmed.aac7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer research. 1997;57:1244–1249. [PubMed] [Google Scholar]
- 10.Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer research. 2000;60:4026–4029. [PubMed] [Google Scholar]
- 11.Herynk MH, Parra I, Cui Y, Beyer A, Wu MF, Hilsenbeck SG, et al. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:3235–3243. doi: 10.1158/1078-0432.CCR-06-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuqua SA. The role of estrogen receptors in breast cancer metastasis. Journal of mammary gland biology and neoplasia. 2001;6:407–417. doi: 10.1023/a:1014782813943. [DOI] [PubMed] [Google Scholar]
- 13.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife. 2016;5 doi: 10.7554/eLife.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, et al. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. Journal of the National Cancer Institute. 2014;106:djt337. doi: 10.1093/jnci/djt337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gendreau S, Spoerke J, Johnston S, Schmid P, Krop I, Qui J, et al. High prevalence and clonal heterogeneity of ESR1 mutations (mt) in circulating DNA (ctDNA) from patients (pts) enrolled in FERGI, a randomized phase II study testing pictilisib (GDC-0941) in combination with fulvestrant (F) in pts that failed a prior aromatase inhibitor (AI). [abstract]; Proceedings of the Thirty-Eighth Annual CTRC-AACR San Antonio Breast Cancer Symposium; 2015 Dec 8–12; San Antonio, TX. 2016. Abstract nr PD6-03. [Google Scholar]
- 16.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England journal of medicine. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandarlapaty S, Sung P, Chen D, He W, Samoila A, You D, et al. cfDNA analysis from BOLERO-2 plasma samples identifies a high rate of ESR1 mutations: Exploratory analysis for prognostic and predictive correlation of mutations reveals different efficacy outcomes of endocrine therapy-based regimens. [abstract]. In: Proceedings of the Thirty-Eighth Annual CTRC-AACR San Antonio Breast Cancer Symposium: 2015 Dec 8–12; San Antonio, TX. Philadelphia (PA): AACR; Cancer Research. 2016;76(4 Suppl) Abstract nr S2-07. [Google Scholar]
- 18.Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Molecular endocrinology. 1999;13:787–796. doi: 10.1210/mend.13.5.0274. [DOI] [PubMed] [Google Scholar]
- 19.Yee D. Insulin-like growth factor receptor inhibitors: baby or the bathwater? Journal of the National Cancer Institute. 2012;104:975–981. doi: 10.1093/jnci/djs258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burris HA., 3rd Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer chemotherapy and pharmacology. 2013;71:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 21.Hasson SP, Rubinek T, Ryvo L, Wolf I. Endocrine resistance in breast cancer: focus on the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin signaling pathway. Breast care. 2013;8:248–255. doi: 10.1159/000354757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston SR, Kilburn LS, Ellis P, Dodwell D, Cameron D, Hayward L, et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. The Lancet Oncology. 2013;14:989–998. doi: 10.1016/S1470-2045(13)70322-X. [DOI] [PubMed] [Google Scholar]
- 23.Massarweh S, Romond E, Black EP, Van Meter E, Shelton B, Kadamyan-Melkumian V, et al. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast cancer research and treatment. 2014;143:325–332. doi: 10.1007/s10549-013-2810-9. [DOI] [PubMed] [Google Scholar]
- 24.Chan JY, LaPara K, Yee D. Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene. 2016 doi: 10.1038/onc.2015.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen-Dien GT, Smith RA, Haupt LM, Griffiths LR, Nguyen HT. Genetic polymorphisms in miRNAs targeting the estrogen receptor and their effect on breast cancer risk. Meta gene. 2014;2:226–236. doi: 10.1016/j.mgene.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soroceanu L, Kharbanda S, Chen R, Soriano RH, Aldape K, Misra A, et al. Identification of IGF2 signaling through phosphoinositide-3-kinase regulatory subunit 3 as a growth-promoting axis in glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3466–3471. doi: 10.1073/pnas.0611271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer cell. 2014;26:479–494. doi: 10.1016/j.ccell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giguere V. Estrogen receptor mutations in breast cancer-an anticipated “rediscovery?”. Molecular endocrinology. 2014;28:427–428. doi: 10.1210/me.2014-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barone I, Iacopetta D, Covington KR, Cui Y, Tsimelzon A, Beyer A, et al. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene. 2010;29:2404–2414. doi: 10.1038/onc.2009.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giordano C, Cui Y, Barone I, Ando S, Mancini MA, Berno V, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast cancer research and treatment. 2010;119:71–85. doi: 10.1007/s10549-009-0334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:2702–2708. doi: 10.1158/1078-0432.CCR-09-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, et al. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. The Journal of steroid biochemistry and molecular biology. 2010;118:219–230. doi: 10.1016/j.jsbmb.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: An oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1 mutant breast tumours in preclinical models. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-15-2357. [DOI] [PubMed] [Google Scholar]
