

 This work is licensed under a
This work is licensed under a Abstract
Cushing’s syndrome (CS) due to ectopic adrenocorticotrophic hormone (ACTH) is associated with a variety of tumours most of which arise in the thorax or abdomen. Prostate carcinoma is a rare but important cause of rapidly progressive CS. To report a case of severe CS due to ACTH production from prostate neuroendocrine carcinoma and summarise previous published cases. A 71-year-old male presented with profound hypokalaemia, oedema and new onset hypertension. The patient reported two weeks of weight gain, muscle weakness, labile mood and insomnia. CS due to ectopic ACTH production was confirmed with failure to suppress cortisol levels following low- and high-dose dexamethasone suppression tests in the presence of a markedly elevated ACTH and a normal pituitary MRI. Computed tomography demonstrated an enlarged prostate with features of malignancy, confirmed by MRI. Subsequent prostatic biopsy confirmed neuroendocrine carcinoma of small cell type and conventional adenocarcinoma of the prostate. Adrenal steroidogenesis blockade was commenced using ketoconazole and metyrapone. Complete biochemical control of CS and evidence of disease regression on imaging occurred after four cycles of chemotherapy with carboplatin and etoposide. By the sixth cycle, the patient demonstrated radiological progression followed by recurrence of CS and died nine months after initial presentation. Prostate neuroendocrine carcinoma is a rare cause of CS that can be rapidly fatal, and early aggressive treatment of the CS is important. In CS where the cause of EAS is unable to be identified, a pelvic source should be considered and imaging of the pelvis carefully reviewed.
Keywords: ectopic ACTH production, Cushing’s syndrome, small cell prostate cancer, neuroendocrine tumour
Introduction
Cushing syndrome (CS) secondary to ectopic adrenocorticotrophic hormone (ACTH)–producing tumours (EAS) accounts for approximately 10% of CS depending on the series (1). A variety of tumours have been reported to be associated with EAS. The most common are those of neuroendocrine origin, namely small-cell lung carcinoma (3.3–50%), bronchial carcinoid (4.8–38.9%), thymic carcinoid (4.7–10.6%), medullary thyroid carcinoma (MTC) (1.9–11.6%), gastroenteropancreatic neuroendocrine tumours (NET) (8.9–14%) and phaeochromocytomas (2.5–5.6%) (1, 2, 3).
Sources of ectopic ACTH production arising in the pelvis are rare although EAS has been described due to gonadal tumours (adenocarcinoma, androblastoma, Sertoli cell tumour, carcinoid tumour and teratoma) (4, 5, 6) and genitourinary sources including the prostate (7) as well as cloacogenic carcinoma of the anal canal (8). Prostate cancer causing CS due to ectopic ACTH or CRH production is rare with <30 published cases.
We describe a patient who presented with severe CS due to an ACTH-producing neuroendocrine carcinoma of the prostate and summarise previously reported cases.
Case report
A 71-year-old Caucasian male was admitted to hospital with profound hypokalaemia, generalised oedema and new-onset hypertension. Further history established a two-week history of weight gain (~6 kg), generalised muscle weakness, labile mood and insomnia.
The patient’s medical history included a transurethral resection of the prostate 10 years earlier (histology not available), a recent colonoscopy and polypectomy of a hyperplastic caecal polyp.
Medications included hydrochlorothiazide. The patient had infrequently used a topical 0.1% triamcinolone dental cream over last 2–3 weeks for mouth ulcers but reported no oral, inhaled or parenteral corticosteroid use.
On examination, the patient was hypertensive (BP 154/74 mmHg), euphoric and grossly oedematous with bilateral pitting oedema to the mid-shins and mild facial oedema. The patient had oral candidiasis, thin skin, multiple petechiae on the chest, a small supraclavicular fat pad and proximal muscle weakness. There was no hyperpigmentation, moon facies, dorsocervical fat pad or abdominal striae.
Laboratory results showed a hypokalaemic metabolic alkalosis (Table 1). Hypercortisolaemia was confirmed with morning cortisol >1655 nmol/L, absence of diurnal cortisol rhythm and markedly increased 24-h urinary free cortisol excretion (24-h UFC).
Table 1.
