

Abstract
Antibody blockade of Programmed Death-1 (PD-1) or its ligand, PD-L1, has led to unprecedented therapeutic responses in certain tumor-bearing individuals, but PD-L1 expression’s prognostic value in stratifying cancer patients for such treatment remains unclear. Reports conflict on the significance of correlations between PD-L1 on tumor cells and positive clinical outcomes to PD-1/PD-L1 blockade. We investigated this issue using genomically-related, clonal subsets from the same methylcholanthrene-induced sarcoma: a highly immunogenic subset that is spontaneously eliminated in vivo by adaptive immunity and a less immunogenic subset that forms tumors in immunocompetent mice, but is sensitive to PD-1/PD-L1 blockade therapy. Using CRISPR/Cas9-induced loss-of-function approaches and overexpression gain-of-function techniques, we confirmed that PD-L1 on tumor cells is key to promoting tumor escape. Additionally, the capacity of PD-L1 to suppresses antitumor responses was inversely proportional to tumor cell antigenicity. PD-L1 expression on host cells, particularly tumor-associated macrophages (TAMs), was also important for tumor immune escape. We demonstrated that induction of PD-L1 on tumor cells was interferon gamma (IFNγ)-dependent and transient, but PD-L1 induction on TAMs was of greater magnitude, only partially IFNγ dependent, and was stable over time. Thus, PD-L1 expression on either tumor cells or host immune cells could lead to tumor escape from immune control, indicating that total PD-L1 expression in the immediate tumor microenvironment may represent a more accurate biomarker for predicting response to PD-1/PD-L1 blockade therapy, compared to monitoring PD-L1 expression on tumor cells alone.
Keywords: PD-1, PD-L1, biomarker, neoantigens, cancer immunoediting
Introduction
Monoclonal antibody (mAb) blockade of PD-1 or its major ligand PD-L1 can provoke durable antitumor responses in some cancer patients and tumor-bearing mice (1–5). Whereas expression of PD-1 is largely restricted to lymphocytes, PD-L1 has been observed on a wide variety of cells present in the tumor microenvironment including tumor cells, lymphocytes, myeloid cells, and cells of epithelial and endothelial origin (6–8). Although high constitutive PD-L1 expression has been noted in a few tumors, it is more commonly induced in tumor and normal cells by cytokines, especially interferon-γ
(IFNγ) (9). The complexity of PD-L1 expression has made it difficult to identify the specific PD-L1–expressing cells that contribute to a tumor’s escape from immune control. This issue has important mechanistic and clinical implications because PD-L1 expression may stratify patients for response to anti-PD-1/PD-L1 immunotherapy (3,5,10,11). Past attempts to resolve this dilemma have been inconclusive (12–23). In addition, PD-L1 on immune cells is expressed more frequently than that on tumor cells in patients with non-small cell lung cancer, urothelial carcinoma, and esophageal squamous cell carcinoma (24–26), suggesting distinct extrinsic regulatory pathway(s) are involved with tumor versus immune cell PD-L1 induction. Here, using our well-characterized, methylcholanthrene (MCA)-induced sarcoma system (27–29), we investigated whether (i) PD-L1 expression on tumor cells known to be sensitive to anti-PD-1/anti-PD-L1 checkpoint blockade in vivo was required for tumor immune escape; (ii) the capacity of PD-L1 to inhibit immune elimination of a tumor was linked to the antigenicity of that tumor; (iii) PD-L1 expression on host cells participated in the process; and (iv) the extrinsic PD-L1 induction on tumor versus host immune cells was regulated in a distinct manner.
Materials and Methods
Mice
Male wild-type (WT) and Rag2−/− mice on a 129S6 background were purchased from Taconic Farms. Female WT mice on a C57BL/6J background were purchased from Jackson Laboratory. Mice used in the study were between 8–12 weeks of age and maintained in accordance with procedures approved by the AAALAC accredited Animal Studies Committee of Washington University in St. Louis.
Tumors
MCA-induced sarcoma cells used in this study were previously generated in male 129S6 WT and Rag2−/− mice (27). Resequencing of d42m1-T3 and d42m1-T9 cells confirmed their genomic stability over time. As variant calling algorithms have become significantly more accurate since our initial reporting of these cell lines (30), we reassessed the mutational landscapes of T3 and T9 cells using the original sequence data and data from tumor cell line resequencing and found that the number of expressed missense mutations in T3 and T9 were 827 and 815, respectively. This modification did not lead to an alteration of either the predicted or found dominant antigenic epitopes. Whereas the genomic landscapes of T3 and T9 were clearly similar to one another, they were completely distinct from that of F244, an independent MCA sarcoma derived from a different 129S6 WT mouse, expressing 943 other missense somatic mutations with the single exception that T3/T9 and F244 cells expressed an identical activating Kras G12C mutation.
Tumor cells were maintained in vitro in RPMI media (Hyclone) supplemented with 10% FCS (Hyclone) for less than 3 weeks prior to use in experiments. 1.0 × 106 tumor cells were injected subcutaneously unless otherwise indicated. Tumor growth was monitored at least two times a week using a digital caliper. The mean of long and short diameters was used for tumor growth curves. Mice were euthanized when tumors were > 2 cm or severely ulcerated. No statistical methods were used to predetermine sample size. However, adequate sample size was chosen based on extensive previous work with this animal model. No randomization or blinding was performed. Ex vivo analyses were performed as previously described (29). Murine glioma cell line GL261 with ectopic expression of murine PD-L2 (GL261-PD-L2) was kindly gifted from G. P. Dunn (Washington University School of Medicine). For detection of PD-L1 and MHC class I expression in vitro, tumor cells were treated with 300 U ml−1 murine IFNγ for 48–72 h unless otherwise indicated. For detection of PD-L2 in vitro, tumor cells were treated with 300 U ml−1 murine IFNγ, 10 μg ml−1 murine TNF-α, or in combination for 48 h. Cell lines were authenticated by NGS in our lab and routinely tested for mycoplasma infection.
Antibodies
Antibodies used for comprehensive flow cytometry analysis of cell subsets in tumors are listed in Supplementary Table S1 (31). Additionally, the following monoclonal antibodies (mAbs) were used and purchased from BD Bioscience, anti-CD16/CD32 (2.4G2), PE-conjugated anti-SiglecF (E50-2440), and anti–H2-Kb (AF6-88.5). FITC-conjugated anti-F4/80 (BM8), anti–PD-L2 (TY25), anti–H2-Db (KH-95), APC-conjugated anti-CD11c (N418), and APC/Cy7-conjugated anti–Ly-6G/Ly-6C (Gr-1) (RB6-8C5) were purchased from Biolegend. Dead cells were stained with Po-Pro-1 or Live/Dead Aqua (Invitrogen). Numbers of cell surface PD-L1 molecules were calculated using Quantibrite PE beads according to the manufacture’s instructions (BD Bioscience). For in vivo checkpoint blockade treatment, chimeric mouse IgG1 anti–PD-1 (4H2) (Bristol-Myers Squibb) (32), chimeric mouse IgG1 anti–PD-L1 (14D8) (Bristol-Myers Squibb) (32), rat IgG2a anti–PD-1 (RMP1-14) (Biolegend) (BioXcell), and rat IgG2b anti–PD-L1 (10F.9G2) (Biolegend) (BioXcell) were used. Hamster anti-IFNγ (H22) (Leinco Technologies) was used to neutralize mouse IFNγ. Mouse IgG2a anti-human CD3 (OKT3) (BioXcell), mouse IgG1 anti-human IFNα receptor (GIR-208) (Leinco Technologies), and hamster IgG anti-bacterial glutathione S-transferase (PIP) (Leinco Technologies) were used as controls. Antibodies (200 μg per dose) were injected i.p. unless otherwise specified. For the mAb clones 4H2 and 14D8, injections were on days 3, 6, and 9. For mAb clones RMP1-14 and 10F.9G2, injections were on days 3, 6, 9, 12, 15, and 18. In vivo CD4+/CD8+ cell depletion was performed as previously described using rat IgG2b anti-mouse CD4 (GK1.5) (Leinco Technologies) and rat IgG1 anti-mouse CD8b (53–5.8) (BioXcell) (28).