- 35.Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, Dann SG, et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:5121–5130. doi: 10.1158/1078-0432.CCR-15-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robertson JF, Ferrero JM, Bourgeois H, Kennecke H, de Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. The Lancet Oncology. 2013;14:228–235. doi: 10.1016/S1470-2045(13)70026-3. [DOI] [PubMed] [Google Scholar]
- 37.Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:5319–5328. doi: 10.1158/1078-0432.CCR-04-2402. [DOI] [PubMed] [Google Scholar]
- 38.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4452–4461. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun M, Paciga JE, Feldman RI, Yuan Z, Coppola D, Lu YY, et al. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer research. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 40.Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 41.Hurvitz SA, Kalous O, Conklin D, Desai AJ, Dering J, Anderson L, et al. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast cancer research and treatment. 2015;149:669–680. doi: 10.1007/s10549-015-3282-x. [DOI] [PubMed] [Google Scholar]
- 42.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, et al. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Translational research : the journal of laboratory and clinical medicine. 2015;166:540–553. e542. doi: 10.1016/j.trsl.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu G, Gelsomino L, Covington KR, Beyer AR, Wang J, Rechoum Y, Huffman K, Carstens R, Andò S, Fuqua SA. Targeting thyroid hormone receptor beta in triple-negative breast cancer. Breast Cancer Res Treat. 2015;150(3):535–45. doi: 10.1007/s10549-015-3354-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effects of ESR1-HBD mutations in modulating the anti-proliferative Tam response in different breast cancer cell lines. a, c, e, immunoblot analysis to detect ERα in MCF-7BK, ZR-75B and T47D stable clones. GAPDH was used as a loading control. b, d, f cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM). Cells were allowed to grow for 14 days, and the number of colonies greater than 50μm were counted graphed. Experiments were performed in triplicate and error bars indicate SD. ns, not significant, *P<0.05.
Figure S2. HBD-mutant growth effects in MCF-7BK. a, Y537N, Y537S, and D538G-ERα showed activation of IGF-1 signaling pathway. Immunoblot analysis to detect phosphorylation and total protein expression of IGF1Rβ and ERα. GAPDH was used as a loading control. b, Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and GSK (1μM). Cells were allowed to grow for 14 days, and the number of colonies greater than 50μm were counted. Experiments were performed in triplicate and error bars indicate SD. *P<0.5. c, Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and everolimus (100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant; **P<0.01 d, fulvestrant in combination with everolimus displayed a greater cell growth reduction in mutant expressing cells. Cells were plated in soft agar assay and then treated with vehicle (-) or with Tam (100nM) and fulvestrant (100nM). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; **P<0.01; ***P<0.001.
Figure S3. Effects of HBD-ESR1 mutations on cell motility. Wound-healing assays in cells treated as indicated. Fields were photographed immediately after wounding (inset, time 0) and 24 hours later for WT and Y537S-MCF-7 expressing cells, respectively. Representative images from each condition are shown (a and c). The histograms represent the percentage of wound closure calculated by image analysis using Scion Image software (b and d). Experiments were performed in triplicate and error bars indicate SD. ns, not significant. *P<0.05; ***P<0.001.
Figure S4. Effects of individual mutations on recurrence free survival or overall survival. a, b, c, d, e and f, Kaplan–Meier analysis for recurrence free survival (a,b,c) and overall survival of each HBD-ESR1 mutations (d, e, f).
Supplementary Table 1. Clinical characteristerics of breast cancer cases and by Y537N/S and D538G mutation status