Pertinent laboratory investigations at initial assessment.
| Analyte | Result | Reference interval |
| Serum potassium | 2.3 | 3.5–5 mmol/L |
| Glucose | 11.6 | <7.8 mmol/L |
| HbA1c | 33 | <41 mmol/mol |
| Serum cortisol (1104 h) | >1655 | 200–700 nmol/L |
| Serum bicarbonate | 36.3 | 22–26 mmol/L |
| Venous pH | 7.498 | 7.35–7.45 |
| PSA | 1.35 | <6.5 ng/mL |
| Initial endocrine evaluation | ||
| Midnight cortisol | >1655 | <50 nmol/L |
| 1 mg overnight dexamethasone suppression test | >1655 | <50 nmol/L |
| 8 mg overnight dexamethasone suppression test | >1655 | |
| 24-h Urinary free cortisol | 36,315 | 35–285 nmol/24 h |
| Plasma ACTH | 72 | 2–11 pmol/L |
| Plasma CRH | 0.7 | <5 pmol/L |
| Chromogranin A | 32 | <20 U/L |
The ACTH was elevated (72 pmol/L), confirming ACTH-dependent CS. MRI of the pituitary was normal, and there was failure to suppress cortisol secretion following both 1 mg and 8 mg overnight dexamethasone suppression tests, suggesting ectopic ACTH or CRH production rather than Cushing’s disease (CD).
Computed tomography (CT) of the chest, abdomen and pelvis was initially reported as normal. As no causative lesion was identified on imaging, the patient was transferred to a university hospital for further evaluation.
Careful review of the CT images detected an enlarged prostate with irregular margins and a soft tissue area in the pelvis felt to be possible abnormal lymph node enlargement (Fig. 1A). Additional history suggested mild symptoms of prostatism, and a hard irregular prostate gland was present on digital rectal examination. The patient’s prostate-specific antigen (PSA) was normal (Table 1).
Figure 1.
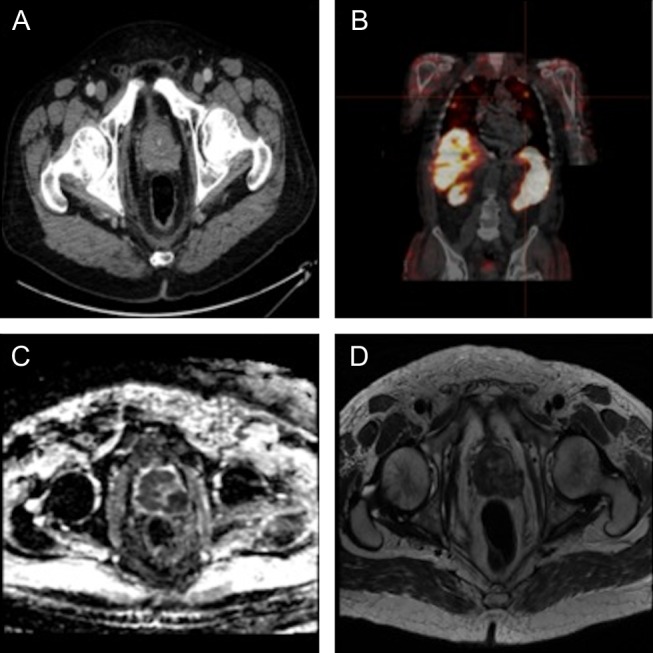
(A) CT imaging showing irregular prostate margins with possible extracapular spread. (B) Octreotide SPECT showing the presence of octreotide-avid pulmonary metastases. (C) Diffusion-weighted MRI imaging with ADC at 550 mm2/s. (D) T2-weighted MRI showing extensive signal abnormality of the prostate with extracapsular spread.
An MRI pelvis revealed an enlarged prostate (52.6 cc volume), extracapsular spread of tumour, extensive T2 signal abnormality, with apparent diffusion coefficient (ADC) values of 550 mm2/s suggestive of high-grade prostatic malignancy (Fig. 1C and D). Abnormal bone marrow signal in pelvis and femur was consistent with bony metastases. A technetium-99m 2,3-dicarboxypropane-1,1-diphosphonate isotope bone scan confirmed the presence of diffuse skeletal metastases and a technetium-99m-HYNIC-[Tyr3]-octreotide scan confirmed the presence of multiple pulmonary octreotide-avid lesions (Fig. 1B).