Cloning murine PD-L1 on a 129S6 background
cDNA was isolated from total RNA extracted from F244 tumor cells treated with 300 U ml−1 IFNγ for 48 h and PD-L1 cDNA amplified by PCR using a forward primer (5′-AGATCTATGAGGATATTTGCTGGCATT-3′) and a reverse primer (5′-CTCGAGTTACGTCTCCTCGAATTGTGTATC-3′). The PD-L1 cDNA was subsequently cloned into the pCR-TOPO-Blunt II vector (Invitrogen). The PD-L1 cDNA cloned from the MCA sarcoma cells showed an identical sequence to that from a spleen in a naïve 129S6 male mouse (data not shown).
Generation of expression transduced tumor cells using the retroviral system
The retroviral vector with GFP (RV-GFP) was a gift of K. Murphy, Washington University. For generation of the retroviral vector without GFP (RV), RV-GFP was digested with SalI and self-ligated. Following digestion of the PD-L1-pCR-TOPO Blunt II vector with BglII and XhoI, PD-L1 cDNA was subcloned into the RV (RV-PD-L1). After 48 h of retroviral production (28), the supernatant was subsequently used for transfection with tumor cells. Tumor clones such as T9-PD-L1ovr and T9-PD-L1phy cells were obtained by limiting dilution.
Mutation-specific RT-PCR and qRT-PCR
The procedures for detection of mutant Spectrin-β2 by RT-PCR followed by restriction enzyme digestion were previously described in detail (28). For detection of mutant Lama4 by qRT-PCR, a forward primer (5′-GGATGCCCAGAGGACTCTCTG-3′) and a reverse primer (5′-GTAATGTTCGGAAATTGAAGCCTA-3′) were used. For detection of mutant Alg8 by qRT-PCR, a forward primer (5′-TCCCGTTTACCTCCTGGAAGC-3′) and a reverse primer (5′-AGCATACAGCCTGGTCCAGGT-3′) were used.
In vitro cytotoxicity assay
The mutant Spectrin-β2-specific T-cell line (C3) was established as previously described (28). Following treatment with 300 U ml−1 IFNγ for 48 h, tumor cells were labeled with eFluor 670 (eBioscience) at 0.5 μM as a target. 10,000 tumor cells and T cells were incubated in a well of a 96 well plate for 12 h at different ratios. Another 10,000 tumor cells labeled with eFluor 670 at 5 μM were used to calculate numbers of tumor cells killed as a reference. Dead cells were stained with Po-Pro-1. Killing efficiency was calculated by the following formula: 100% × {1− [(% tumor cells with 5 μM)control × (% tumor cells with 0.5 μM)target] / [(% tumor cells with 0.5 μM)control × (% tumor cells with 5 μM)target]}.
ELISA
Following treatment with 300 U ml−1 IFNγ for 48 h, tumor cells were irradiated at 10,000 rads. T cells and tumor cells were cocultured in a well of a 96-well plate for 48 h. IFNγ concentration in the supernatant was measured by mouse IFN gamma ELISA Ready-SET-Go! (eBioscience).
Generation of PD-L1 knockout tumor lines using CRISPR-Cas9
To generate T3 lines lacking PD-L1 expression (T3ΔPDL1.1-7) and a F244 line lacking PD-L1 expression (F244ΔPDL1.1), we designed the single guide RNAs (sgRNA) at http://crispr.mit.edu in June 2014. The sgRNA targeting mouse PD-L1 (5′-GTATGGCAGCAACGTCACGA-3′) was subcloned into the pX330 (px330-PD-L1) (Addgene plasmid 42230). Tumor cells were transiently transfected with pX330-PD-L1 and pmaxGFP (Lonza) using FuGENE HD (Promega) according to the manufacture’s instruction. GFP positive cells were subsequently sorted 72 h after transfection. Following generation of clones by limiting dilution, we performed targeting deep sequencing of the PD-L1 genomic locus and confirmed the presence of premature stop codons in all alleles. For detection of Cas9 cDNA, a forward primer (5′-CCGAAGAGGTCGTGAAGAAG-3′) and a reverse primer (5′- TCGCTTTCCAGCTTAGGGTA-3′) were used. PD-L1 wild-type parental tumor cells treated with pX330 and pmaxGFP, and the PD-L1 knockout cells were subsequently transduced with either the RV or the RV-PD-L1 to generate T3WT-Mock, T3ΔPDL1-Mock, and T3ΔPDL1-PDL1. For the other F244 tumor lines lacking PD-L1 expression (F244ΔPDL1.2 and F244ΔPDL1.3), the extracellular domain of murine PD-L1 was genetically deleted by two step CRISPR-dCas9. The sgRNAs targeting 5′-GAAAGAACGCATGATACATA-3′ (g7) and 5′-TTCTACTACAGCAGCCCGGG-3′ (g43) were used for the first step. Subsequently those targeting 5′-CACACTTGCAAATCGGTTGT-3′ (g29) and 5′-AGTCATTGAGTATTCGTGGC-3′ (g2) were used for the second. Gene deletion was confirmed by diagnostic PCR. For generation of MC38 lines lacking PD-L1 expression (MC38ΔPDL1.1 and MC38ΔPDL1.2), MC38 cells were transiently transfected with a vector encoding Cas9-2A-GFP and the guide oligo (5′-GCCAGGGCAAAACCACACAG-3′) derived from exon IV of the mouse PD-L1 gene. Cells were sorted for GFP, sequenced by NGS and single-cell cloned. MC38ΔPDL1.1 had one allele with a 2-bp deletion in exon IV and the other allele with a single bp deletion, both of which introduced in-frame stop codons. Both alleles of the PD-L1 gene in MC38ΔPDL1.2 had the same single bp insertion that also caused premature translation termination.
cDNA-CapSeq and mutation calling
Following data generation and alignment of Illumina paired-end reads to the mouse reference genome sequence, somatic variant analysis was done comparing tumor cDNA-CapSeq data to matched normal exome data. We used a combination of three variant callers – Samtools (33,34), Sniper (35), and VarScan (36,37) as previously described (30). Missense mutations were then translated into a 17-mer amino acid FASTA sequence and analyzed through pVAC-Seq (30) to identify and shortlist potential high-affinity neoantigens. Briefly, to only target variants in the expressed genes, we restricted our subsequent analysis to genes with expression levels (in fragments per kilobase of exon per million reads mapped (FPKM)) values of > 1, and wherein we could identify evidence that the mutant allele was expressed. Also, we filtered out any variants with normal coverage ≤ 5× and normal VAF of ≥ 2%. Additionally, only variants with tumor coverage of ≥ 10× with a VAF of ≥ 25% were considered.
Statistical analysis
Prism 6 (GraphPad Software, Inc) was used for statistical analysis. No samples or animals were excluded from the analysis. Comparison between samples were performed using an unpaired, two-tailed Student’s t-test or one-way ANOVA followed by multiple comparison test. Welch’s corrections were used when variances between groups were unequal. P < 0.05 was considered as statistically significant.
