

Abstract
Synucleins (α, β, γ-synuclein) are a family of abundant presynaptic proteins. α-Synuclein is causally linked to the pathogenesis of Parkinson disease (PD). In an effort to define their physiological and pathological function(s), we investigated the effects of deleting synucleins and overexpressing α-synuclein PD mutations, in mice, on synapse architecture using electron microscopy (EM) and Cryo-Electron Tomography (Cryo-ET). We show that synucleins are regulators of presynapse size and synaptic vesicle (SV) pool organization. Using Cryo-ET, we observed that deletion of synucleins increases SV tethering to the active zone but decreases the inter-linking of SVs by short connectors. These ultrastructural changes were correlated with discrete protein phosphorylation changes in αβγ-synuclein−/− neurons. We also determined that α-synuclein PD mutants (PARK1/hA30P, PARK4/hα-syn) primarily affected presynaptic cytomatrix proximal to the active zone, congruent with previous findings that these PD mutations decrease neurotransmission. Collectively, our results suggest that synucleins are important orchestrators of presynaptic terminal topography.
eTOC blurb
Synucleins (α, β, γ-synuclein) are abundant presynaptic proteins, with α-synuclein linked to the pathogenesis of Parkinson’s disease. Vargas et al. investigate the effects of deleting synucleins and overexpressing mutated α-synuclein on synapse architecture using electron microscopy. They find that synucleins regulate presynaptic terminal size and synaptic vesicle distribution.
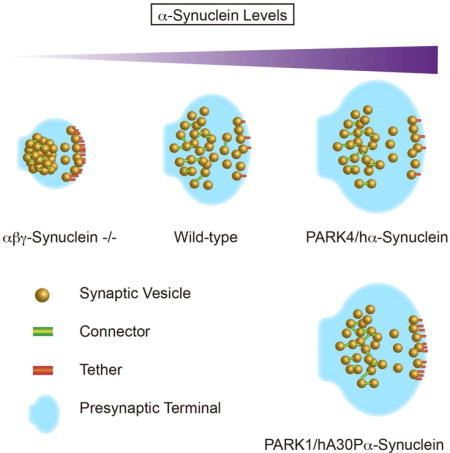
Introduction
Synapses are exquisite examples of the axiom ‘form follows function’. Subtle alterations in synapse structure can have profound functional consequences (Atasoy et al., 2007; Ho et al., 2003; Altrock et al., 2003; Gundelfinger et al., 2016; Jiang and Ehlers, 2013). This is underscored by the fact that many neurological and psychiatric illnesses are associated with alterations in synapse structure (van Spronsen and Hoogenraad, 2010; Penzes et al., 2011).
The presynaptic terminal has a complex architecture with SVs being located at different distances from the active zone (AZ) and functionally organized in three main pools: reserve, recycling and readily releasable pool. Cryo-ET has been recently used for the visualization of SV interacting proteins, due to its superior preservation of labile structures. Short filaments tethering SVs to the AZ, known as tethers, and filaments linking SVs to one another, known as connectors, were shown by Cryo-ET to be the most prominent elements of the presynaptic cytomatrix. Tethers and connectors play important roles in controlling the distribution of SVs and the vesicle progression towards release (Fernandez-Busnadiego et al., 2010; Fernandez-Busnadiego et al., 2011). However, the proteins that regulate presynaptic structure, supramolecular organization, and canonical SV distribution, especially the molecular constituents of tethers and connectors, are not well defined, even though the presynaptic proteome has been cataloged (Takamori et al., 2006; Wilhelm et al., 2014) and molecular functions assigned to a large fraction of proteins.
Synucleins are a family of abundant presynaptic proteins (α, β, and γ-synuclein). They are 14 KDa proteins, with conserved N-termini that bind acidic lipids and divergent C-termini (Vargas and Chandra, 2015). Synucleins can sense and generate membrane curvature (Westphal and Chandra, 2010; Middleton and Rhoades, 2013), and this property implies functions in either SV exocytosis or endocytosis (Merrifield and Kaksonen 2014; Martens et al., 2007; Hui et al., 2009), the two steps that require membrane curvature. We previously showed that a conserved function of the synuclein family is to regulate the kinetics of SV endocytosis (Vargas et al., 2014). In addition, recent literature supports a role for α-synuclein in SV exocytosis (Burré et al., 2010). These properties suggest that synucleins are likely to have an impact on presynaptic structure.
Mutations in the α-synuclein gene cause familial Parkinson’s disease (PD), including 6 point mutations (A30P, E46K, H50Q, G51D, A53T, A53E—PARK1) (Polymeropoulos et al., 1997) and gene multiplications (duplication and triplication—PARK4) (Singleton et al., 2003). Moreover, sequence variants that enhance α-synuclein expression increase risk for developing PD. In addition, α-synuclein is the main component of Lewy bodies, the signature pathology of PD, prompting many to consider α-synuclein as the key gene in the pathophysiology of PD. As with other neurodegenerative diseases, synapse dysfunction and loss are early events in the pathogenesis of PD (Calo et al., 2016; Bellucci et al., 2016), therefore there is great interest in understanding how α-synuclein and its pathological variants impact synaptic structure and function.
To understand how synucleins influence synapse structure, we performed conventional EM, immunoEM as well as Cryo-ET on wild type, synuclein null (αβγ-Syn−/−) and α-synuclein PD mutant (PARK1/hA30P, PARK4/hα-syn) overexpressing synaptosomes and neurons. Through these different modes of visualization, we discovered that synucleins are important regulators of synapse size as well as SV distribution. We also determined that PD mutants have select effects on the presynaptic cytomatrix.
Results
Synucleins Are Associated With SVs Throughout the Terminal
Synucleins are highly abundant at presynaptic termini (Nakajo et al., 1996, Westphal and Chandra, 2013; Wilhelm et al., 2014). In standard biochemical fractionation experiments, synucleins appear to be mainly cytosolic and absent from the SV fraction (Takamori et al., 2006). This is due to the characteristic of synucleins to come off the membrane in low ionic conditions. Fractionation experiments that preserves physiological salt conditions suggest that synucleins are SV associated proteins. To investigate the native distribution of synucleins in the physiological setting of the presynaptic terminal, we purified synaptosomes and quickly fixed them and carried out immunoEM (De Camilli et al, 1983; Fig. 1A). As a positive control, we tested the location of the SV protein synaptobrevin 2 and found that 82.2% of the gold particles were found on SVs and 11.6% on the synaptic membrane. To confirm the specificity of the α-synuclein antibody, we stained αβγ-Syn−/− synaptosomes. There was a minimal background signal (Fig. 1C) on mitochondria and synaptic plasma membrane, but none on SVs. When we probed for the localization of α-synuclein in wild type synaptosomes, the majority of the labelling, 74.2% of gold particles were found on SVs and 16.7% on the synaptic membrane (Fig. 1C–D). This is consistent with previously published EM images localizing α-synuclein to SVs (Clayton and George, 1999; Boassa et al., 2013). From these electron micrographs, we conclude that under native conditions a significant fraction of α-synuclein is associated with SVs and found throughout the terminal (Fig. 1). Biophysical studies have shown that synucleins have high affinity for curved membranes with the strongest binding to 40–50 nm vesicles (Middleton and Rhoades, 2010). This is the precise diameter range of SVs, and our findings can thus be explained by this biophysical property of synucleins.
Figure 1. Synucleins are Peripheral SV Associated Proteins.