A CT-guided lymph node biopsy was inconclusive. Transrectal ultrasound-guided prostate biopsy was performed. Histological examination of the prostatic biopsy was consistent with a poorly differentiated prostate carcinoma with features of both high-grade (Gleason 5 + 5) conventional acinar adenocarcinoma (10–20% of the tumour) and a small-cell neuroendocrine carcinoma. Immunohistochemistry markers for neuroendocrine differentiation were positive (CD56, synaptophysin, neuron-specific enolase (NSE)), as was TTF-1. The Ki-67 (MIB-1) proliferation index was estimated as 35–45% (WHO grade 3) and weak positive staining for ACTH was present. Staining for CRH was not available. Plasma CRH levels were within the reference interval (Table 1). Sections of the tumour that did not demonstrate positive staining for neuroendocrine markers were positive for PSA.
While awaiting histological confirmation of the diagnosis of prostate neuroendocrine carcinoma, urgent treatment with inhibitors of adrenal steroidogenesis was initiated (ketoconazole, 1.2 g daily in divided doses and metyrapone 6 g daily in divided doses), along with mineralocorticoid blockade (spironolactone) and potassium supplementation (Fig. 2A). The patient’s hypokalaemia was corrected and the 24-h urinary free cortisol normalised within one week (Fig. 2C).
Figure 2.

(A) Response of 08:00 h cortisol to adrenal blockade and chemotherapy. Cortisol reference interval 200–700 nmol/L (shown in hatched box). (B) Dramatic ACTH response to carboplatin and etoposide chemotherapy. ACTH reference interval 2–11 pmol/L (shown in hatched box). (C) 24-h urinary free cortisol response to adrenal blockade and chemotherapy. UFC = urinary free cortisol. 24-h UFC reference interval 35–285 nmol/24 h (shown in hatched box).
The patient was commenced on chemotherapy targeting small-cell neuroendocrine carcinoma (carboplatin and etoposide) immediately after confirmation of histological diagnosis. Biochemical control of hypercortisolaemia was maintained and both ketoconazole and metyrapone were able to be discontinued. The patient completed four cycles of chemotherapy with radiographic evidence of disease regression and normalisation of ACTH, 08:00 h cortisol and 24-h UFC levels (Fig. 2A, B and C). By the end of six cycles, at five months following initial presentation, the patient developed evidence of further metastatic spread with increasing liver and bone metastases. Second-line treatment with paclitaxel was commenced, but recurrent hypercortisolism was evident by six months with a rise in morning cortisol to 722 mmol/L and ACTH to 19 pmol/L and 24-h urinary free cortisol to 2146 nmol/24 h (RR 35–285 nmol/day) requiring reinstitution of adrenal blockade. Due to continued disease progression, the patient was transferred to palliative care and died nine months after initial presentation. Ethical approval for case publication was provided by the institutional review board.
Discussion
This case illustrated both diagnostic and treatment challenges. The patient presented with classical features of severe hypercortisolism typical of EAS with myopathy, hypertension, alkalosis, hypokalaemia and oedema. It was important to initiate urgent blockade of adrenal steroidogenesis plus blockade of the mineralocorticoid receptor while undertaking further investigations to determine the cause of the CS. Management of severe CS has recently been reviewed and while the optimal order of drug treatment for medical therapy is yet to be established, rapid blockade with ketoconazole and/or metyrapone, as used in this case, is recommended if parenteral therapy (etomidate) is not required (9). Other potential medical therapies to control hypercortisolism include mitotane, although the onset is slow, and mifepristone (not currently available in New Zealand) (10). Regular monitoring, both clinical and biochemical, is required to assess the efficacy of adrenal blockade and watch for the development of hypoadrenalism (10). In a multicentre study of 195 patients including 37 patients with EAS, where 164 patients received metyrapone monotherapy, biochemical monitoring showed improvement in all cases with approximately half of the patients developing normal cortisol levels and most of the adverse events mild and reversible (11). However, there were limited details on those with EAS in this paper making the relevance to this case unclear. Importantly, metyrapone may result in an increase in 11-deoxycortisol, which may cross-react with cortisol in cortisol immunoassays (12). Mineralocorticoid blockade was performed using spironolactone, as eplerenone was not available. Spironolactone may worsen androgen-sensitive prostate cancer as it may act as a selective androgen receptor modulator (13); however, in this case, the benefit of mineralocorticoid receptor blockade was considered to outweigh any potential harm.