Results
We previously reported that the d42m1 sarcoma line, derived from an MCA treated immunodeficient 129S6 strain Rag2−/− mouse, comprises two genomically related tumor cell subsets that display distinct immunogenicities (28,29) (Fig. 1). The major subset, comprising ~80% of d42m1 cells (exemplified by d42m1-T9 (T9) cells), represents the highly immunogenic, unedited (38) variant because it expresses an R913L somatic point mutation in Spectrin-β2 (mSβ2) that functions as a strong rejection neoantigen responsible, at least in part, for the spontaneous elimination of T9 cells when transplanted into naïve syngeneic wild-type (WT) recipients. The minor subset, comprising ~20% of d42m1 cells (exemplified by d42m1-T3 (T3) cells), represents edited variants of d42m1 that emerge following T cell–dependent immunoselection of parental d42m1 sarcoma cells. T3 cells do not express mutant mSβ2, are capable of forming progressively growing tumors in WT mice, but can be immunologically eliminated when tumor-bearing mice are treated with mAbs that block the PD-1/PD-L1 axis (Fig. 1) or CTLA-4 (29). Such checkpoint blockade–induced immune rejection of T3 tumors is the result of reinvigoration of T cells with specificities against two dominant neoantigens specifically expressed in T3 cells derived from somatic point mutations in Laminin α subunit 4 (mLama4) and Asparagine-linked glycosylation 8 (α-1,3-glucosyltransferase) (mAlg8) (29).
Figure 1. Highly related T9 and T3 sarcoma cells show distinct tumor growth patterns but similar PD-L1 expression kinetics in vivo.
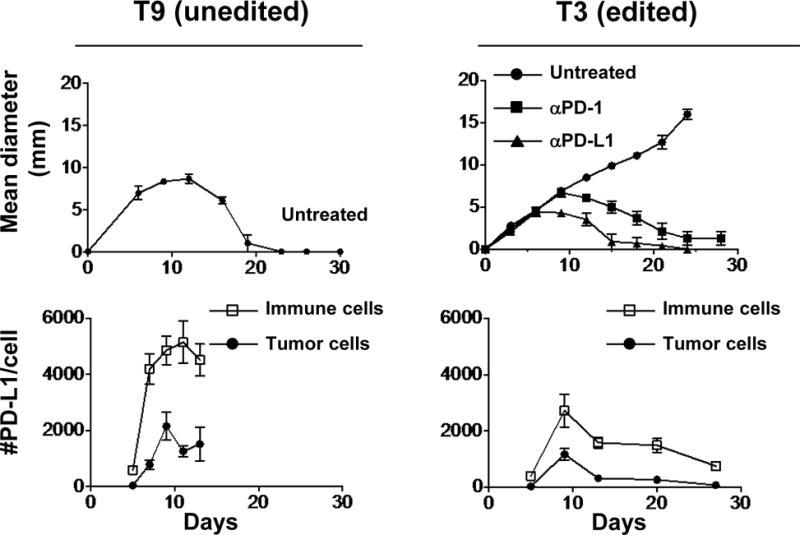
Top: In vivo tumor growth of unedited T9 and edited T3 sarcoma clones in naïve syngeneic 129S6 strain WT mice. Groups of 5 mice bearing T9 sarcoma cells were left untreated. Groups of 5 mice bearing T3 sarcoma cells were left untreated or treated with anti-PD-1 (RMP1-14) or anti-PD-L1 mAb (10F.9G2). Data are shown by mean ± s.e.m from at least two independent experiments. Bottom: Numbers of PD-L1 molecules on tumor (CD45−) and immune cells (CD45+) in vivo. Data are shown by mean ± s.e.m from two independent experiments (n = 4).
We began this study by asking whether PD-L1 expression on T3 sarcoma cells plays an important role in preventing their immune elimination in vivo. Using a PD-L1-guided CRISPR-Cas9 gene editing approach, we generated seven T3-based sarcoma lines lacking PD-L1 (T3ΔPDL1.1- T3ΔPDL1.7). Deep sequencing of the PD-L1 genomic locus of each line showed the presence of premature stop codons in all alleles (Supplementary Fig. S1A). Expression of the Cas9 protein should be transient but since the Cas9 protein is predicted to express strong MHC class I epitopes that could influence tumor cell growth in vivo (39), we tested each T3ΔPDL1 line for the presence of residual Cas9 expression by reverse transcriptase (RT)-PCR and verified no Cas9 mRNA was present (Supplementary Fig. S1B). All of the T3 ΔPDL1 lines retained expression of mLama4 and mAlg8 as detected by quantitative (q) RT-PCR (Supplementary Fig. S1C and D). Functionally, whereas wild-type T3 cells (T3WT) constitutively expressed low amounts of PD-L1 that were significantly upregulated when the cells were exposed to IFNγ, PD-L1 expression on T3ΔPDL1 cells was undetectable either before or after IFNγ treatment (Fig. 2A). Each T3 ΔPDL1 line upregulated MHC class I expression in response to IFNγ to an extent that was indistinguishable from T3WT cells (Fig. 2A).
Figure 2. Ablation of PD-L1 in edited T3 sarcoma cells leads to augmented growth inhibition in WT mice.
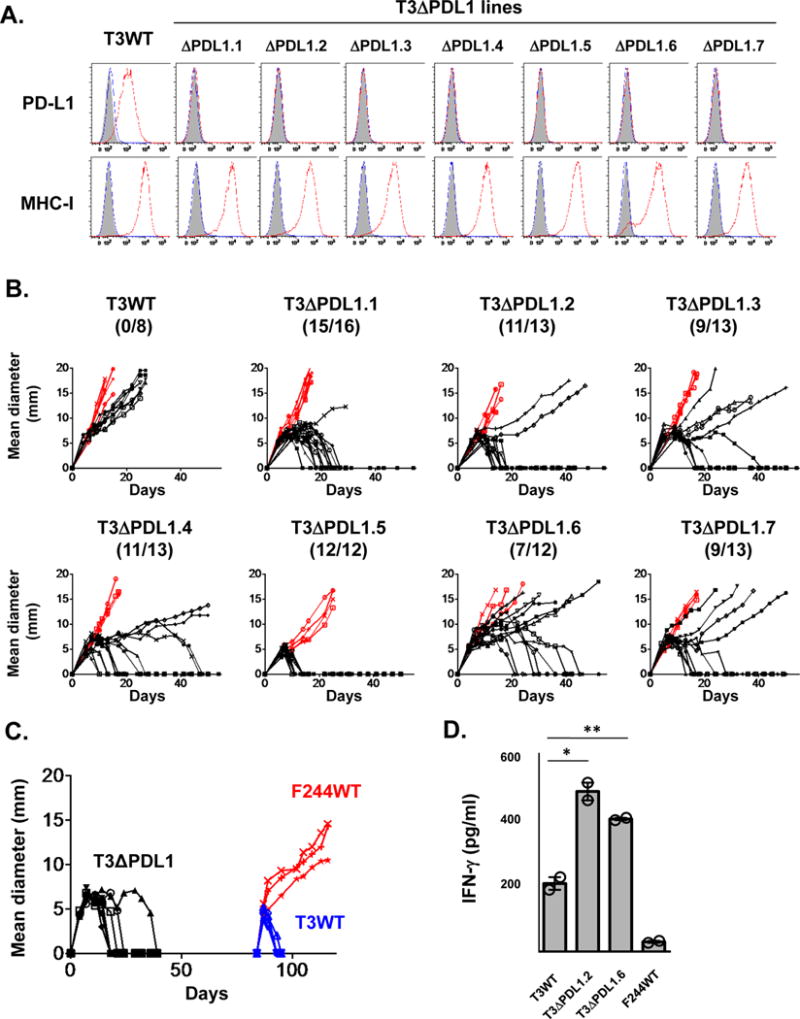
A) In vitro PD-L1 and MHC class I (H2-Kb) expression on cells treated with IFNγ were analyzed by flow cytometry. Black: isotype control, blue: untreated, red: IFNγ treated. Data are shown from at least three independent experiments. B) In vivo tumor growth of T3WT and T3ΔPDL1 lines in syngeneic WT (black) or Rag2−/− mice (red). T3WT are parental T3 sarcoma cells. T3ΔPDL1.1-T3ΔPDL1.7 are T3 lines treated with CRISPR-Cas9 + sgRNA that lack PD-L1 expression (T3ΔPDL1). Each panel represents data from 2–3 independent experiments. Numbers in parentheses show tumor-free WT mice / total WT mice on day 50 post-transplantation. C) Mice rejecting T3ΔPDL1 cells mount a memory response to parental T3 cells. Seven naïve syngeneic WT mice were challenged with T3ΔPDL1.1 cells on day 0. After in vivo rejection, mice were rested for 45 days and then challenged with T3 (n = 4) or F244 (n = 3) sarcoma cells. Data are shown from at least two independent experiments. D) In vitro IFNγ secretion from mutant Lama4-specific T cells (CTL74.17) against T3WT, T3ΔPDL1.2, T3ΔPDL1.6 cells. Data are shown by mean ± s.e.m of technical duplicates from two independent experiments. Samples were compared using an unpaired, two-tailed Student’s t test. **P < 0.01, *P < 0.05.
