

Summary
The molecular mechanisms underlying the opposing functions of glucocorticoid receptors (GR) and estrogen receptor α (ERα) in breast cancer development remain poorly understood. Here, we report that in breast cancer cells liganded GR represses a large ERα-activated transcriptional program by binding, in trans, to ERα-occupied enhancers. This abolishes effective activation of these enhancers and their cognate target genes, and leads to inhibition of ERα-dependent binding of components of the MegaTrans complex. Consistent with the effects of SUMOylation on other classes of nuclear receptors, dexamethasone (Dex)-induced trans-repression of the estrogen (E2) program appears to depend on GR SUMOylation, which leads to stable trans-recruitment of the GR-NCoR/SMRT-HDAC3 co-repressor complex on these enhancers. Together, these results uncover a mechanism by which competitive recruitment of DNA-binding nuclear receptors/transcription factors in trans to “hot spot” enhancers serves as an effective biological strategy for trans-repression with clear implications for breast cancer and other diseases.
Graphical abstract
Introduction
Regulation of gene transcription is largely orchestrated by enhancers, based on the recruitment of specific DNA binding transcription factors in response to signals or ligands (Heinz et al., 2010; Spitz and Furlong, 2012; Li et al., 2016). Indeed, regulation of enhancer activity is particularly well characterized for the actions of the large family of nuclear receptors (Carroll et al., 2006; Ghisletti et al., 2010; Sever and Glass, 2013; Wang et al., 2007). It is noteworthy that multiple members of the nuclear receptor family are co-expressed in many tissues and cell types. The ability of nuclear receptors to coordinately regulate transcriptional programs via positive and negative crosstalk regulation is of particular importance for homeostasis and disease development (Liu et al., 2014; Ogawa et al., 2005). For example, peroxisome proliferator-activated receptors (PPARs), a type II nuclear receptor known to regulate lipid homeostasis, can compete with thyroid hormone receptors (TRs) for the interaction with retinoid X receptors (RXRs), and thus inhibiting TRs function (Ogawa et al., 2005). In addition, GR, PPARγ, and liver X receptors (LXRs) agonists were found to repress both common and distinct subsets of toll like receptor (TLR) target genes through the use of nuclear receptor- and TLR-specific trans-repression mechanisms (Bensinger and Tontonoz, 2008; Glass and Ogawa, 2006; Nagy et al., 2012).
ERα is a ligand-dependent sex steroid-regulated transcription factor that mediates most of the biological effects of estrogens, primarily at the level of gene transcription (Heldring et al., 2007; Liang and Shang, 2013). Following ligand-induced nuclear entry, ERα binds to ~30,000 estrogen response elements (EREs), a subset of which harbor the histone/epigenetic marks associated to enhancers (Carroll et al., 2006; Li et al., 2013; Hah et al., 2013). At putative functional enhancers, ERα promotes the recruitment of co-factors on these enhancers to activate the transcription of enhancer RNA (eRNA) and target coding genes (Li et al., 2015; Liu et al., 2014; Nagarajan et al., 2014). In addition to the coactivator complexes, it appears that additional factors can be critically required. Recently, we found that liganded-ERα stimulates the in situ nucleation of a complex of DNA-binding transcription factors, which involves the binding of GATA3 and RARα/γ that we refer to as MegaTrans transcription factors (Liu et al., 2014). This event occurs on ERα-occupied enhancers and is required to regulate gene expression.
GR is another well-characterized member of the nuclear receptor superfamily of ligand-activated transcription factors. In addition to activating a large number of enhancers harboring glucocorticoid response elements (GREs), GR also inhibits the actions of other transcription factors, including AP1 and NFκB (Glass and Saijo, 2010; Reichardt et al., 2001). In ERα positive breast cancer, GR expression has been associated with good clinical outcomes (Abduljabbar et al., 2015; Pan et al., 2011) and glucocorticoids have been reported to antagonize E2-induced genes expression in breast cancer cells (Gong et al., 2008; Karmakar et al., 2013). Diverse mechanistic models have been proposed for specific effects of GR repression, including: GR mediating gene trans-repression through inhibiting AP1/NF-ҡB activity, GR directly binding to the negative DNA binding sites, and GR recruitment of the coprepressor GRIP1 (Chinenov et al., 2012; De Bosscher et al., 2003; Glass and Saijo, 2010; Gupte et al., 2013; Rogatsky et al., 2002; Rogatsky et al., 2003; Surjit et al., 2011). However, a general mechanism by which glucocorticoids negatively regulate the ERα signaling pathway remains unclear.