From both the literature (14) and our personal experience, somatostatin analogues may be of benefit in EAS, but despite the tumour demonstrating uptake on somatostatin receptor scintigraphy, there was no biochemical response. A partial response to somatostatin analogue has previously been reported in CS secondary to prostate neuroendocrine carcinoma (15). While somatostatin receptor scintigraphy can be helpful to identify neuroendocrine tumours such as bronchial carcinoids causing CS, given the likely aggressive underlying tumour in this setting, FDG-PET/CT would have been more appropriate (16, 17). FDG-PET/CT was due to be performed in this case, but the diagnosis was confirmed prior to the scan being performed so the scan was cancelled as it was considered unlikely to change management.
Pelvic sources of CS are uncommon and the CT scan was initially reported as normal. This case demonstrates the importance of carefully reviewing imaging with an experienced radiologist. In addition, if a digital rectal examination was performed at initial assessment, it is probable that the diagnosis would have been made earlier; however, this would have been unlikely to alter the outcome. The prostate is not typically thought of as a cause for CS, as neuroendocrine tumours of the prostate are rare. Prostate neuroendocrine carcinoma of small cell type (SCPC) comprise approximately 0.5–2% of men with prostate carcinoma (18). Currently, it is thought that SCPC has a common origin with adenocarcinoma of the prostate (18). Approximately 40–50% of men with SCPC have a prior or concurrent history of prostatic adenocarcinoma (18).
Patients with SCPC typically present with lower urinary tract symptoms but may also have neurological symptoms due to brain metastases and/or pain from lytic bone lesions (19). Not only is SCPC associated with CS but also other paraneoplastic syndromes have been reported including hypercalcaemia, Eaton-Lambert and SIADH (18). PSA levels are typically normal, and SCPC is not responsive to androgen deprivation therapy (18). Given the rarity and lack of randomised trial data, treatment is typically based on that for small-cell lung cancer with platinum and etoposide-based chemotherapy (18). The molecular markers however differ with almost half of patients with SCPC demonstrating ERG (ETS transcription factor) rearrangements (similar to that of prostate adenocarcinoma) compared to an absence of this change in small-cell lung cancer. As such, assessment of ERG rearrangements may be helpful in determining a prostatic origin (20) but was not available in this case.
SCPC has an aggressive clinical course with median survival of 10–19 months despite platinum-based chemotherapy (18), which compares to approximately 60 months for patients with metastatic prostate adenocarcinoma (21). In patients who have CS associated with SCPC the prognosis is abysmal and, based on the currently published cases, the median survival is only two months (Table 2). Our patient survived for nine months from presentation, which is surprising given the markedly elevated levels of cortisol at presentation with peak 24-h urinary free cortisol >150-fold the upper limit of the reference range, and an extremely poor functional status when first transferred to our centre. The patient’s survival may be contributed to a prompt response to aggressive adrenal blockade and excellent initial response to chemotherapy. Of cases where the cause of death is reported, most patients die from sepsis, likely secondary to uncontrolled CS (Table 2). As such, control of the hypercortisolism is urgent and should not be delayed by waiting to identify the source of the CS. Patients who received both adrenal blockade and chemotherapy had a longer median survival compared to cases that only received adrenal blockade (including bilateral adrenalectomy) or no specific treatment (nine months vs two months and less than one month, respectively) (Table 2). In the current case, given the severity of the hypercortisolaemia, bilateral adrenalectomy was discussed, but was initially declined by the patient and then deferred due to a rapid response to chemotherapy. Bilateral adrenalectomy has been shown to be a potentially lifesaving procedure in patients critically unwell with CS (10, 22), although in the three cases of SCPC treated with bilateral adrenalectomy, death occurred at four weeks, three months and three weeks postoperatively due to metastatic disease, sepsis and liver failure from extensive metastatic disease, respectively (23, 24, 25).
Table 2.
Summary of previous published cases of small-cell (neuroendocrine) prostate cancer.