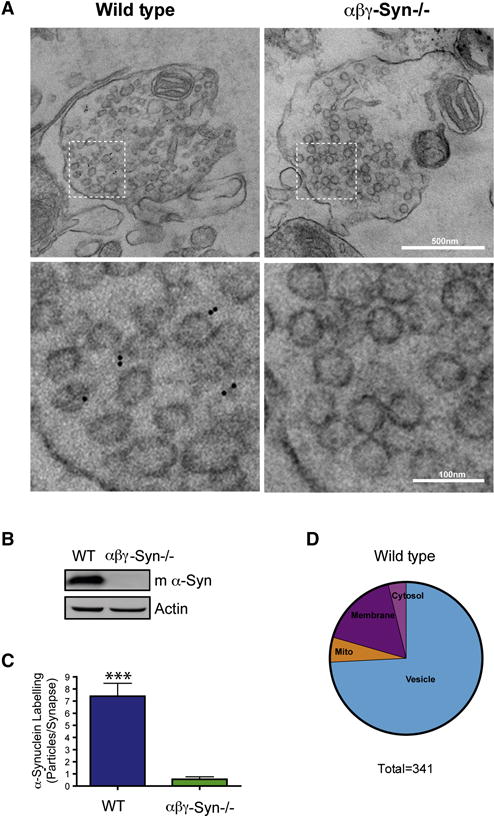
A. Representative electron micrographs of wild type and αβγ-Syn−/− synaptosomes after hypotonic fixation and immunoEM with an α-synuclein specific antibody and secondary gold particles (top panels; Scale bar = 500 nm). The bottom panels are zoomed in regions noted in the top panels (Scale bar = 100 nm). The black dots denote the location of the gold particles. B. Western blotting of wild type and αβγ-Syn−/− synaptosomes with a mouse α-synuclein antibody to confirm genotypes. C. Quantification of α-synuclein labeling in micrographs such as shown in A. N = experiments; 46–25 synapses/2–3 mice/genotype. D. Pie chart of the localization of α-synuclein gold particles (n=341). *** p<0.001.
Deletion of Synucleins Results in Smaller Presynaptic Termini
To examine whether deletion of synucleins results in structural alterations, in addition to the documented vesicle cycling deficits (Burré et al. 2010; Vargas et al., 2014), we performed conventional EM on wild type and αβγ-Syn−/− dissociated hippocampal neuronal cultures (Fig. 2). In wild type CA1 hippocampal neurons in culture, the average presynaptic terminal area was 112 μm2 and has on average 90 vesicles per bouton, consistent with literature (Greten-Harrison et al., 2010; Fig. 2C–D). These features are altered when synucleins are absent. In αβγ-Syn−/− neurons, the mean presynaptic area is decreased by ~16% to 94 μm2 (Fig. 2C) while maintaining the number of SVs (Fig. 2D). Consequently, SV density is increased relative to the wild type (Fig. 2E). The same phenotype was observed in brain tissue, specifically in CA1 and CA3 regions of the hippocampus (Greten-Harrison et al. 2010), strongly suggesting that this phenotype is cell-autonomous.
Figure 2. EM of Wild type and αβγ-Syn−/− Synapses.
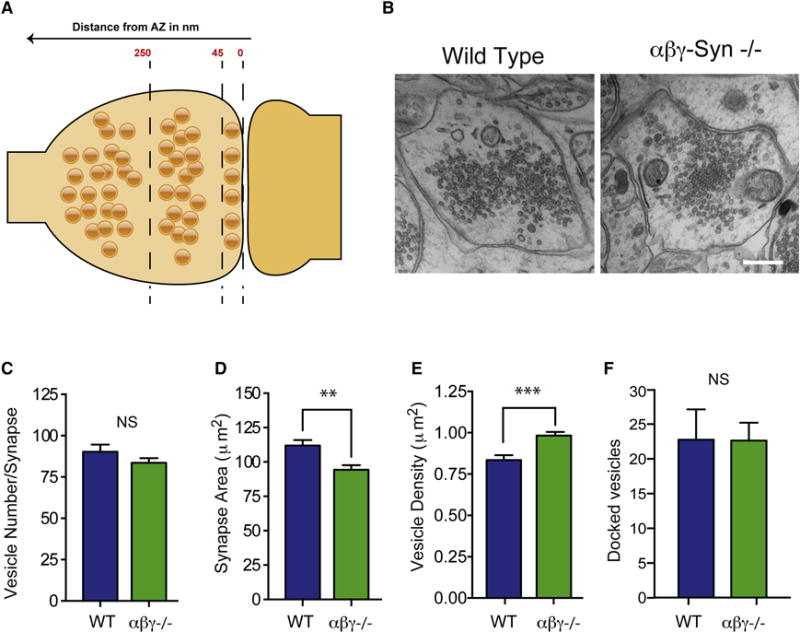
A. Cartoon of a wild type synapse with the three zone of SVs indicated: 45 nm from AZ, docked/proximal SVs; 45–250 nm, intermediate SVs; >250 nm, distal SVs. B. Electron micrograph of wild type and αβγ-Syn−/− synapses. Quantification in wild type (blue) and αβγ-Syn−/− (green) synapses of C. presynaptic terminal area; D. SV number; E. SV Density/Presynaptic Area F. Docked vesicle number. N = 3 independent cultures, with a minimum of 56 micrographs/neuronal culture. N= 3 independent neuronal cultures, 50–150 synapses/genotype/culture were analyzed. Scale Bar for Bi and ii = 400 nm. NS, not significant, ** p<0.01, *** p<0.001.
In wild type synapses, SV distribution is not homogenous and three SV pools have been defined both functionally and spatially in reference to the AZ. Docked/proximal vesicles are found adjacent to the AZ (Fig. 2A–B). In high-pressure frozen and freeze dehydrated samples, docked vesicles that are attached to the plasma membrane at the AZ constitute the readily releasable pool (RRP) that is primed for release (Fig. 2A–B) (Imig et al 2014). The recycling pool is used to sustain normal neurotransmission, while the reserve pool is used under conditions of high stimulation. The recycling and reserve pool SVs are now recognized to be interspersed, though the reserve pool, which makes up a majority of the total SV pool, is physically located more distally (Rizzoli and Betz et al 2005). To investigate the organization of SVs from a structural standpoint by EM, the presynaptic terminal was divided into three regions: 0–45 nm from AZ, 45–250 nm and >250 nm (Fig. 2A). We characterized the overall distribution of SVs in wild type and αβγ-Syn−/− synapses by both EM and cryo-ET. In the case of cryo-ET, as in previous studies, a 250 nm limit was imposed because of the large variability of the number of SVs and the decreased image contrast beyond that distance. By conventional EM, we observe that wildtype and αβγ-Syn−/− synapses, have a similar SV distribution (Fig. 2A–B). SVs were visible in all three regions in αβγ-Syn−/− synapses, even though they are smaller. By Cryo-ET, the overall number and distribution of SVs in the first 250 nm from the AZ was also similar in wild type and αβγ-Syn−/− (Fig. 3A–B).
Figure 3. Cryo-ET of Wild type and αβγ-Syn−/− Synapses Reveals Increased Tethering Upon Deletion of Synucleins.
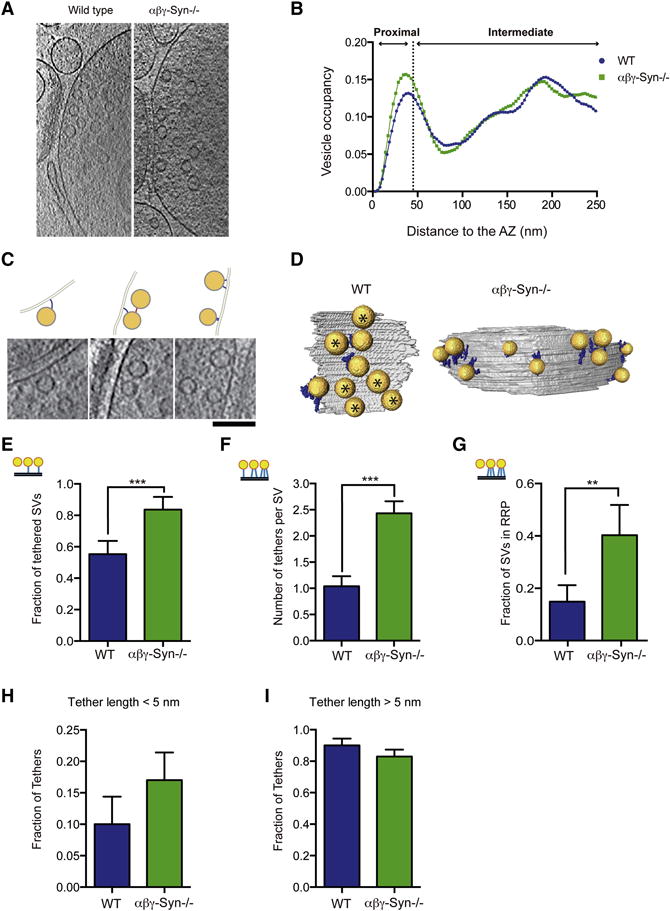
A. Representative computationally extracted 2 nm thick cryo-ET slices of wild type and αβγ-Syn−/− synaptosomes. B. The SV distribution calculated as the fraction of cytoplasmic volume occupied by SVs in the first 250nm from the AZ. C. Examples of tethered SVs along with line diagrams of images. Computationally extracted tomographic slices are 2nm thick. Scale Bar = 100nm. D. 3D visualization of SVs (yellow) tethers (blue) and AZ (grey). Asterisks are proximal vesicles that are not tethered. E. Fraction of proximal SVs that have at least one tether in wild type and αβγ-Syn−/−. F. The mean number of tethers per proximal SV per synapse in the two genotypes. p<0.001 by Kruskal-Wallis (K-W) test). G. The fraction of proximal SVs with two or more tethers (structural RRP). p<0.01 by χ2 test. H. Fraction of tethers shorter than 5 nm. I. Fraction of tethers longer than 5 nm. N = 3 independent experiments. ** p<0.01, *** p<0.001.