In this study, we have investigated the molecular mechanisms by which GR represses the transcriptional program directed by ERα. By using global genomic data generated in breast cancer cells, we find that, unexpectedly, glucocorticoids significantly repress the expression of a large group of estrogen-activated genes by inhibiting the recruitment of the MegaTrans complex to ERα-bound enhancers. The MegaTrans complex (e.g. GATA3 and RARα/γ) is required for ERα-dependent enhancer and target gene activation.
We show that the repressive effects of liganded-GR on estrogen-activated enhancers occur via ERα-dependent trans-recruitment of GR to these sites, blocking the recruitment of the MegaTrans complex. This event is associated with poorer metastasis-free outcomes in breast cancer patients.
Unexpectedly, the effective trans-recruitment of GR to the ERα-bound enhancers depends on its association with the NCoR/SMRT-HDAC3 complex, which also apparently requires the SUMOylation of GR. Together, these results have revealed a previously unappreciated, enhancer-based mechanism underlying glucocorticoids repression of a large ERα-mediated transcriptional program.
RESULTS
The E2-Activated Transcriptome Is Dramatically Altered by Glucocorticoids in Breast Cancer Cells
Ligand-dependent translocation of GR from cytoplasm to nucleus in MCF-7 breast cancer cells occurs rapidly after addition of the synthetic glucocorticoids, dexamethasone (Dex) (Figure S1A). By analyzing the effects of Dex on the ERα transcriptional program with qPCR, we showed that target genes and eRNAs expression were significantly inhibited in MCF7 cells co-treated with E2 and Dex (E2+Dex) in comparison with cells treated with E2 only (Figure 1A). In order to determine the direct effects of glucocorticoids on the estrogen-dependent transcriptional response, we performed global run-on sequencing (GRO-seq) in E2, Dex, or E2+Dex-treated MCF-7 cells. For each treatment condition, we observe the transcriptional induction and repression of many transcription units as reported in Figure S1B. Here, we focus primarily on the E2-activated genes that are repressed by addition of Dex, which represents a major component of the Dex effect on the E2-regulated transcriptional program in MCF7 cells. In fact, we identified 465 coding target genes that were highly up-regulated in response to E2, but strongly repressed following addition of Dex (Figure 1B). The levels of mRNA or protein of ERα and GR themselves were not perturbed by these treatments (Figure S1C), suggesting that the transcriptional repression of E2-activated genes in E2+Dex-treated MCF-7 cells was due to the crosstalk between ERα and GR. In the absence of E2, Dex treatment alone did not significantly change the transcription of estrogen-target genes (Figure 1B). A similar effect of E2+Dex treatment, compared with E2 treatment only, was observed for the transcriptional levels of transcribed enhancer elements. This indicates that Dex-activated GR dramatically repressed the E2-dependent activation of eRNAs transcription at ERα-bound enhancers (Figure 1C). These functional enhancers exhibited a strong binding of the MegaTrans complex (Figure S1D). Two representative examples of the repressive effect of Dex on the transcriptional activation of coding genes and cognate enhancers by E2 are shown in Figure 1D.
Figure 1. Dex significantly repress the E2-mediated activation of target genes and enhancers.