| Author | Year | Patient age | Stage | Prior hx adenoCa | MR blockade | CS treatment | Chemotherapy | Survival from CS dx | Cause of death |
|---|---|---|---|---|---|---|---|---|---|
| Webster (7) | 1959 | 68 | Metastatic | 2 months prior | No | No | No | <1 week | Sepsis |
| Wise (23) | 1965 | 58 | Metastatic | 38 months prior | No | Bilateral adrenalectomy | No | Approx. 2 months | Not stated |
| Hall (37) | 1968 | 63 | Metastatic | 36 months prior | No | No | No | 2 months | Coma, hypotension |
| Newmark (26) | 1973 | 57 | Metastatic | 48 months prior | Yes | Mitotane, metyrapone | No | 8 days | Sepsis |
| Lovern (24) | 1975 | 66 | Metastatic | 48 months prior | No | Bilateral adrenalectomy | No | 3 months | Liver failure secondary metastatic disease |
| Wenk (27) | 1977 | 62 | Locally advanced | 8 months prior | No | No | No | 23 days | Sepsis |
| Molland (38) | 1978 | 76 | Locally advanced | No | Not stated | Not stated | No | 4 weeks | Coma? cause |
| Statham (39) | 1981 | 63 | Metastatic | 36 months prior | No | Metyrapone | No | 2 months | Renal failure |
| Vuitch (28) | 1981 | 70 | Metastatic | 23 months prior | Not stated | Not stated | Not stated | 2 months | Aspergillus |
| Carey (35) | 1984 | 66 | Metastatic | No | Not stated | Not stated | Doxorubicin, cyclophosphamide* | 22 days | Bronchopneumonia |
| Ghali (29) | 1984 | 76 | Metastatic | No | No | Mitotane | No | 6 weeks | Bronchopneumonia |
| Slater (30) | 1985 | 69 | Locally advanced | No | No | No | No | 1 week | Sepsis |
| Fjellestad-Paulsen (36) | 1988 | 63 | Metastatic | No | No | Ketoconazole | No | 6 days | Not stated |
| Haukaas (40) | 1999 | 74 | Metastatic | 4 months prior | Yes | Metyrapone | No | 80 days | Sepsis |
| Rickman (41) | 2001 | 69 | Metastatic | 24 months prior | Yes | Ketoconazole | No | 1 month | Not known |
| Hussein (31) | 2002 | 53 | Metastatic | 9 months prior | Yes | Ketoconazole | No | 2.5 months | Not stated |
| Nimalasena (42) | 2008 | 64 | Locally advanced | 26 months prior | No | Ketoconazole, metyrapone | No | 3.5 months | Not stated |
| Nimalasena (42) | 2008 | 52 | Metastatic | 10 months prior | No | Ketoconazole, metyrapone, octreotide | Epirubicine, carboplatin, 5FU | 1 month | Not stated |
| Alwani (25) | 2009 | 62 | Metastatic | No | No | Mifepristone, bilateral adrenalectomy | No | 3 weeks | Sepsis |
| Alshaikh (32) | 2010 | 70 | Locally advanced | No | No | Ketoconazole, metyrapone | Cisplatin, etoposide | 6 months | Sepsis |
| Rueda-Camino (17) | 2016 | 63 | Metastatic | No | No | Ketoconazole | Cisplatin, etoposide | 12 months | Brain metastases |
| Shrosbree (33) | 2016 | 71 | Metastatic | Not stated | No | Metyrapone | Carboplatin, etoposide | Not stated | Not stated |
| Ramalingam (34) | 2016 | 51 | Metastatic | 2 months prior | Yes | Ketoconazole | Cisplatin, etoposide | 12 months | Not stated |
| Balestrieri (15) | 2016 | 75 | Locally advanced | No | Yes | Ketoconazole, metyrapone, octreotide | No | 3 months | Intestinal perforation |
| Current case | 71 | Metastatic | No | Yes | Ketoconazole, metyrapone, octreotide | Carboplatin, etoposide | 9 months | Metastatic disease |
Died shortly after first dose.
AdenoCa, prostate adenocarcinoma; CS, Cushing’s syndrome; dx, diagnosis; hx, history; MR, mineralocorticoid.
Of the reported cases of CS associated with SCPC most are due to ectopic ACTH production (24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34), but two cases of ectopic CRH production have also been reported (35, 36). As expected, based on the SCPC literature, in 14 of 25 cases, conventional prostate adenocarcinoma had been diagnosed a median of 23.5 months prior to the presentation with CS-associated SCPC (Table 2). While the current case also had concurrent prostate adenocarcinoma, the patient reported previous prostate surgery elsewhere and despite extensive efforts, the histology result was not available.
Conclusions
SCPC is a rare but important cause of EAS. If not diagnosed and treated aggressively it is rapidly fatal. This case reminds us of the importance of detailed radiology review looking for evidence of ACTH-secreting lesions, consideration of a pelvic source and the need for aggressive treatment of hypercortisolism.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Acknowledgements
The authors would like to thank Dr Peter Mwamure, Endocrinologist, Dr Nadir Hassan, Pathologist, Pathlab and Mr Michael Holmes, Urologist, Waikato Hospital for their contribution to this case.