Alterations in Presynaptic Cytomatrix of Synapses Lacking Endogenous Synucleins
We systematically characterized presynaptic, ultrastructural elements starting from the AZ and going outwards through the three regions. First, we examined the number of docked SVs (0–45 nm from AZ) in the two genotypes by EM and showed that they were largely unaltered αβγ-Syn−/− synapses (Fig. 2F). Similarly, by Cryo-ET, the SV distribution traces showed the characteristic proximal peak (related to docked SVs in EM), indicating that synapses do not suffer from serious release defects. The number of proximal SVs/AZ area (wild type = 99.58±18.43, αβγ-Syn−/− = 100.81±17.3 per 1 μm2), and the minimum distance of proximal SVs to the AZ (wild type = 9.95±0.65 nm, αβγ-Syn−/− = 9.29±0.54 nm) were the same in both genotypes.
Cryo-ET has been used to detect and visualize tethers, protein components that link SVs to the AZ, as these delicate structures are preserved during vitrification but not in conventional EM glutaraldehyde fixation (Fernández-Busnadiego et al., 2010; 2011; Fig. 3). We examined tethering of proximal SVs (< 45 nm from AZ) in wild type and αβγ-Syn−/− synaptosomes by Cryo-ET, following procedures we have established (Fernández-Busnadiego et al. 2010; 2011; 2013). Tomographic slices and 3D renderings of tethers and vesicles are shown in Fig. 3C–D. In wild type synapses, we found that 55 ± 8.4% of proximal SVs were tethered (have at least one tether), similar to our previously published results (Fernández-Busnadiego et al., 2010; Fig. 3E), while in synapses lacking endogenous synucleins, 80± 8.1 % of proximal vesicles were tethered (Fig. 3E). The number of tethers per proximal SVs was determined and found to be 1.04 ± 0.19 in wild type synapses and 2.43 ± 0.23 in αβγ-Syn−/− synapses (Fig. 3F; p<0.001). When the length of the tethers was measured, the increase in tethering in αβγ-Syn−/− synapses was largely accounted by short tethers (<5 nm) (Fig. 3F, H–I). Our previous work has established that proximal SVs in synaptosomes treated by hypertonic sucrose have fewer and longer tethers, leading to the proposal that SVs that have three or more tethers are termed the structurally defined RRP (Fernandez-Busnadiego et al., 2010). Therefore, we calculated the fraction of multiply tethered proximal SVs to determine the structurally defined RRP. We found that the fraction of proximal vesicles that belong to structural RRP was increased ~3–4 fold in αβγ-Syn−/−synapses as compared to the wild type (WT = 0.149, αβγ-Syn−/− = 0.403; p<0.01) (Fig. 3G). These results showed that neurons lacking mouse synucleins have a larger structurally defined RRP.
Next, we examined connectors that interlink SVs located within the 250 nm from the AZ, by Cryo-ET. We analyzed the fraction of connected SVs and the number of connectors per SV for proximal (<45 nm) and intermediate (45–250 nm) SVs in wild type and αβγ-Syn−/− synapses (Fig. 4A–B). In both the proximal and intermediate zones, the fraction of connected SVs (Fig. 4C–D) and the number of connectors per SV (Fig. 4E–F) were decreased in αβγ-Syn−/− synaptosomes. Significantly, we could revert the loss of connectivity by expressing human α-synuclein in αβγ-Syn−/− background (Fig. 4). Specifically, both the fraction of connected SVs and the number of connectors per SV were rescued to wild type levels by expression of human α-synuclein (Fig. 4). Because connectors were still detected in αβγ-Syn−/− synaptosomes by Cryo-ET, synucleins are unlikely to be uniquely required for the formation of connectors. However, our findings indicate that synucleins regulate connectors, leading to altered SV connectivity.
Figure 4. Decreased Connectivity of SVs in αβγ-Syn−/−.
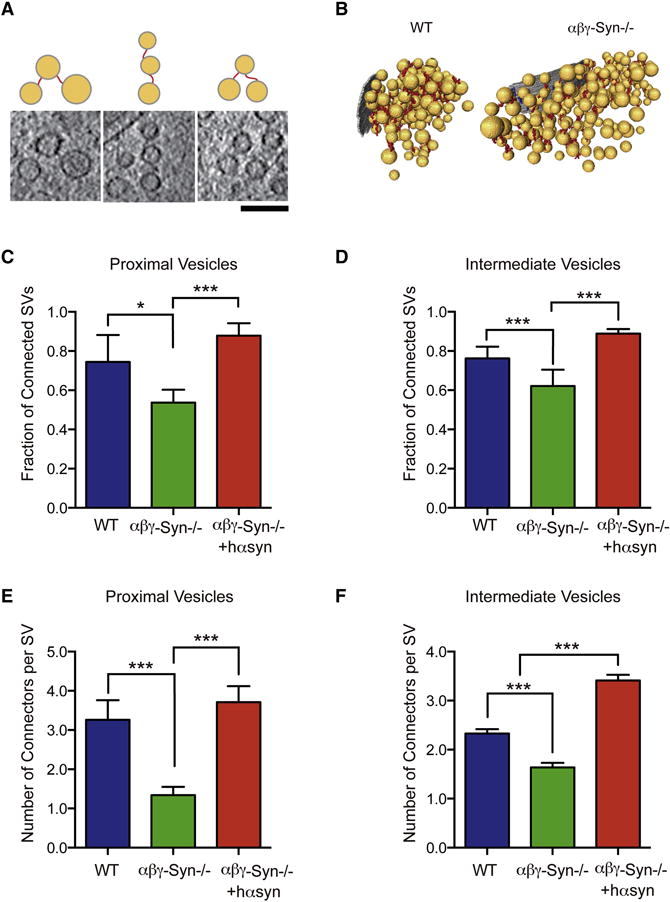
A. Examples of connectors imaged by Cryo-ET and corresponding line diagrams. Computationally extracted tomographic 2nm thick, slices. Scale Bar = 100 nm. B. 3D visualization of connectors in wild type and αβγ-Syn−/−samples by Cryo-ET. C. Fraction of proximal SVs that have at least one connector in wild type (blue), αβγ-Syn−/− (green) and synuclein nulls rescued by human α-Syn expression (red). D. Fraction of intermediate SVs that have at least one connector in wild type, αβγ-Syn−/− and rescued synapses. p<0.05 and p<0.001 respectively by χ2 test E. The mean number of connectors per proximal SV in the three genotypes. F. The mean number of connectors per intermediate SV in synapses of the three genotypes. p<0.001 for both zones by χ2 test. N = 3 independent experiments. NS, not significant, ** p<0.01, *** p<0.001.
We finally examined the distal vesicles at >250 nm from the AZ, in wildtype and αβγ-Syn−/−synapses by EM. We observe a striking increase in the frequency of clustered SVs in αβγ-Syn−/−synapses. Typical examples of this phenotype are shown in Figs. 2B and 5A. We confirmed that these vesicles belong to the reserve pool, as they can be dispersed upon strong stimulation (Fig. 5B). This phenotype is also seen in vivo in both the CA1 and CA3 regions of the hippocampus (Fig. 5C–D). These experiments show that deletion of synuclein leads to clustering of distal pool vesicles at >250 nm from the AZ both in vitro and in vivo. This would suggest that mobilization of reserve pool SVs is different in αβγ-Syn−/− despite similar numbers of SVs to wild type (Fig. 2D). Due to the 250 nm imposed Cryo-ET limit, we were unable to determine if the increased distal SV clustering in αβγ-Syn−/− (Fig. 2B; Fig. 5) is due to altered connectivity.
Figure 5. Distal SV Clusters in αβγ-Syn−/− Synapses.