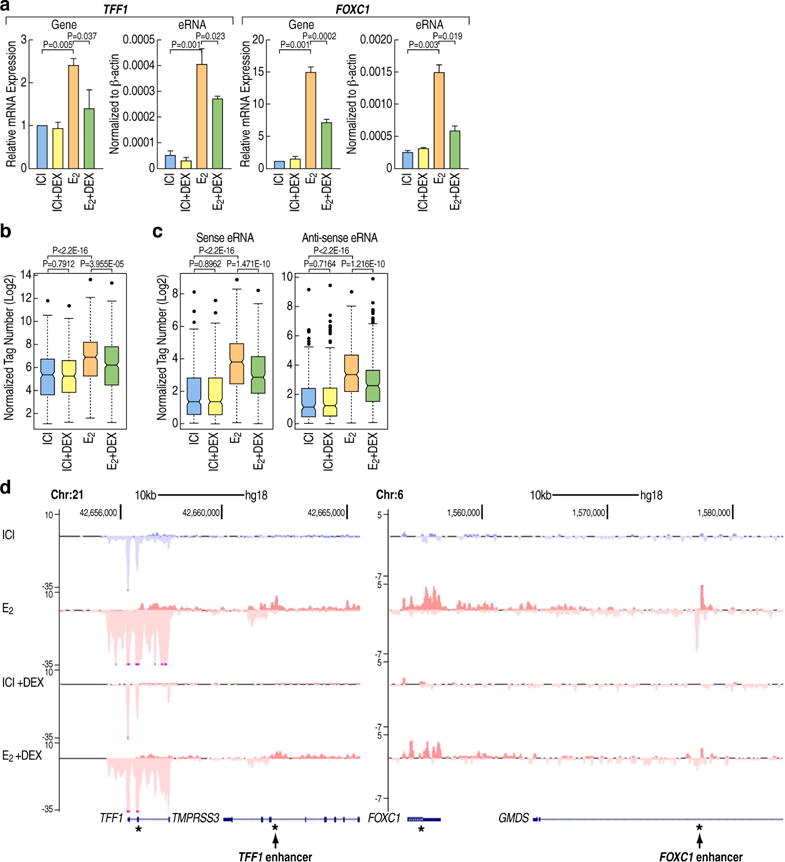
(A). E2+Dex treatment of MCF7 cells significantly represses E2-activated genes (TFF1, FOXC1) and eRNAs expression. qRT-PCR results are presented as mean ± SD. N≥3, two-tailed Student’s t test.
(B). Boxplot showing normalized GRO-seq tags (Log2) for E2-induced genes under different treatment conditions (ICI, ICI+DEX, E2, E2+Dex) in MCF7 cells. (P value is calculated by Wilcoxon rank sum test)
(C). Boxplots showing normalized GRO-seq tags (Log2) for eRNAs transcribed at active enhancers under different treatment conditions (ICI, ICI+DEX, E2, E2+Dex) in MCF7 cells. Sense and antisense eRNA transcripts are shown separately. (P value is calculated by Wilcoxon rank sum test).
(D) Genome browser image showing normalized GRO-seq tag counts in MCF7 cells under different ligands treatment (ICI, ICI+DEX, E2, E2+Dex). TFF1 and FOXC1 loci are shown. “*” indicates the position of primers used for qPCR detection. See also Figure S1
To determine if the repressive actions of GR on the ERα-dependent transcriptional program might infer a broad clinical significance, we mined breast cancer outcome-linked gene expression data using the Gene Expression-Based Outcome for Breast Cancer Online (GOBO) tool, with outcomes shown by Kaplan-Meier survival plots. We found that the expression levels of the 465 E2-activated genes are strong predictors of clinical outcomes. For example, high expression levels of these genes more significantly predict poor outcomes for patients affected by ERα-positive lymph node-negative breast cancer and Luminal A breast cancer, compared to an identical number of randomly selected E2-activated genes (Figure S1E). A soft agar colony formation assay also consistently revealed that E2+Dex-treated MCF7 cells exhibited strong defects in growth compared with E2 treatment alone (Figure S1F). Collectively, these results indicated that the crosstalk between E2 and Dex signaling regulates the transcription of specific subsets of coding genes and cognate enhancers, which underlies important biological outcomes (e.g. growth of breast cancer cells) and also clinical outcomes in some breast cancer types.