References
- 1.Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. Journal of Clinical Endocrinology and Metabolism 2005. 90 4955–4962. ( 10.1210/jc.2004-2527) [DOI] [PubMed] [Google Scholar]
- 2.Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, Hu MI, Waguespack SG, Jimenez C, Ying AK, Cabanillas M, Abbara M, Habra MA. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center Experience. Cancer 2011. 117 4381–4389. ( 10.1002/cncr.26029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liddle GW, Nicholson WE, Island DP, Orth DN, Abe K, Lowder SC. Clinical and laboratory studies of ectopic humoral syndromes. Recent Progress in Hormone Research 1969. 25 283–314. ( 10.1016/b978-0-12-571125-8.50009-0) [DOI] [PubMed] [Google Scholar]
- 4.Sworczak K, Blaut K, Malecha M, Lewczuk A. Ectopic ACTH syndrome associated with ovarian steroid-cell tumor. Journal of Endocrinological Investigation 2002. 25 369–372. ( 10.1007/BF03344020) [DOI] [PubMed] [Google Scholar]
- 5.Kasperlik-Zaluska AA, Sikorowa L, Ploch E, Soszynski P, Jeske W, Migdalska B, Wysocki M. Ectopic ACTH syndrome due to bilateral ovarian androblastoma with double, gynandroblastic differentiation in one ovary. European Journal of Obstetrics, Gynecology, and Reproductive Biology 1993. 52 223–228. ( 10.1016/0028-2243(93)90077-P) [DOI] [PubMed] [Google Scholar]
- 6.Huang B, Wu X, Zhou Q, Hu Y, Zhao H, Zhu H, Zhang Q, Zheng F. Cushing’s syndrome secondary to ectopic ACTH secretion from carcinoid tumor within an ovarian mature teratoma: a case report and review of the literature. Gynecological Endocrinology 2014. 30 192–196. ( 10.3109/09513590.2013.871518) [DOI] [PubMed] [Google Scholar]
- 7.Webster GD, Jr, Touchstone JC, Suzuki M. Adrenocortical hyperplasia occurring with metastatic carcinoma of the prostate: report of a case exhibiting increased urinary aldosterone and glucocorticoid excretion. Journal of Clinical Endocrinology and Metabolism 1959. 19 967–979. ( 10.1210/jcem-19-8-967) [DOI] [PubMed] [Google Scholar]
- 8.Bankole DO, Bertino JR, Coyne EA, Koonce M. Ectopic Cushing’s syndrome in cloacogenic carcinoma of the anal canal: a case demonstrating APUD characteristics. Yale Journal of Biology and Medicine 1980. 53 543–553. [PMC free article] [PubMed] [Google Scholar]
- 9.Alexandraki KI, Grossman AB. Therapeutic strategies for the treatment of severe Cushing’s syndrome. Drugs 2016. 76 447–458. ( 10.1007/s40265-016-0539-6) [DOI] [PubMed] [Google Scholar]
- 10.Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO. & Tabarin A. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 2015. 100 2807–2831. ( 10.1210/jc.2015-1818) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniel E, Aylwin S, Mustafa O, Ball S, Munir A, Boelaert K, Cuthbertson DJ, Daousi C, Rajeev SP, Davis J, et al. Effectiveness of metyrapone in treating Cushing’s syndrome: a retrospective multicenter study in 195 patients. Journal of Clinical Endocrinology and Metabolism 2015. 100 4146–4154. ( 10.1210/jc.2015-2616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owen LJ, Halsall DJ, Keevil BG. Cortisol measurement in patients receiving metyrapone therapy. Annals of Clinical Biochemistry 2010. 47 573–575. ( 10.1258/acb.2010.010167) [DOI] [PubMed] [Google Scholar]
- 13.Sundar S, Dickinson PD. Spironolactone, a possible selective androgen receptor modulator, should be used with caution in patients with metastatic carcinoma of the prostate. BMJ Case Reports 2012. 2012 ( 10.1136/bcr.11.2011.5238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doi M, Imai T, Shichiri M, Tateno T, Fukai N, Ozawa N, Sato R, Teramoto K, Hirata Y. Octreotide-sensitive ectopic ACTH production by islet cell carcinoma with multiple liver metastases. Endocrine Journal 2003. 50 135–143. ( 10.1507/endocrj.50.135) [DOI] [PubMed] [Google Scholar]