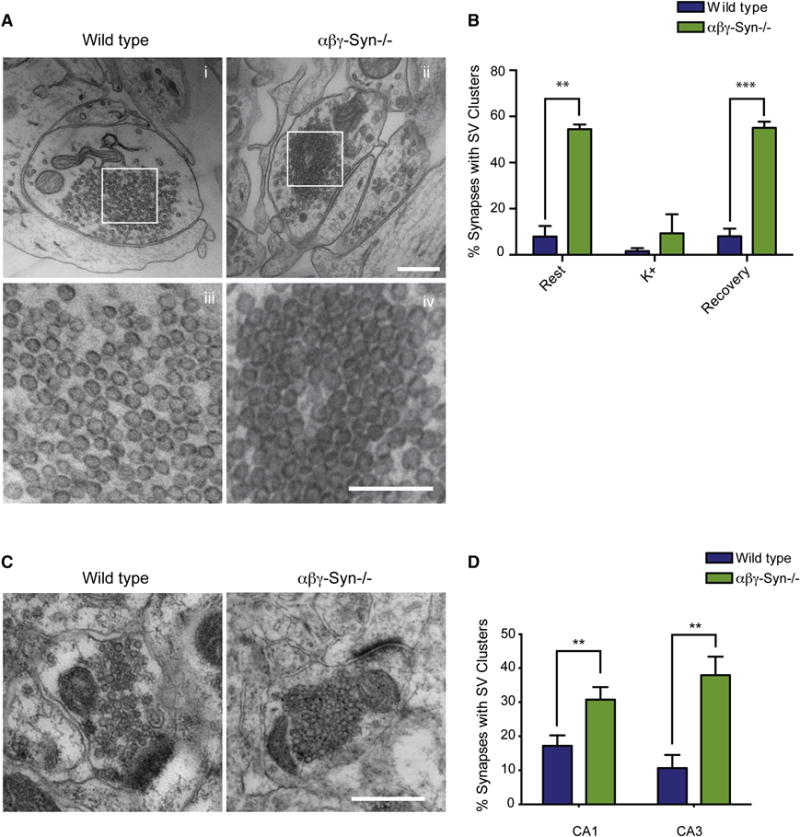
A. Representative micrographs of dissociated hippocampal neurons showing distal SV clusters are present in αβγ-Syn−/− synapses compared to wild type. iii–iv, Zoomed in regions from i and ii respectively, showing distal vesicles. Note that SVs are clustered and arrayed touching each other in αβγ-Syn−/− synapse. B. Quantification of percent of synapses with SV clusters in wild type (blue) and αβγ-Syn−/− (green) neurons at rest, after 90s stimulation with 45 mM K+ and subsequent recovery for 10 minutes. Note that the SV clusters disperse upon high K+ stimulation. N= 3 independent neuronal cultures, 50–150 synapses/genotype/culture were analyzed. C. Micrographs of CA1 synapses from wild type and αβγ-Syn−/− mice. D. Quantification of synapses with SV clusters in CA1 and CA3 regions of the brain. N= 2–3 mice/genotype Scale bar = 400nm; Scale Bar for Aiii–iv = 200 nm ** p<0.01, *** p<0.001.
Biochemical Changes in Synuclein Null Synapses: Clues to SV Pool Organization
In order to be able to ascribe the ultrastructural changes in αβγ-Syn−/− synapses to molecular changes, we need to catalog the proteomic changes induced by the absence of synucleins. We have previously systematically compared the entire synaptic proteome of wild type and αβγ-Syn−/− brains, allowing us to begin to make such ultrastructural-molecular associations. αβγ-Syn−/− synapses showed select changes (Greten-Harrison et al., 2010; Westphal and Chandra, 2013; Vargas et al., 2014; Burre et al., 2010). The upregulated (Endophilin A1, A2, Synapsin IIb, Annexin V) and downregulated proteins (synaptobrevin 2 and AP180) may mediate SV tethering, connectivity as well as distal clustering changes seen in αβγ-Syn−/−.
It has been proposed that connectors are peripheral SV proteins that behave differentially upon phosphorylation. Even though the molecular identity of the connectors remains to be determined, several candidates have been put forward based on these properties (Supplementary Table I). We analyzed the phosphorylation of most prominent candidates in wild type and αβγ-Syn−/− synaptosomes by immunoblotting with phospho-site specific antibodies. Synapsins, which are known to regulate reserve pool SVs via their phosphorylation status (Rosahl et al., 1995; Gaffield and Betz, 2007; Benfenati et al., 1993), were proposed to interlink SVs (Hirokawa et al., 1989). Synapsin I sites 1, 2, and 3 were unchanged in comparing wild type and αβγ-Syn−/− (Fig. 6A, D–E). However, the baseline phosphorylation of synapsin I at sites 4 and 5 was increased (148 %; Fig. 6A, F). Next, we tested other candidate proteins implicated in SV pool regulation, namely amphiphysins, epsins and dynamin. We observed changes in amphiphysin-2, with a greater percent of dephosphorylated amphiphysin-2 present in αβγ-Syn−/−samples. In contrast, there was no change in the level of phospho-amphiphysin-1 (Fig. 6A–B). There were no changes in phosphorylation of epsin-1 or dynamin-1 at position 774 (Fig. 6G–I), but we did observe an increase in dynamin-1 phosphorylation at position 778. Since phosphorylation of synapsin I at 4, 5 sites, amphyphysin 2 and dynamin-1 at 778 are regulated by the phosphatase calcineurin (Supplementary Table I), we tested the effect of its inhibitor Cyclosporin A. As expected, the drug increases phosphorylation of these proteins, thus ameliorating the decreased phosphorylation of amphiphysin-2 in αβγ-Syn−/− and further increasing phosphorylation of synapsin I 4, 5 and dynamin-1 at position 778 (Supplementary Fig. 1). Collectively, the pattern of phosphorylation events that occurs upon deletion of synucleins is known to promote SV endocytosis, but may contribute to alter SV clustering and decreased connectivity as well.
Figure 6. Biochemical Changes in αβγ-Syn−/− in Candidate Proteins That Regulate SV Connectivity.
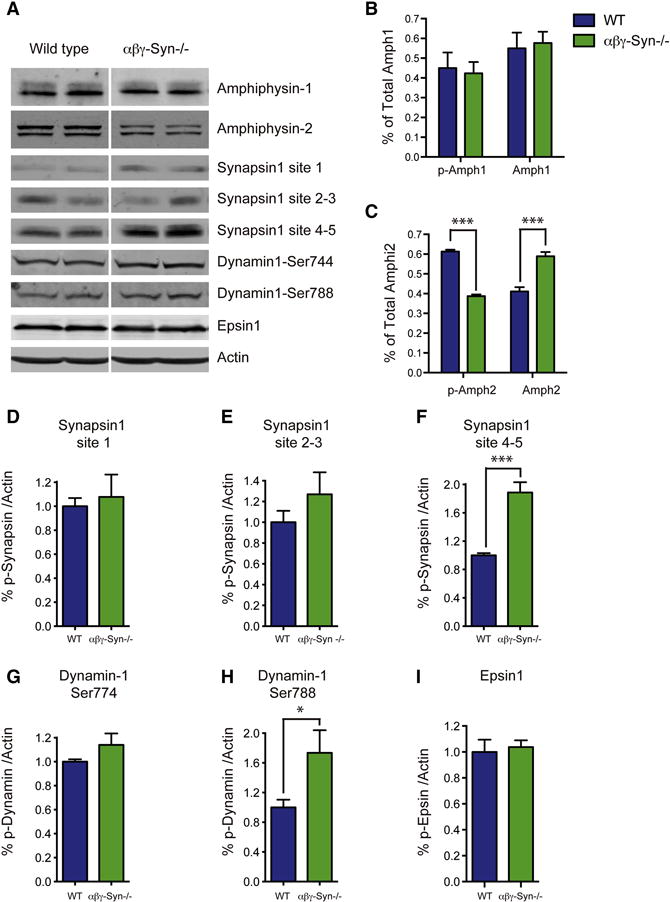
A. Representative western blots of wild type and αβγ-Syn−/− synaptosomes for the denoted proteins. B. Quantification of levels of total and phosphorylated Amphiphysin-1. C. Quantification of phospho and dephospho amphipysin-2 in the two genotypes. D–I. Phosphorylation of synapsin I at site 1 (D), site 3 (E) and sites 4, 5 (F), Dynamin 1 site 774 (G), site 788 (H), Epsin1 (I) in wild type and αβγ-Syn−/− synaptosomes. N = 3 independent experiments. NS, not significant, * p<0.05; *** p<0.001.