We next monitored the in vivo growth behavior of T3ΔPDL1 cells. By day 50 post-injection, 80% (74/92) of naïve syngeneic WT mice had spontaneously rejected the different T3ΔPDL1 cells, whereas growing tumors were observed in all of the Rag2−/− mice (Fig. 2B). T3ΔPDL1 tumors escaping rejection neither expressed PD-L1 in response to IFNγ stimulation in vitro nor lost expression of immunogenic neoantigens (Supplementary Fig. S2A and B). These data suggest that additional sources of PD-L1 may participate in the process of tumor immune escape. WT mice that rejected T3ΔPDL1 cells resisted rechallenge with T3WT, but not challenge with unrelated F244 MCA sarcoma cells, demonstrating the induction of tumor specific T-cell memory in T3ΔPDL1 experienced mice (Fig. 2C). When the capacities of parental T3 and two representative T3ΔPDL1 lines to stimulate IFNγ production from CTL74.17 mLama4-specific cytotoxic T-lymphocyte (CTL) clones were compared, T3ΔPDL1 cells stimulated significantly more IFNγ than T3WT cells, revealing that PD-L1 expression on T3 sarcoma cells functionally suppresses activation of tumor-specific CTL (Fig. 2D). To validate the causality of tumor cell-expressed PD-L1 in immune escape of T3 sarcoma cells, we enforced PD-L1 expression in T3ΔPDL1.1 cells by retroviral transduction (T3ΔPDL1-PDL1) (Fig. 3A) and showed that T3ΔPDL1-PDL1 cells regained the capacity to form progressively growing tumors in WT mice (Fig. 3B). In contrast, T3ΔPDL1.1 cells transduced with empty virus (T3ΔPDL1-Mock) were rejected. When mice bearing T3ΔPDL1-PDL1 tumors were treated with anti-PD-1, the tumors were rejected (Fig. 3B). Because PD-1 can also engage a second inhibitory ligand, PD-L2 (40–42), we also tested T3 cells for PD-L2 expression. We did not detect PD-L2, either constitutively or following exposure to IFNγ or TNF-α, either alone or in combination (Supplementary Fig. S3A and B). Similar results were obtained when PD-L1 expression was extinguished in a second, independent MCA sarcoma line (F244) and in the MC38 colorectal carcinoma cell line (Figs. 4A–D). Thus, the importance of PD-L1 on tumor cells in promoting tumor immune escape is generalizable not only to different edited MCA sarcoma lines but also to at least one other tumor type as well.
Figure 3. PD-L1 expressed on edited T3 sarcoma cells prevents their immune elimination.
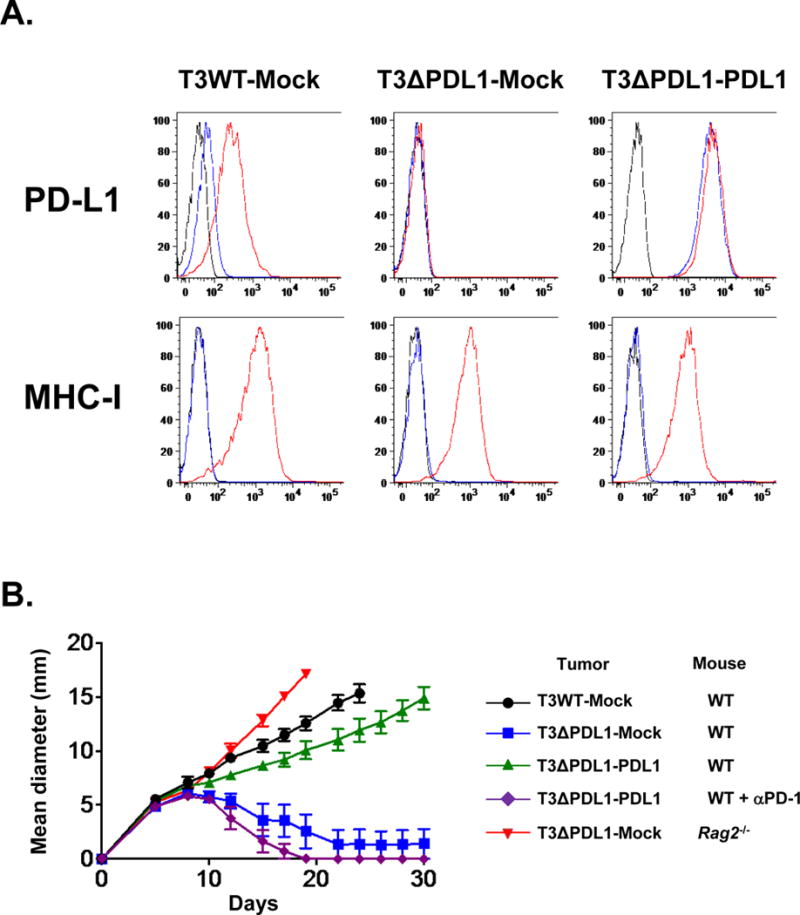
A) T3ΔPDL1-PDL1 cells express high levels of PD-L1 constitutively. In vitro PD-L1 and MHC class I (H2-Db) expression on cells treated with IFNγ. Black: isotype control, blue: untreated, red: IFNγ-treated. Data are shown from at least two independent experiments. B) In vivo tumor growth of mock-transduced T3WT cells (T3WT-Mock), mock-transduced T3ΔPDL1.1 cells (T3ΔPDL1-Mock), and T3ΔPDL1.1 cells with enforced PD-L1 expression (T3ΔPDL1-PDL1) in WT or Rag2−/− mice. Tumor cells (0.5 × 106) were injected subcutaneously on day 0 in the mice. Mice bearing T3ΔPDL1-PDL1 cells were left untreated or treated with anti-PD-1 (4H2). Data are shown by mean ± s.e.m from at least two independent experiments (n = 5).
Figure 4. Ablation of PD-L1 in the edited F244 MCA sarcoma and MC38 colorectal carcinoma leads to augmented growth inhibition in WT mice.