- 15.Balestrieri A, Magnani E, Nuzzo F. Unusual Cushing’s syndrome and hypercalcitoninaemia due to a small cell prostate carcinoma. Case Reports in Endocrinology 2016. 2016 6308058 ( 10.1155/2016/6308058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santhanam P, Taieb D, Giovanella L, Treglia G. PET imaging in ectopic Cushing syndrome: a systematic review. Endocrine 2015. 50 297–305. ( 10.1007/s12020-015-0689-4) [DOI] [PubMed] [Google Scholar]
- 17.Rueda-Camino JA, Losada-Vila B, De Ancos-Aracil CL, Rodriguez-Lajusticia L, Tardio JC, Zapatero-Gaviria A. Small cell carcinoma of the prostate presenting with Cushing syndrome. A narrative review of an uncommon condition. Annals of Medicine 2016. 48 293–299. ( 10.3109/07853890.2016.1168936) [DOI] [PubMed] [Google Scholar]
- 18.Nadal R, Schweizer M, Kryvenko ON, Epstein JI, Eisenberger MA. Small cell carcinoma of the prostate. Nature Reviews Urology 2014. 11 213–219. ( 10.1038/nrurol.2014.21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and underappreciated: small cell carcinoma of the prostate. Seminars in Oncology 2007. 34 22–29. ( 10.1053/j.seminoncol.2006.10.026) [DOI] [PubMed] [Google Scholar]
- 20.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, Hicks JL, Meeker AK, Bieberich CJ, De Marzo AM,, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Modern Pathology 2011. 24 820–828. ( 10.1038/modpathol.2011.7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gravis G, Fizazi K, Joly F, Oudard S, Priou F, Esterni B, Latorzeff I, Delva R, Krakowski I, Laguerre B, et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. Lancet Oncology 2013. 14 149–158. ( 10.1016/S1470-2045(12)70560-0) [DOI] [PubMed] [Google Scholar]
- 22.Han JY, Mirsadraei L, Yeh MW, Suh JD, Yong WH, Bergsneider M, Heaney AP. Bilateral adrenalectomy: lifesaving procedure in severe Cushing syndrome. Endocrine Practice 2012. 18 e85–e90. ( 10.4158/EP11315.CR) [DOI] [PubMed] [Google Scholar]
- 23.Wise HM, Jr, Pohl AL, Gazzaniga A, Harrison JH. Hyperadrenocorticism associated with ‘reactivated’ prostatic carcinoma. Surgery 1965. 57 655–664. [PubMed] [Google Scholar]
- 24.Lovern WJ, Fariss BL, Wettlaufer JN, Hane S. Ectopic ACTH production in disseminated prostatic adenocarcinoma. Urology 1975. 5 817–820. ( 10.1016/0090-4295(75)90365-9) [DOI] [PubMed] [Google Scholar]
- 25.Alwani RA, Neggers SJ, van der Klift M, Baggen MG, van Leenders GJ, van Aken MO, van der Lely AJ, de Herder WW, Feelders RA. Cushing’s syndrome due to ectopic ACTH production by (neuroendocrine) prostate carcinoma. Pituitary 2009. 12 280–283. ( 10.1007/s11102-008-0100-z) [DOI] [PubMed] [Google Scholar]
- 26.Newmark SR, Dluhy RG, Bennett AH. Ectopic adrenocorticotropin syndrome with prostatic carcinoma. Urology 1973. 2 666–668. ( 10.1016/0090-4295(73)90333-6) [DOI] [PubMed] [Google Scholar]
- 27.Wenk RE, Bhagavan BS, Levy R, Miller D, Weisburger W. Ectopic ACTH, prostatic oat cell carcinoma, and marked hypernatremia. Cancer 1977. 40 773–778. () [DOI] [PubMed] [Google Scholar]
- 28.Vuitch MF, Mendelsohn G. Relationship of ectopic ACTH production to tumor differentiation: a morphologic and immunohistochemical study of prostatic carcinoma with Cushing’s syndrome. Cancer 1981. 47 296–299. () [DOI] [PubMed] [Google Scholar]