Previous studies have established that strong stimulation of synapses leads to phosphorylation changes (Supplementary Table I) and dispersion of the reserve pool SVs. To test if this is also the case in αβγ-Syn−/−, we strongly stimulated synaptosomes to disperse SV clusters as in Fig. 5B and monitored the changes in phosphorylation of amphiphysin-2 and synapsin site 4 and 5. As anticipated, we could dephosphorylate amphiphysin-2 and synapsin I with high K+, though the magnitude of the changes was different in the two genotypes (Supplementary Fig. 2), indicating that these two processes are correlated.
Effect of Familial PD Mutations on Presynaptic Structure
Several EM studies have examined the overexpression of human α-synuclein and consistently show an enlargement of presynaptic termini all the way to hypertrophy as well as a greater dispersion of reserve pool SVs (Boassa et al., 2013, Nemani et al., 2010, Scott et al., 2012; Busch et al., 2014). To further investigate how PD mutants impact synapse structure, we focused on how PD mutants influence the presynaptic cytomatrix. We previously developed and characterized transgenic human α-synuclein mice that either carry the A30P mutation, like a subset of PARK1 patients, or overexpress wild type human α-synuclein, like PARK4 patients (Chandra et al., 2005; Gallardo et al., 2008). These transgenic mice have ~5 fold α-synuclein in the brain compared to wild type mice, consistent with published data (Chandra et al., 2005; Fig. 7B). The α-synuclein transgenics develop age-dependent PD-like phenotypes, accompanied by α-synuclein aggregation and neuronal loss (Chandra et al., 2005; Gallardo et al., 2008).
Figure 7. α-Synuclein PD Mutants Primarily Alter Tethering.
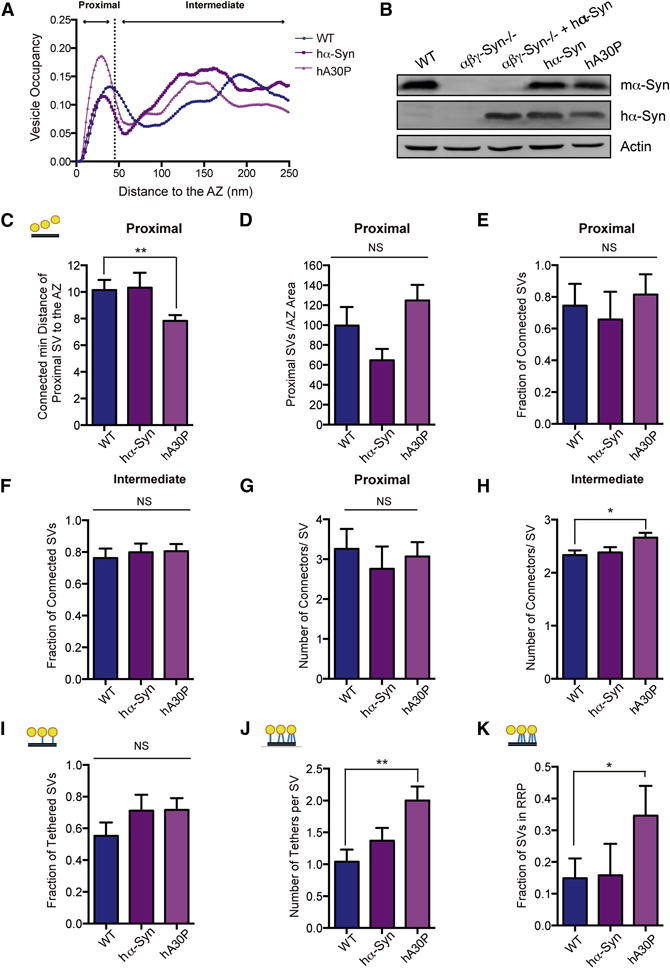
A. The SV distribution calculated as the fraction of cytoplasmic volume occupied by SVs in the first 250nm from the AZ of wild type (WT; blue), human α-synuclein overexpressing (hα-Syn; purple) and human A30P mutant α-synuclein (mauve) overexpressing transgenics. B. Western blotting of synaptosomes of the denoted genotypes. C. The mean distance of proximal SVs to the AZ, where the distance between a SV and the AZ is calculated as the minimal distance between them. p<0.05 by t-test. D. The mean number of proximal SVs/AZ area in wild type, hα-Syn and hA30P E. Fraction of proximal SVs that have at least one connector in WT, hα-Syn and hA30P overexpressing transgenic synapses. F. Fraction of intermediate SVs that have at least one connector in in wild type, hα-Syn and hA30P synapses. G. The mean number of connectors per proximal SV in the three genotypes. H. The mean number of connectors per intermediate SV in the three genotypes. I. Fraction of proximal SVs that have at least one tether in wild type, hα-Syn and hA30P. J. The mean number of tethers per proximal SV in the three genotypes. K. The number of proximal SVs with two or more greater tethers (structural RRP). p<0.01 by K-W test and p<0.05 by χ2 test, respectively. N = 3 independent experiments. NS, not significant, * p<0.05, ** p<0.01, *** p<0.001.
Cryo-ET data showed that the distribution of SVs was normal in synaptosomes of PD mutant (PARK1/hA30P and PARK4/hα-syn) mice (Fig. 7A), though there were 35% fewer proximal vesicles/AZ area in PARK4/hα-syn and 25% more in PARK1/hA30P synaptosomes (n.s.) (Fig. 7A, D). The mean distance of proximal SVs to the AZ was significantly reduced in the PARK1/hA30P mutant in respect to the wild type (p<0.05). This distance was further reduced when only the connected proximal SVs were considered (p<0.01), while there was no significant difference for any other genotypes (Fig. 7C).
We then examined both tethers (Fig. 7I–K) and connectors (Fig. 7D–H) in wild type and α-synuclein transgenic synaptosomes. The fraction of proximal SVs that were tethered was not significantly changed (Fig. 7I), but the number of tethers per proximal SV (Fig. 7J), as well as the fraction of proximal vesicles with multiple tethers, i.e. structurally defined RRP, were all significantly increased in PARK1/hA30P α-synuclein transgenic compared to wild type samples (Fig. 7K; p<0.01 by K-W test and p<0.05 by χ2 test, respectively), while there were no significant changes in PARK4/hα-syn synapses. The increased tethering (Fig. 7I–K) and decreased mean distance of connected proximal SVs to AZ (Fig. 7D) suggest that PARK1/hA30P synapses have specific structural alterations in the proximal zone, which may be related to neurotransmitter release alterations.
The inverse correlation between the mean number of tethers per proximal SV and the vesicle distance to the AZ, a sensitive indicator of synaptic release (Fernandez-Busnadiego et al., 2013) was present in wild type and PARK1/hA30P but absent in PARK4/hα-syn synapses (Supplementary Table II). Taking into account that tethering was not altered, our data suggest that PARK4 synapses show a mild release deficit. This finding is consistent with published electrophysiological studies that show overexpression of human wild type α-synuclein decreases neurotransmission (Nemani et al., 2010; Janezic et al., 2013; Scott and Roy, 2012).
Regarding SV connectivity, there was only a modest increase in the mean number of connectors per SV in PARK1/hA30P synaptosomes (Fig. 7E–H). Interestingly, the connectivity of the proximal SVs showed a significant dependence on the interaction between the human α-synuclein expression and the level of endogenous synucleins (p<0.05, factorial design ANOVA). These data, along with those for αβγ-Syn−/− synapses (Fig. 5), indicate that the SV interconnectivity is primarily sensitive to the loss of synucleins. To determine the phosphorylation changes associated with PARK1/hA30P and PARK4/hα-syn synapses, we immunoblotted for the three phosphoproteins changed in αβγ-Syn−/− synapses. We find the phosphorylation of amphiphysin-2 in PARK1/hA30P and PARK4/hα-syn synapses is similar to wildtype; while the phosphorylation status of synapsin I site 4–5 is decreased in PARK4/hα-syn only (Supplementary Fig. 3A–B). Dynamin-1 778 phosphorylation is increased in both PD mutants relative to wildtype (Supplementary Fig. 3C)
Collectively, these observations indicate there are select disturbances in SV tethering and connectivity depending on the α-synuclein PD mutation.