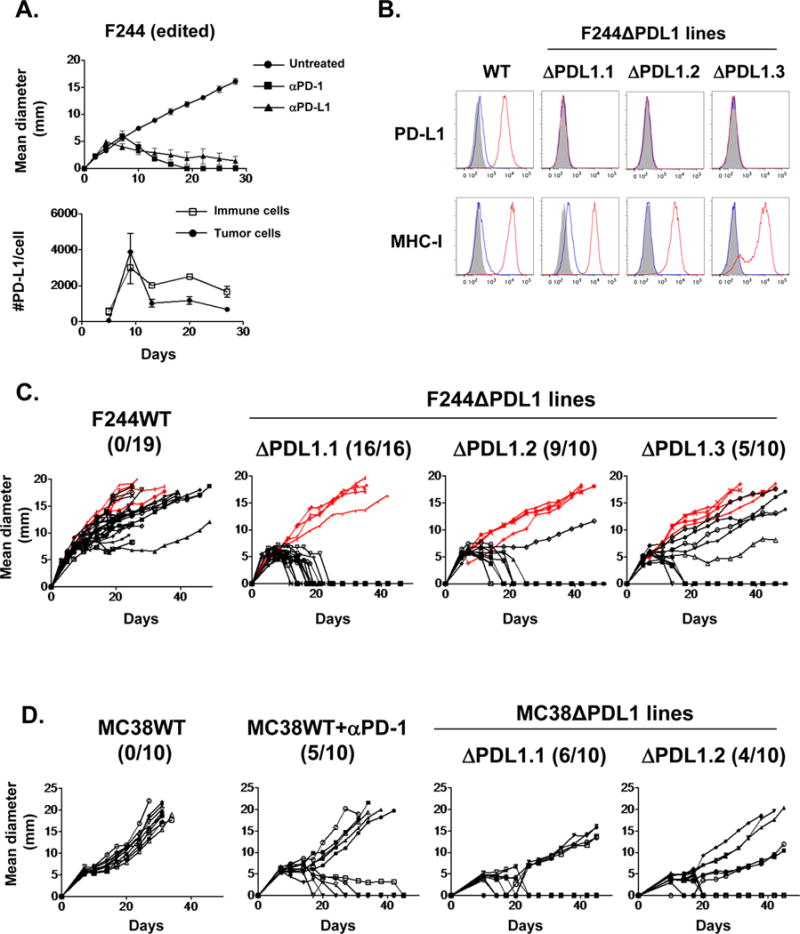
A) Top: In vivo tumor growth of edited F244 MCA sarcoma cells in WT mice. Tumor-bearing mice were left untreated or treated with anti-PD-1 (RMP1-14) or anti-PD-L1 mAb (10F.9G2). Data are shown by mean ± s.e.m from at least two independent experiments (n=5). Bottom: Numbers of PD-L1 molecules on tumor (CD45−) and immune cells (CD45+) in vivo. Data are shown by mean ± s.e.m from two independent experiments (n = 4). B) In vitro PD-L1 and MHC class I (H2-Kb) expression on F244 tumor lines lacking PD-L1 expression (F244ΔPDL1.1-F244ΔPDL1.3) treated with IFNγ were analyzed by flow cytometry. Black: isotype control, blue: untreated, red: IFNγ treated. Data are shown from at least two independent experiments. C) In vivo tumor growth of F244WT and F244ΔPDL1 lines in syngeneic 129S6 WT (black) or Rag2−/− mice (red). Each panel represents data from 2–3 independent experiments. Numbers in parentheses show tumor-free WT mice / total WT mice on day 50 post-transplantation. D) In vivo tumor growth of MC38WT and MC38ΔPDL1 lines in C57BL/6J WT mice. Tumor cells (0.5 × 106) were injected subcutaneously on day 0 in the mice. Mice injected with MC38WT cells were treated on day 7 post-implantation with control or anti-PD-1 mAb (4H2). Numbers in parentheses show tumor-free WT mice / total WT mice on day 45 post-transplantation. MC38WT, but not MC38ΔPDL1, cells upregulated PD-L1 expression in vitro after IFNγ stimulation as evidenced by flow cytometry. Data shown in this figure is representative of at least two independent experiments (n = 10).
Having validated a causal role in escape for tumor cell-expressed PD-L1 on edited sarcoma cells, we then investigated whether PD-L1 played a similar role on highly immunogenic, unedited variants from the same tumor (Fig. 1 and Supplementary Fig. S4A). We enforced PD-L1 expression in T9 tumor cells and monitored in vivo growth of the bulk-transduced T9 cell line in naïve WT recipients. Most PD-L1-transduced T9 cells (T9-PDL1 cells) in the bulk population expressed much more PD-L1 than either untreated or IFNγ treated parental T9 cells (T9WT) (Supplementary Fig. S4B). T9-PDL1 cells, but not mock transduced T9 cells, formed progressively growing tumors in WT mice (Supplementary Fig. S4C). All progressively growing T9-PDL1 tumors retained expression of mSβ2 (Supplementary Fig. S4D and E) thereby ruling out the possibility that tumor cell outgrowth was due to loss of the major rejection antigen.
To examine the quantitative requirements of PD-L1 expression on the immunogenicity of unedited sarcomas, we isolated T9-PDL1 clones that ectopically expressed PD-L1 at levels either similar to (T9-PDL1phy) or 20-fold above (T9-PDL1ovr) those induced on T9WT cells by IFNγ (Figs. 5A and B). When injected into WT mice, T9-PDL1phy cells were spontaneously rejected. In contrast T9-PDL1ovr cells formed progressively growing tumors (Fig. 5C). However, T9-PDL1ovr cells were eliminated when tumor-bearing mice were treated with mAbs to PD-1 or PD-L1 (Fig. 5D). Similar results were obtained using either a blocking, but nondepleting, anti-PD-L1 (mAb 14D8) or a blocking and depleting anti-PD-L1 (mAb 10F.9G2) (32).
Figure 5. Physiologic levels of PD-L1 are not sufficient to prevent immune elimination of highly immunogenic unedited T9 sarcoma cells.
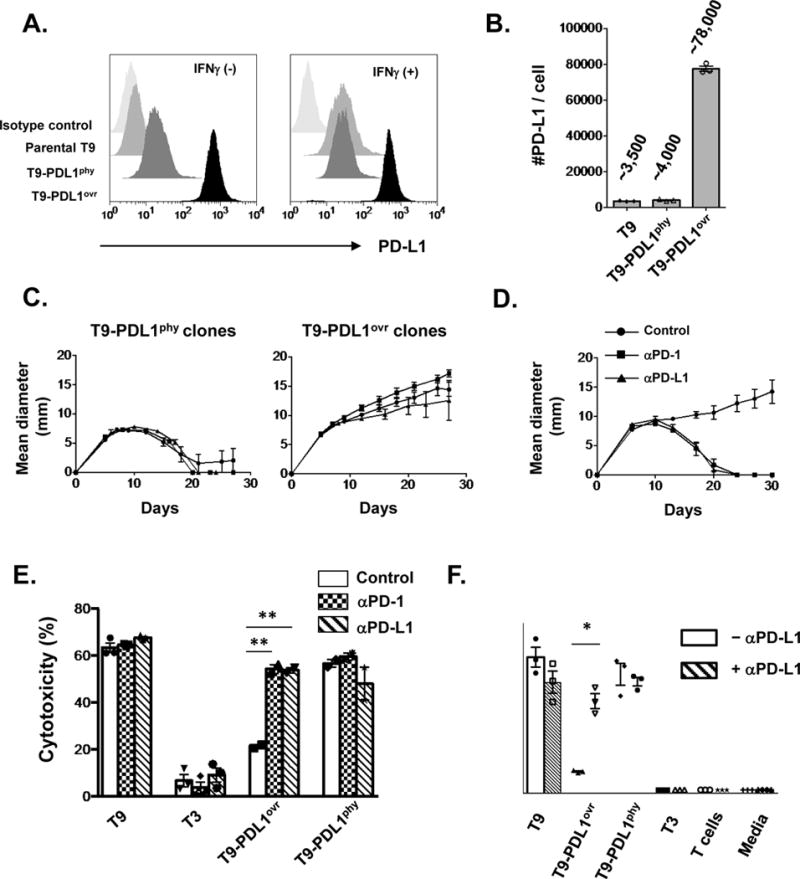
A) In vitro PD-L1 expression with or without IFNγ on T9 sarcoma cells constitutively expressing ectopic PD-L1 either at physiologic levels comparable to that induced by IFNγ on parental T9 cells (T9-PDL1phy) or those at high level over-expression (T9-PDL1ovr). Data are shown from at least three independent experiments. B) Numbers of PD-L1 molecules expressed on cells after IFNγ treatment in vitro. Data are shown by mean ± s.e.m of technical triplicates from at least three independent experiments. C) In vivo growth of three clones of T9-PDL1phy or T9-PDL1ovr cells in WT mice. Data are shown by mean ± s.e.m from at least two independent experiments (n = 5). D) Anti-PD-1 (4H2) or anti-PD-L1 (14D8) leads to tumor rejection in T9-PDL1ovr-bearing mice. Data are shown by mean ± s.e.m from two independent experiments (n = 5). E) In vitro cytotoxicity assay of mutant Spectrin-b2-specific CTL (C3) against tumor cells with anti-PD-1(4H2)/anti-PD-L1(14D8) blockade. Data are shown by mean ± s.e.m of technical triplicates (T9, T3) and duplicates (T9-PDL1ovr, T9-PDL1phy) from at least two independent experiments. F) In vitro IFNγ secretion from mutant Spectrin-b2-specific CTL (C3) against tumor cells with or without anti-PD-L1 (14D8). Data are shown by mean ± s.e.m of technical triplicates from at least two independent experiments. Samples in E, F were compared using an unpaired, two-tailed Student t test. **P < 0.01, *P < 0.05.