- 29.Ghali VS, Garcia RL. Prostatic adenocarcinoma with carcinoidal features producing adrenocorticotropic syndrome. Immunohistochemical study and review of the literature. Cancer 1984. 54 1043–1048. () [DOI] [PubMed] [Google Scholar]
- 30.Slater D. Carcinoid tumour of the prostate associated with inappropriate ACTH secretion. British Journal of Urology 1985. 57 591–592. ( 10.1111/j.1464-410X.1985.tb05878.x) [DOI] [PubMed] [Google Scholar]
- 31.Hussein WI, Kowalyk S, Hoogwerf BJ. Ectopic adrenocorticotropic hormone syndrome caused by metastatic carcinoma of the prostate: therapeutic response to ketoconazole. Endocrine Practice 2002. 8 381–384. ( 10.4158/EP.8.5.381) [DOI] [PubMed] [Google Scholar]
- 32.Alshaikh OM, Al-Mahfouz AA, Al-Hindi H, Mahfouz AB, Alzahrani AS. Unusual cause of ectopic secretion of adrenocorticotropic hormone: Cushing syndrome attributable to small cell prostate cancer. Endocrine Practice 2010. 16 249–254. ( 10.4158/EP09243.CR) [DOI] [PubMed] [Google Scholar]
- 33.Shrosbree J, Pokorny A, Stone E, Epstein R, McCormack A, Greenfield JR. Ectopic Cushing syndrome due to neuroendocrine prostatic cancer. Internal Medicine Journal 2016. 46 630–632. ( 10.1111/imj.13063) [DOI] [PubMed] [Google Scholar]
- 34.Ramalingam S, Eisenberg A, Foo WC, Freedman J, Armstrong AJ, Moss LG. & Harrison MR. Treatment-related neuroendocrine prostate cancer resulting in Cushing’s syndrome. International Journal of Urology 2016. 23 1038–1041. ( 10.1111/iju.13225) [DOI] [PubMed] [Google Scholar]
- 35.Carey RM, Varma SK, Drake CR, Jr, Thorner MO, Kovacs K, Rivier J, Vale W. Ectopic secretion of corticotropin-releasing factor as a cause of Cushing’s syndrome. A clinical, morphologic, and biochemical study. New England Journal of Medicine 1984. 311 13–20. ( 10.1056/NEJM198407053110103) [DOI] [PubMed] [Google Scholar]
- 36.Fjellestad-Paulsen A, Abrahamsson PA, Bjartell A, Grino M, Grimelius L, Hedeland H, Falkmer S. Carcinoma of the prostate with Cushing’s syndrome. A case report with histochemical and chemical demonstration of immunoreactive corticotropin-releasing hormone in plasma and tumoral tissue. Acta Endocrinologica 1988. 119 506–516. [PubMed] [Google Scholar]
- 37.Hall TC. Symptomatic hypokalemic alkalosis in hyperadrenocorticism secondary to carcinoma of the prostate. Cancer 1968. 21 190–192. () [DOI] [PubMed] [Google Scholar]
- 38.Molland EA. Prostatic adenocarcinoma with ectopic ACTH production. British Journal of Urology 1978. 50 358 ( 10.1111/j.1464-410X.1978.tb03653.x) [DOI] [PubMed] [Google Scholar]
- 39.Stathem BN, Pardoe TH, Mir MA. Response of ectopic prostatic ACTH production to metyrapone. Postgraduate Medical Journal 1981. 57 467–468. ( 10.1136/pgmj.57.669.467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haukaas SA, Halvorsen OJ, Nygaard SJ, Paus E. Cushing’s syndrome in prostate cancer. An aggressive course of prostatic malignancy. Urologia Internationalis 1999. 63 126–129. ( 10.1159/000030431) [DOI] [PubMed] [Google Scholar]
- 41.Rickman T, Garmany R, Doherty T, Benson D, Okusa MD. Hypokalemia, metabolic alkalosis, and hypertension: Cushing’s syndrome in a patient with metastatic prostate adenocarcinoma. American Journal of Kidney Diseases 2001. 37 838–846. ( 10.1016/S0272-6386(01)80134-7) [DOI] [PubMed] [Google Scholar]
- 42.Nimalasena S, Freeman A, Harland S. Paraneoplastic Cushing’s syndrome in prostate cancer: a difficult management problem. BJU International 2008. 101 424–427. ( 10.1111/j.1464-410X.2007.07294.x) [DOI] [PubMed] [Google Scholar]