Discussion
In this study, we systematically investigated the impact of deleting synucleins as well as expressing PD mutants of α-synuclein on synapse architecture. We find multiple effects of synucleins on presynaptic termini, consistent with the fact that they are abundantly expressed and have conformational plasticity.
Synucleins are important determinants of synapse size
We observed smaller synapses in αβγ-Syn−/− cultured neurons (Fig. 2D), a phenotype first seen in synuclein null brain sections (Greten-Harrison et al., 2010). Interestingly, several groups have reported that overexpression of wild type human α-synuclein leads to increased synapse size, suggesting that synucleins can bidirectionally control synapse size. The linear relationship between synuclein levels and synaptic size also indicates that synucleins are influencing insertion and removal of membrane. This is in line with ascribed functions of synucleins in SV exo- and endocytosis (Burré et al., 2010; Vargas et al., 2014; Busch et al., 2014), although the precise relationship between synapse size and trafficking is unknown.
αβγ-Syn−/− synapses have increased short tethers in the proximal zone (<45 nm)
Our Cryo-ET results showed that wild type and αβγ-Syn−/− synapses have similar number and distribution of SVs, arguing against major disturbances in the synaptic release. However, we observe a robust increase in the SV tethering, as well as the fraction of SVs in structurally defined RRP pool in αβγ-Syn−/− synapses (Fig. 3C–I). Tethers are hypothesized to be SNAREs or constituents of AZ such as Rim 1, based on their lengths and tetanus toxin susceptibility. By our analysis of αβγ-Syn−/− synapses, we can clearly establish that synucleins themselves are not tethers, but that endogenous synucleins negatively regulate short tethers. Possibly because deletion of synucleins stabilizes certain SNARE assemblies on the synaptic membrane, perhaps due to the absence of the α-synuclein-synaptobrevin 2 interaction (Burré et al., 2010) or decreased levels of AP180, the endocytic adaptor for synaptobrevin 2 (Koo et al., 2015) in αβγ-Syn−/− synapses (Vargas et al., 2014). Because the number of docked SVs and their distribution in the proximal zone were unaltered (Figs. 2F, 3B) but the structural RRP was increased in αβγ-Syn−/− synapses (Fig. 3G), it suggests a larger number of docked SVs are primed. This interpretation is consistent with our previous work characterizing the electrophysiology of αβγ-Syn−/− showing increased basal transmission (Greten-Harrison et al., 2010). However, this proposition is not in line with the finding that the amount of SNARE complexes is reduced in αβγ-Syn−/− brain homogenate (Burré et al., 2010); we did not find the amount of SNAREs is reduced in the brain homogenates of young αβγ-Syn−/− (Greten-Harrison et al., 2010; unpublished results). Regardless, the increased tethers in αβγ-Syn−/− synapses could mean that the number of short tethers are not directly proportional to the number of SNARE complexes in the brain. In keeping with this idea, Imig et al, 2014 have suggested that SV docking is mediated by Munc13. With our result that synucleins negatively regulate short tethers, future studies can begin to investigate the exact nature of SNARE machinery that may constitute short tethers.
SV connectivity within 250 nm from AZ and the characteristics of connectors
Deletion of endogenous synucleins caused a decrease in SV connectivity, which was rescued upon human α-synuclein overexpression (Fig. 4C–F), suggesting that both mouse and human synucleins support SV connectivity. On the surface, this loss of connectivity appears at odds with our EM data showing increased clustering (>250 nm) in αβγ-Syn−/− (Fig. 5). However, the connectivity decrease was detected at closer distances to the AZ (<250 nm), suggesting that there are distinct connectors for regions of the presynaptic terminal. Furthermore, our data agree with the observation that α-synuclein clusters SVs in reconstituted systems (Diao et al., 2013) and suggests that the increase in connectivity is structural correlate of the synuclein-dependent increase in clustering and decrease of SV trafficking in vitro (Wang et al., 2014).
The molecular constituents of connectors in the 0–250 nm zone are not yet established. Our previous Cryo-ET studies had revealed that these connectors are dynamic structures that respond to phosphorylation. As we observed phosphorylation changes in synapsin 1, amphiphysin-2 and dynamin 1 in αβγ-Syn−/−, we can categorize these proteins as potential connectors. It was proposed that synapsins mediate SV connectivity, but recent analysis suggest that other proteins are involved (Benfenati et al., 1993; Hirokawa et al., 1989; Gaffield and Betz 2007, Siksou et al, 2007, Siksou et al, 2011). Our finding that synapsin I 4, 5 phosphorylation is decreased in PARK4/hα-syn but connectivity was unchanged in this genotype, also suggests proteins other than synapsin I may form connectors. The levels of phosphorylated amphiphysin-2 are in line with the changes we observe in connectors, raising the possibility that phospho-amphiphysin-2 make up or regulate connectors (Table III). The finding that the phosphatase calcineurin dephosphorylates these proteins (Suppl. Fig. 1) strongly implies it is a key player in the regulation of connectors.
Distal SV organization (>250 nm from the AZ)
Using EM, the most striking phenotype we observe in αβγ-Syn−/− synapses is that distal SVs are clustered (Fig. 2, 5). As SV density is higher and it is possible that the SVs are clustered together because the volume of each synapse is smaller. However, this is not a likely explanation, as we often find clustered SVs even in areas devoid of larger structures that could constrain the SVs (Fig. 5). This complements an earlier EM result showing that overexpression of α-synuclein disperses reserve pool SVs (Nemani et al., 2010), hinting at bidirectional effect of synapse structure by synuclein levels.
Biochemically, we observed changes in phosphorylation of synapsin 1 (sites 4–5), amphiphysin-2 and dynamin-1 in αβγ-Syn−/− synapses (Fig. 6). The changes in synapsin sites 4–5 have been hypothesized to control synaptic vesicle endocytosis (Cesca et al., 2010), similar to other endocytic proteins that are dephosphorylated upon stimulation, the so-called ‘dephosphins’ (Dynamin-1, Epsin-1, Amphiphysin). Collectively, the changes in phosphorylation of peripheral SV proteins in αβγ-Syn−/− (Fig. 6), could serve to compensate for the endocytic regulation that synucleins normally exert (Vargas et al., 2014). This conclusion is strengthened by the previously reported upregulation of Synapsin IIb (Westphal and Chandra, 2013), the isoform that lacks phosphorylation sites 2 and 3, but contains sites 4 and 5. But more intriguingly, these results open the possibility that these proteins also regulate distal SV connectors to control SV clustering and mobility.
PARK1 and PARK4 mutants: How do they influence synaptic structure and function?
Structurally, the PARK mutants were not opposite of the synuclein nulls, even though some phenotypes such as synapse size and distal SV clustering are reversed in these two set of mice. The PARK 1 and 4 mutants have distinct phenotypes that set them apart from wildtype mice.
The PARK1/hA30P mutant shows increased tethering (Fig. 7J–K) that mirrors that in αβγ-Syn−/−synapses (Fig. 3). This is consistent with previous predictions that the A30P mutant is both a loss-of-function mutation as well as a gain-of-function mutation (Chandra et al., 2005). The loss-of-function is likely due to the fact that the PARK1/hA30P mutant does not fold completely in the presence of acidic lipids and targeted inefficiently to synapses. Functionally, the A30P mutant is associated with decreased neurotransmitter release only in some but not all types of dopaminergic synapses (Taylor et al., 2014), and not in hippocampal synapses (Nemani et al., 2010), quite different from the αβγ-Syn−/− synapses. These differences might be due to the increased connectivity of SVs in the intermediate zone of PARK1/hA30P and the decreased connectivity in αβγ-Syn−/− transgenic synapses (Fig. 7H).
In contrast to PARK1/hA30P, the PARK4/hα-syn mutation had no significant effect on tethering. Yet, the absence of the inverse correlation between the mean number of tethers per proximal SV and the vesicle distance to the AZ (Supplementary Table II) indicates that PARK4 synapses show a mild release-related structural deficit. This is consistent with the work from many groups that showed that overexpression of human wild type α-synuclein leads to decreased neurotransmission (Nemani et al., 2010; Janezic et al., 2013; Taylor et al., 2014; Wang et al., 2014; Scott and Roy, 2012). Our data indicates that this effect has a structural basis and that the disruption of lipid binding properties induced by A30P mutation of human α-synuclein causes significant cytoskeletal alterations.