To further assess the functional consequences of differences in PD-L1 expression on T9-PDL1 cells we compared their relative sensitivities to in vitro killing by the C3 mSβ2-specific CTL clone. T9-PDL1ovr cells were poorly killed by C3 CTL compared to T9-PDL1phy cells or parental T9 sarcoma cells (Supplementary Fig. S5A and B). However, when anti-PD-1 or anti-PD-L1 was added into the in vitro culture, killing efficiency against T9-PDL1ovr cells was restored to levels similar to those against T9-PDL1phy or parental T9 sarcoma cells (Fig. 5E). Similar results were obtained when IFNγ secretion from C3 CTL was used as the read-out (Fig. 5F).
To confirm the generality of these findings, we enforced PD-L1 expression in a second, unedited highly immunogenic MCA sarcoma line (H31m1) (28,43,44) (Fig. 6A). As was the case for T9-PDL1 cells, levels of ectopically expressed PD-L1 on H31m1-PDL1 were considerably higher than those on the parental H31m1 cells treated with IFNγ (Fig. 6B). Progressively growing H31m1-PDL1 tumors were observed in 17/20 challenged WT mice compared to 0/20 WT mice injected with control H31m1 tumor cells (Fig. 6C). Together, these results show that highly immunogenic unedited sarcoma cells require abnormally high expression of PD-L1 to escape immune control and form progressively growing tumors in immunocompetent mice. Thus, levels of PD-L1 expression that can be induced on tumor cells under physiologic conditions are not sufficient to prevent immune elimination of highly immunogenic unedited MCA sarcoma cells that express strong neoantigens.
Figure 6. H31m1-PDL1 cells form progressively growing tumors in WT mice.
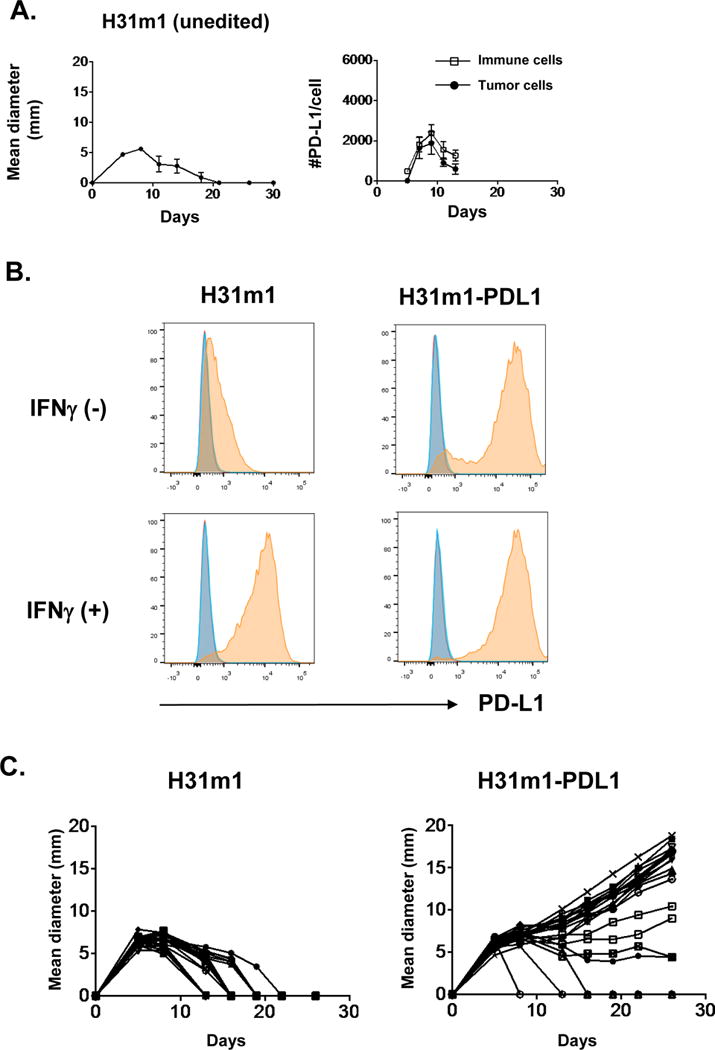
A) Left: In vivo tumor growth of unedited H31m1 MCA sarcoma cells in WT mice. Mice bearing H31m1 cells were left untreated. Data are shown by mean ± s.e.m from at least two independent experiments (n = 5). Right: Numbers of PD-L1 molecules on tumor (CD45−) and immune cells (CD45+) in vivo. Data are shown by mean ± s.e.m from two independent experiments (n = 4). B) In vitro PD-L1 expression on cells treated with or without IFNγ (100 ng ml−1) for 24 h. Data are shown from at least two independent experiments. Red: unstained, blue: isotype control, orange: anti-PD-L1. C) In vivo tumor growth of H31m1 parental and H31m1-PDL1 tumor cells in WT mice. Tumor cells (10 × 106) were injected on day 0. Data shown in this figure is representative of at least two independent experiments (n = 20).
Although our experiments supported a critical role for PD-L1 on tumor cells in mediating tumor escape, they did not rule out the possibility that PD-L1 on host cells also contributed to the process. Unexpectedly, when WT mice were challenged with increasing numbers of T3ΔPDL1 tumor cells, the number of mice with progressively growing tumors increased (Figs. 7A and B). Because the only source of PD-L1 in these experiments was from host cells, we treated mice with progressively growing T3ΔPDL1 tumors with anti-PD-L1 or control mAb and found that therapeutic administration of the former but not the latter induced tumor rejection (Figs. 7A and B). Thus, PD-L1 expression on host cells also participates in preventing immune elimination of edited MCA sarcoma cells.
Figure 7. Host PD-L1 participates in inhibiting immune elimination of T3 sarcoma cells through distinct regulatory machineries.
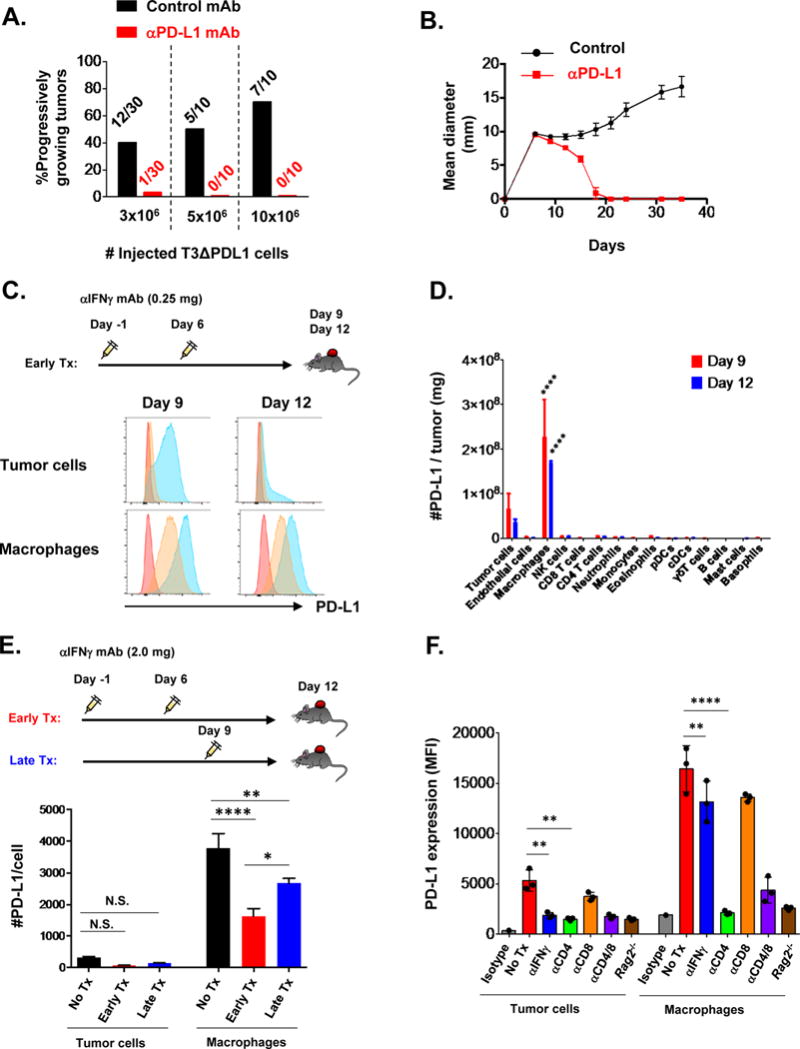
A) Percentage of progressively growing T3ΔPDL1.1 tumors in WT mice treated either with control mAb or anti-PD-L1. Numbers represent mice with progressively growing tumors / all mice injected with the indicated number of tumor cells on day 0. Data are shown from at least two independent experiments. B) Treatment of WT mice with anti-PD-L1 (10F.9G2) leads to rejection of 10 × 106 T3ΔPDL1.1 cells. Data are shown by mean ± s.e.m from two independent experiments (n=5). C) PD-L1 expression on tumor cells and TAMs in T3 tumors on days 9 and 12. Mice were treated with anti-IFNγ neutralizing mAb (0.25 mg/mouse) on days -1 and 6, and injected with T3 sarcoma cells on day 0. Red: isotype, blue: untreated mice, orange: mice treated with anti-IFNγ. Data are shown from three independent experiments. D) Absolute numbers of PD-L1 molecules expressed on cell types in pooled three T3 tumors on days 9 and 12. Data are shown by mean ± s.e.m from three independent experiments (n = 3). E) A large proportion of PD-L1 molecules on TAMs are IFNγ independent. Mice were treated with anti-IFNγ neutralizing mAb (2.0 mg/mouse) either on days -1 and 6, or on day 9, and injected with T3 sarcoma cells on day 0. PD-L1 expression on tumor cells and TAMs was analyzed on day 12. Data are shown by mean ± s.e.m from three independent experiments (n = 6). F) CD4+ T cells contribute to PD-L1 expression on TAMs in the absence of IFNγ. F244 sarcoma cells were injected into either WT or Rag2−/− mice. WT Mice were left untreated or treated with anti-IFNγ neutralizing mAb, anti-CD4 mAb, anti-CD8 mAb, or the combination. PD-L1 expression on tumor cells and macrophages in the tumors was analyzed on day 8. Data are shown by mean ± s.e.m from at least two independent experiments (n = 3). Data in D, E, F were compared using one-way ANOVA followed by multiple comparison test. N.S., not significant; ****P < 0.0001, **P < 0.01, *P < 0.05.
To obtain mechanistic insight into the roles of host versus tumor PD-L1 in preventing immune elimination of sarcomas, we measured the kinetics and magnitude of PD-L1 induction and retention on tumor cells and various populations of host cells in the tumor microenvironment. PD-L1 was upregulated on T3 tumor cells in vivo in a transient and time-dependent manner with peak expression occurring at ~9-days post-injection, but returning to baseline by ~12 days (Figs. 1 and 7C, and Supplementary Fig. S6A). Upregulation of PD-L1 on tumor cells was tightly linked to IFNγ exposure in vivo, because it was completely inhibited when mice were treated with IFNγ neutralizing mAb (Fig. 7C). The IFNγ dependency of PD-L1 induction on tumor cells was also observed in vitro (Supplementary Fig. S6B). Similarly, PD-L1 expression on host immune cells in the tumor microenvironment reached maximal values at 9 days and declined thereafter although at a slower rate than seen on tumor cells (Fig. 1 and Supplementary Fig. S6A).
This slow decay of PD-L1 expression on host immune cells was particularly noteworthy. We therefore defined three distinctive features about host cell-expressed PD-L1 expression. First, a detailed analysis of the cellular composition of the tumor microenvironment revealed that tumor-associated macrophages (TAMs) were not only the major host cell population in the tumor microenvironment, but they also expressed the vast majority of PD-L1 in the tumor (72% of total PD-L1 on tumor and host cells was expressed on TAMs at day 9) (Fig. 7D, Supplementary Figs. S7A–B and S8A–D). Second, PD-L1 expression on TAMs was retained for long periods of time, well after PD-L1 on tumor cells was completely extinguished (Fig. 7C). Third, PD-L1 expression on TAMs was increased in tumor-bearing mice treated with either saturating doses of neutralizing anti-IFNγ (Fig. 7C) or even with amounts that were 8-fold higher (Fig. 7E), although the magnitude of increased PD-L1 expression was less than when IFNγ was present. This observation was not due to changes in the tumor microenvironment by the early mAb injection protocol, because PD-L1 expression on TAMs at day 12 remained high even when IFNγ mAb was injected at day 9 (Fig. 7E). Similar results were obtained when the experiments were repeated with the unrelated F244 sarcoma cell line (Figs. 4A and 7F). Finally, when CD4+ or CD8+ cells were depleted from WT mice, we found that the IFNγ-independent induction/retention of PD-L1 on TAMs required the presence of CD4+, but not CD8+, T cells (Fig. 7F). Thus, PD-L1 on TAMs is induced by two alternative cell-extrinsic pathways involving CD4+ T cells, one that is IFNγ-dependent and one that is IFNγ-independent. Taken together, these results show that TAMs are the major host cell type that contributes PD-L1 in our sarcoma tumor model both quantitatively and temporally.