Compared to tethering, we find very few effects from both PD mutations on connectors and proximal intermediate SV interconnectivity. However, decreased reserve pool SV clustering has been previously seen by EM (Nemani et al., 2010; Scott and Roy et al., 2010). This again suggests that the nature of connectors varies within the presynaptic terminal.
Significantly, structural changes in the presynaptic cytomatrix are already observed in synaptic samples derived from 3 month old PARK1/hA30P, the PARK4/hα-syn mice, suggesting that with increasing age, it may lead to more pronounced synaptic dysfunction and eventually synapse loss seen in PD.
Overall, our data point to synucleins as a major orchestrator of presynaptic structure.
Materials and Methods
Animals
αβγ-Syn−/− mice as well as human wild type and A30P α-synuclein transgenics have been previously described (Chandra et al., 2005; Greten-Harrison et al., 2010). The mice are maintained under the aegis of an approved IACUC protocol.
Antibodies
α-Synuclein antibody was purchased from BD Biosciences, amphiphysin-1, amphiphysin-2 (bin1) from Upstate, synapsin antibodies were a kind gift by Dr. Angus Nairn, Yale University. Synapsin I site 4,5, Dynamin-1 Ser-774 from Phosphosolutions and Epsin antibody kindly provided by Dr. Pietro De Camilli, Yale University.
Synaptosome Preparation
Synaptosomes are an established model for neurotransmitter release that can sustain multiple exocytic cycles and are susceptible to pharmacological manipulations (Nicholls and Sihra, 1986; Whittaker, 1993; Nicholls, 2003). Synaptosomes were prepared from 2–3 month old mice as described (Dunkley et al, 1988; Godino et al, 2007). Brains from mice of the denoted genotypes were homogenized in sucrose buffer (2 mM HEPES, pH 7.4, 320 mM Sucrose, 5 mM EDTA, 2 mM DTT). The homogenates were centrifuged 3000g for 2 minutes. The supernatant (S1) was centrifuged twice at 9200g for 15 min to obtain a crude synaptosomal fraction (P3). The P3 fraction was separated on a Percoll gradient (3%, 10%, 23%) by centrifugation (18,700g × 12 minutes). The synaptosomes were taken from the interface between the 10% and 23% layers, washed and pelleted using a modified Tyrode buffer (140 mM NaCl, 5 mM KCl, 5 mM NaHCO3, 1.2 mM NaHPO4, 1 mM MgCl2, 10 mM Glucose, 10 mM HEPES, pH 7.4).
Immuno-EM
Purified synaptosomes (N =46 images from 3 WT and 25 images from 2 KO mice) were fixed using a hypotonic fixative (3% paraformaldehyde, 0.25% glutaraldehyde in 5mM phosphate buffer, pH 7.4). Because of the hypotonic fixation step, we primarily see membrane associated proteins in this procedure. The sample was embedded in gelatin, solidified and cut in little cubes. The cubes were incubated with primary and secondary antibodies in incubation buffer (5% BSA, 0.5 M NaCl, 0.02M Phosphate Buffer). Samples were then dehydrated and embedded in Epon and sectioned. Uranyl acetate staining was performed, dehydrated and then samples were imaged using a Phillip CM10 transmission electron microscope. The resulting electron micrographs were analyzed for the distribution of gold particles. Gold articles within an 9 nm distance of a vesicle, membrane or mitochondria were assigned to these categories, while the remaining particles were assigned to the cytosol.
Conventional EM of Neuronal Cultures
Primary hippocampal neurons grown on coverslips were incubated in Tyrode’s buffer for 5 min. Subsequently, the coverslips were (1) fixed immediately, (2) stimulated for 2 min by adding high K+ Tyrode’s buffer (45 mM KCl) before fixing, or (3) allowed to recover poststimulation in Tyrode buffer for 10 min before fixing. All incubations were done at 37°C. Fixation was perform ed for 1 h with 1.2% glutaraldehyde in 66 mM sodium cacodylate. Postfixation was done for 1h in 1% OsO4 and 1% KFe(CN)6–0.1 M sodium cacodylate. Neurons were stained overnight with 2% uranyl magnesium acetate, then dehydrated in increasing ethanol concentrations, and embedded in Epon. Samples were processed for imaging by standard procedures. Pictures were captured using a Phillip CM10 transmission electron microscope. Synapses (50–150) were analyzed for each condition and repeated using three independent cultures.
Morphometric analysis was done on glutamergic synapses as described in Greten Harrison et al 2010. Analysis was blind to genotype. We defined as distal SV clusters as 20 or more SVs in physical contact with each other as well as arrayed.
Cryo-ET
Vitrification of synaptosomes, tomographic acquisition and analysis were done as described previously (Fernandez-Busnadiego et al 2010). Briefly, a 3 l drop of 10 nm colloidal gold (Sigma-Aldrich) was deposited on plasma-cleaned holey carbon copper EM grids (Quantifoil) and allowed to dry. A 3μl drop of synaptosomal suspension was placed onto the grid, blotted with filter paper (GE Healthcare), and plunged into liquid ethane. Vitrified grids were stored in liquid nitrogen.
Vitrified synaptosomes were imaged on a FEI Tecnai G2 Polara microscope equipped with an FEG operated at 300 keV and a post column energy filter (Gatan) operated in zero loss mode. Images were recorded on Gatan MegaScan CCD camera at the pixel size at the specimen level of 0.47–0.57 nm at 6μm underfocus. Tilt series were acquired at 1.5°–2° angular increment, typically from −60° to +60° using the FEI tomography software. Between 6 and 9 tomograms were processed for each condition. Tomograms were aligned using gold beads as fiducial markers, binned twice (final pixel size 1.9–2.3 nm) and reconstructed using IMOD and TOM packages (Kremer et al 1996; Nickell et al., 2005). Analytical weighted back projection (WBP) algorithm was used for reconstruction.
Tomograms were slightly denoised using anistotropic nonlinear diffusion (Frangakis and Hegerl 2001; Fernandez and Li 2003). SVs and the AZ membrane were manually segmented in Amira software (FEI). Tethers and connectors were automatically segmented (using hierarchical connectivity segmentation) and analyzed using Pyto software (Lucic et al., in press; available on demand) as previously described (Fernandez-Busnadiego et al, 2013). Tether lengths were estimated from the positions of voxels that contact the vesicle membrane or the AZ. Consequently, connector and tether lengths calculated membrane to membrane would be approximately 1 pixel longer.
The number of mice, synapses, SVs, proximal SVs, tethers and connectors analyzed are included in Supplementary Table IV.
Data Analysis
All Data are presented as mean ± sem. Statistical tests used are indicated in figure legends. Means and sems were calculated over all measurements of a specific property. We used K-W test (nonparametric) for values deviating from the normal distribution (e.g., number of tethers/connectors per vesicle). When values fell into discrete bins (e.g., fraction of connected and nonconnected vesicles), the χ2 test was used and the sem was calculated between synapses. We used Pearson’s coefficient for correlation analysis, and its significance was determined using t test. In all cases, confidence levels were calculated using two-tailed tests.
Supplementary Material
Highlights.
-
-
Synucleins are determinants of synapse size.
-
-
Connectors and tethers are altered in αβγ-Synuclein −/−.
-
-
PARK mutations primarily affect tethering of synaptic vesicles.
-
-
Phospho-Amphiphysin-2 may constitute connectors linking synaptic vesicles.
Acknowledgments
We thank Pietro De Camilli for advice and for kindly providing reagents and Wolfgang Baumeister for providing access to cryo-ET instrumentation. We also thank our colleagues Mirko Messa and Christopher Westphal for help with biochemistry, and Andrew Williams, Yumei Wu, and Summer Paradise for help with EM. This work was supported by NIH grants R01NS083846, R01NS064963 and a Bumpus Foundation Grant (to S.S.C.). R.F.-B. is the recipient of a Feodor Lynen postdoctoral fellowship from the Humboldt Foundation. K. J. V. is the recipient of a fellowship from the Pew Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
KJV, NS, TD, RF-B, YVT, and UL performed experiments and analyzed data. SSC and VL analyzed data. SSC, VL and KJV wrote the paper and all authors edited the text.
References
- Altrock WD, tom Dieck S, Sokolov M, et al. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003;37(5):787–800. doi: 10.1016/s0896-6273(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Ambroso MR, Hegde BG, Langen R. Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc Natl Acad Sci USA. 2014;111(19):6982–6987. doi: 10.1073/pnas.1402233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Schoch S, Ho A, et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci USA. 2007;104(7):2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Mercuri NB, Venneri A, et al. Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42(1):77–94. doi: 10.1111/nan.12297. [DOI] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rossi MC, et al. Interactions of synapsin I with phospholipids: possible role in synaptic vesicle clustering and in the maintenance of bilayer structures. J Cell Biol. 1993;123(6 Pt 2):1845–1855. doi: 10.1083/jcb.123.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, et al. Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis. Journal of Neuroscience. 2013;33(6):2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Oliphint PA, Walsh RB, et al. Acute increase of α-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol Biol Cell. 2014;25(24):3926–3941. doi: 10.1091/mbc.E14-02-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo L, Wegrzynowicz M, Santivañez-Perez J, Grazia Spillantini M. Synaptic failure and α-synuclein. Mov Disord. 2016;31(2):169–177. doi: 10.1002/mds.26479. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Harris SM, Huttner WB, Greengard P. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol. 1983;96(5):1355–1373. doi: 10.1083/jcb.96.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91(4):313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123(3):383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen H, Slepnev VI, Di Fiore PP, De Camilli P. The interaction of epsin and Eps15 with the clathrin adaptor AP-2 is inhibited by mitotic phosphorylation and enhanced by stimulation-dependent dephosphorylation in nerve terminals. J Biol Chem. 1999;274(6):3257–3260. doi: 10.1074/jbc.274.6.3257. [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58(1):120–129. [PubMed] [Google Scholar]
- Clayton EL, Sue N, Smillie KJ, et al. Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat Neurosci. 2010;13(7):845–851. doi: 10.1038/nn.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czernik AJ, Pang DT, Greengard P. Amino acid sequences surrounding the cAMP-dependent and calcium/calmodulin-dependent phosphorylation sites in rat and bovine synapsin I. Proc Natl Acad Sci USA. 1987;84(21):7518–7522. doi: 10.1073/pnas.84.21.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Burré J, Vivona S, et al. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife. 2013;2:e00592. doi: 10.7554/eLife.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring M, Loos A, Schrader N, Pfander B, Bauerfeind R. Nerve growth factor-induced phosphorylation of amphiphysin-1 by casein kinase 2 regulates clathrin-amphiphysin interactions. J Neurochem. 2006;98(6):2013–2022. doi: 10.1111/j.1471-4159.2006.04037.x. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Heath JW, Harrison SM, et al. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: homogeneity and morphology of subcellular fractions. Brain Research. 1988;441(1–2):59–71. doi: 10.1016/0006-8993(88)91383-2. [DOI] [PubMed] [Google Scholar]
- Fernández JJ, Li S. An improved algorithm for anisotropic nonlinear diffusion for denoising cryotomograms. J Struct Biol. 2003;144(1–2):152–161. doi: 10.1016/j.jsb.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Fernández-Busnadiego R, Asano S, Oprisoreanu A-M, et al. Cryo-electron tomography reveals a critical role of RIM1α in synaptic vesicle tethering. J Cell Biol. 2013;201(5):725–740. doi: 10.1083/jcb.201206063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Busnadiego R, Schrod N, Kochovski Z, et al. Insights into the molecular organization of the neuron by cryo-electron tomography. J Electron Microsc (Tokyo) 2011;60(Suppl 1):S137–48. doi: 10.1093/jmicro/dfr018. [DOI] [PubMed] [Google Scholar]
- Fernández-Busnadiego R, Zuber B, Maurer UE, et al. Quantitative analysis of the native presynaptic cytomatrix by cryoelectron tomography. J Cell Biol. 2010;188(1):145–156. doi: 10.1083/jcb.200908082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangakis AS, Hegerl R. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. J Struct Biol. 2001;135(3):239–250. doi: 10.1006/jsbi.2001.4406. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27(50):13691–700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11(3):301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- Godino MDC, Torres M, Sánchez-Prieto J. CB1 receptors diminish both Ca(2+) influx and glutamate release through two different mechanisms active in distinct populations of cerebrocortical nerve terminals. J Neurochem. 2007;101(6):1471–1482. doi: 10.1111/j.1471-4159.2006.04422.x. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci USA. 2010;107(45):19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundelfinger ED, Reissner C, Garner CC. Role of Bassoon and Piccolo in Assembly and Molecular Organization of the Active Zone. Front Synaptic Neurosci. 2015;7:19. doi: 10.3389/fnsyn.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108(1):111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Morishita W, Hammer RE, Malenka RC, Südhof TC. A role for Mints in transmitter release: Mint 1 knockout mice exhibit impaired GABAergic synaptic transmission. Proc Natl Acad Sci USA. 2003;100(3):1409–1414. doi: 10.1073/pnas.252774899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell. 2009;138(4):709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Greengard P. Multiple phosphorylation sites in protein I and their differential regulation by cyclic AMP and calcium. Proc Natl Acad Sci USA. 1979;76(10):5402–5406. doi: 10.1073/pnas.76.10.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig C, Min S-W, Krinner S, et al. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron. 2014;84(2):416–431. doi: 10.1016/j.neuron.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci USA. 2013;110(42):E4016–25. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y-H, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78(1):8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, et al. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. Journal of Neuroscience. 2001;21(20):7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, et al. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93(8):3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SJ, Kochlamazashvili G, Rost B, et al. Vesicular Synaptobrevin/VAMP2 Levels Guarded by AP180 Control Efficient Neurotransmission. Neuron. 2015;88(2):330–344. doi: 10.1016/j.neuron.2015.08.034. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Liang S, Wei F-Y, Wu Y-M, et al. Major Cdk5-dependent phosphorylation sites of amphiphysin 1 are implicated in the regulation of the membrane binding and endocytosis. J Neurochem. 2007;102(5):1466–1476. doi: 10.1111/j.1471-4159.2007.04507.x. [DOI] [PubMed] [Google Scholar]
- Lučić V, Fernández-Busnadiego R, Laugks U, Baumeister W. Hierarchical detection and analysis of macromolecular complexes in cryo-electron tomograms using Pyto software. J Struct Biol. 2016;196(3):503–514. doi: 10.1016/j.jsb.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316(5828):1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, et al. Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem. 1996;271(35):21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Kaksonen M. Endocytic accessory factors and regulation of clathrin-mediated endocytosis. Cold Spring Harb Perspect Biol. 2014;6(11):a016733. doi: 10.1101/cshperspect.a016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys J. 2010;99(7):2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo S, Tsukada K, Kameyama H, Furuyama Y, Nakaya K. Distribution of phosphoneuroprotein 14 (PNP 14) in vertebrates: its levels as determined by enzyme immunoassay. Brain Research. 1996;741(1–2):180–184. doi: 10.1016/s0006-8993(96)00914-6. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65(1):66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Sihra TS. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321(6072):772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem Res. 2003;28(10):1433–1441. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- Nickell S, Förster F, Linaroudis A, et al. TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol. 2005;149(3):227–234. doi: 10.1016/j.jsb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375(6531):488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6(1):57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Scott D, Roy S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. Journal of Neuroscience. 2012;32(30):10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihra TS, Wang JK, Gorelick FS, Greengard P. Translocation of synapsin I in response to depolarization of isolated nerve terminals. Proc Natl Acad Sci USA. 1989;86(20):8108–8112. doi: 10.1073/pnas.86.20.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep. 2010;10(3):207–214. doi: 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127(4):831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Tan TC, Valova VA, Malladi CS, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5(8):701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Potgieter D, Anwar S, et al. Region-specific deficits in dopamine, but not norepinephrine, signaling in a novel A30P α-synuclein BAC transgenic mouse. Neurobiology of Disease. 2014;62:193–207. doi: 10.1016/j.nbd.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, et al. Synucleins regulate the kinetics of synaptic vesicle endocytosis. Journal of Neuroscience. 2014;34(28):9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Chandra SS. Synucleins. In: Caplan Michael., editor. Reference Module in Biomedical Sciences. Elsevier; 2015. [DOI] [Google Scholar]
- Wang L, Das U, Scott DA, et al. α-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol. 2014;24(19):2319–2326. doi: 10.1016/j.cub.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J BiolChem. 2013;288(3):1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker VP. Thirty years of synaptosome research. J Neurocytol. 1993;22(9):735–742. doi: 10.1007/BF01181319. [DOI] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344(6187):1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- Wong ASL, Lee RHK, Cheung AY, et al. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat Cell Biol. 2011;13(5):568–579. doi: 10.1038/ncb2217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.