Discussion
This study provides novel functional and fundamental insights into the roles of PD-L1 in facilitating tumor escape from immune control. On edited tumor cells, whose antigenicity has been tempered by the tumor sculpting power of immunity, IFNγ-dependent induction of PD-L1 expression initiates the establishment of the immunosuppressive force that facilitates tumor outgrowth. However, PD-L1 expression on tumor cells is transient and expression is rapidly extinguished. This downregulation of PD-L1 most likely occurs, at least in part, as a consequence of PD-L1’s ability to inhibit IFNγ production by tumor-infiltrating lymphocytes. TAMs are the major cellular source that maintain expression of PD-L1, long after PD-L1 on tumor cells is extinguished. The temporal dichotomy between PD-L1 expression on tumor cells versus TAMs results in the establishment of the immunosuppressive tumor microenvironment in which the majority of PD-L1 is contributed by TAMs as opposed to tumor cells. The prolonged PD-L1 expression on TAMs is cell extrinsic, IFNγ-independent, but requires CD4+ T cells. The eventual discovery of the molecule(s) responsible for the chronic PD-L1 expression by TAM may well provide new opportunities for cancer immunotherapy.
Our findings also are consistent with two mechanisms by which TAMs could exert their immunosuppressive activity in progressively growing tumors. It is likely that PD-L1–expressing TAMs in tumors are recognized by tumor-specific T cells and deliver their PD-L1–dependent inhibitory signal to these T cells just as if they were tumor cells themselves. This scenario suggests that PD-L1–expressing TAMs function in cis to prolong the immunosuppressive state in the microenvironment of a progressively growing tumor. This mechanism is consistent with that observed in vitro with antigen-presenting cells (APCs) that also bear high amounts of PD-L1 (7,45). The second scenario is one in which TAMs, or even other host cell types, repress T-cell function by supplying the high levels of PD-L1 in trans. The latter mechanism is consistent with data from in vitro studies in which T-cell activation was assessed following coculture of PD-L1–expressing monocytes with T cells stimulated with anti-CD3/anti-CD28 (46,47). It is certainly possible that different types of tumors could display distinct distributions of PD-L1 between cancer cells and host cells, or that differences in the nature, quantity, or kinetics of tumor-infiltrating hematopoietic cells determine the ultimate distribution of tumor- versus host cell-expressed PD-L1. Future studies will need to explore these two scenarios in more depth to determine the conditions where one or the other predominates. Therefore, the results of this study argue strongly that PD-L1 expression on either tumor cells or host cells should be used as the biomarker for determining whether a patient is a good candidate for PD-1/PD-L1 blockade therapy, presumably when tumors retain high mutation burden (18,48).
Finally, this study also provides the fundamental insight that PD-L1 expression on edited tumor cells (such as T3), whose antigenicities have undergone immunologic sculpting, versus highly antigenic unedited tumor cells, such as those that have not undergone cancer immunoediting (38) (e.g., T9), results in very different outcomes. This report thus demonstrates the inverse relationship between tumor antigenicity and the capacity of PD-L1 to promote tumor escape. This observation leads to the logical conclusion that adaptive immune resistance (49), the process wherein immune attack on a tumor results in the upregulation of immunosuppressive moieties that inhibit immune control of the tumor, is relevant predominantly to tumors of either inherently low antigenicity or to tumors that have gone through the cancer immunoediting process, resulting in generation of antigen-loss tumor variants with reduced antigenicities (38,50). The important implication, then, is that cancer immunoediting and adaptive immune resistance are not separate processes but rather part of the same continuum of immune system-tumor interactions.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants to R. D. Schreiber from the National Cancer Institute (RO1 CA043059, RO1 CA190700), the Cancer Research Institute and the WWWW Foundation, and Bristol-Myers Squibb Inc.
Research supported by a Stand Up To Cancer – Lustgarten Foundation Pancreatic Cancer Convergence Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT14-14). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
M.M.G. is supported by a postdoctoral training grant (Irvington Postdoctoral Fellowship) from the Cancer Research Institute. J. P. W. is supported by a T32 training grant in hematology (5T32HL007088-40) from the National Heart, Lung, and Blood Institute and by the Paul Calabresi Career Development Award in Clinical Oncology (PCACO) K12 (4K12CA167540-05).
We are grateful to G.P. Dunn, Y. Fu, and D. Kobayashi for providing GL261-PD-L2 tumor cells. We thank S. Miller and Z. Zhang at the Genome Engineering and iPSC Center (GEiC) for technical support with CRISPR-Cas9 genome editing (T3ΔPDL1, F244ΔPDL1) and, D. Zhao and J. Feder for generation of MC38ΔPDL1 lines. We thank E. Lantelme and D. Brinja at the Flow Cytometry Core for technical support with FACS cell sorting. We also thank K. Sheehan and G. P. Dunn for constructive criticisms and comments, J. M. White, A. Miceli, E. Alspach, D. Lussier, D. Runci, R. Medrano, and C. Wilson in the Schreiber laboratory for discussions, and H. Schmidt for assistance with visual inspection of NGS somatic variants. We also thank M. Welsh, T. Chen, P. McCabe, H Zhang, E. Saravia, and D. Walker for assistance in the H31m1-PDL1 experiments. Aspects of studies at Washington University were performed with assistance by the Immunomonitoring Laboratory of the Center for Human Immunology and Immunotherapy Programs and the Siteman Comprehensive Cancer Center.
Footnotes
Conflict of Interest: R.D. Schreiber has ownership interest in, and is a consultant/advisory board member for, Igenica, Jounce, and Neon. M. J. Selby, R.F. Graziano and A.J. Korman are employees of Bristol-Myers Squibb. The other authors report no conflicts of interest.
References
- 1.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nature Medicine. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 2.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 5.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. The Lancet. 2016 doi: 10.1016/S0140-6736(16)32517-X. online first. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proceedings of the National Academy of Sciences. 2004;101:10691–6. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology. 2015;16:375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 11.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 12.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 13.Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chow LQM, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J Clin Oncol. 2016 doi: 10.1200/JCO.2016.68.1478. JCO681478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374:2542–52. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. The Lancet. 2016 doi: 10.1016/S0140-6736(16)32455-2. online first. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nature Medicine. 2003;9:562–7. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y-H, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nature Communications. 2014;5:5241–12. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Victor CT-S, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–6. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 23.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 25.Bellmunt J, Mullane SA, Werner L, Fay AP, Callea M, Leow JJ, et al. Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol. 2015;26:812–7. doi: 10.1093/annonc/mdv009. [DOI] [PubMed] [Google Scholar]
- 26.Hatogai K, Kitano S, Fujii S, Kojima T, Daiko H, Nomura S, et al. Comprehensive immunohistochemical analysis of tumor microenvironment immune status in esophageal squamous cell carcinoma. Oncotarget. 2016;7:47252–64. doi: 10.18632/oncotarget.10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 28.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–4. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8:337–11. doi: 10.1186/s13073-016-0264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Y-RA, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, et al. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS ONE. 2016;11:e0150606. doi: 10.1371/journal.pone.0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28:285–95. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–5. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 39.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–7. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 41.Mathios D, Ruzevick J, Jackson CM, Xu H, Shah S, Taube JM, et al. PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J Neurooncol. 2014;121:251–9. doi: 10.1007/s11060-014-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–26. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 43.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science. 2008;322:1097–100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hobo W, Maas F, Adisty N, de Witte T, Schaap N, van der Voort R, et al. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116:4501–11. doi: 10.1182/blood-2010-04-278739. [DOI] [PubMed] [Google Scholar]
- 46.Kuang D-M, Zhao Q, Peng C, Xu J, Zhang J-P, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding Z-C, Lu X, Yu M, Lemos H, Huang L, Chandler P, et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Research. 2014;74:3441–53. doi: 10.1158/0008-5472.CAN-13-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verdegaal EME, de Miranda NFCC, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536:91–5. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.