GR Trans-Binding to a Subset of Estrogen Activated Genes Enhancers
To explore the molecular crosstalk between estrogen and dexamethasone signaling pathways at a genome wide level, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for GR and ERα, the two key transcription factors underlying these events. For ERα ChIP-seq we used a specific antibody against ERα (Carroll et al., 2006; Li et al., 2013). However, for GR ChIP-seq, we used a biotin-based approach to overcome technical limitations due to the lack of a robust anti-GR antibody that has precluded robust ChIP-seq analyses (Table S4). To this aim, we engineered the MCF7 cells to express a bacterial biotin ligase (BirA) that can biotinylate a biotin ligase recognition peptide (BLRP)-tagged protein in vivo (Heinz et al., 2010; Liu et al., 2014). Under the control of a Tet-On promoter, BLRP-tagged wild-type GR was expressed at a level comparable with the endogenous protein upon doxycycline induction (Figure S2A).
Here, we focused primarily on 423 enhancers, activated by liganded-ERα, and corresponded to. 465 proximal coding genes, which were strongly activated by E2, but repressed following the addition of Dex, as discussed above. The binding of ERα on these enhancers did not significantly change upon E2+Dex treatment when compared to E2 treatment alone. In contrast, the binding of GR exhibited a dramatic increase (Figure 2A, 2B). Motif analyses of the 423 enhancer sites co-bound by ERα and GR in cells treated with E2+Dex showed an expected enrichment of ERα and FOXA1 motifs. Interestingly, we did not find any GRE motifs, including the variant motif suggested by a recent study (Surjit et al., 2011) (Figure 2C). This result suggested that GR recruitment to these ER-bound enhancers might occur in trans.
Figure 2. GR is recruited in trans on the E2-activated enhancers following E2+Dex treatment.
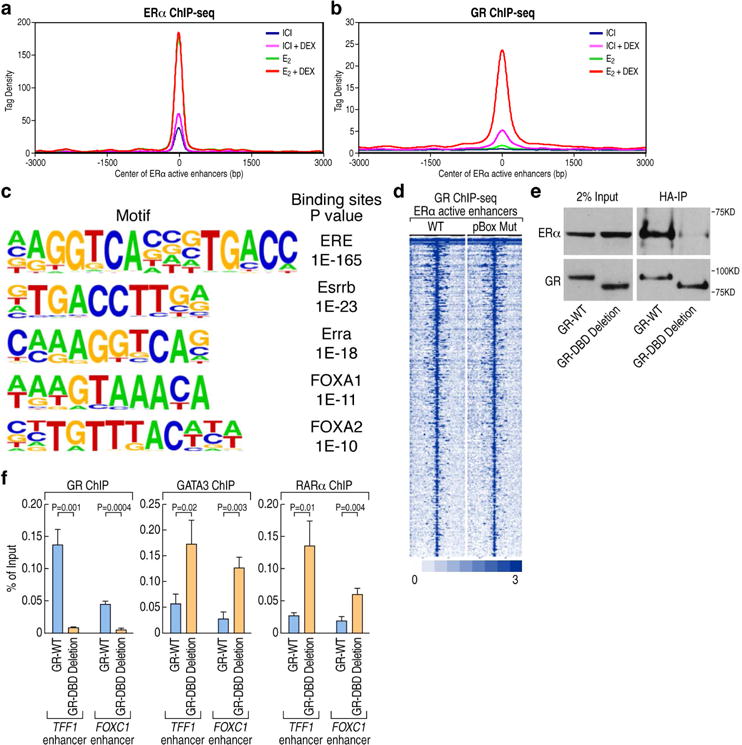
(A). ChIP-seq tag density profile plot showing ERα binding on 423 ERα-activated enhancers upon different ligands treatment (ICI, ICI+DEX, E2, E2+Dex). The center of the plot is based on the center of ERα binding.
(B). ChIP-seq tag density profile plot showing the binding of GR to 423 ERα-activated enhancers with different ligands treatment (ICI, ICI+DEX, E2, E2+Dex). The center of the plot is based on the center of ERα binding.
(C) De novo motif analysis of GR binding sites based on the 423 ERα-activated enhancers.
(D) Heatmaps of ChIP-seq tag counts for 423 ERα-activated enhancers occupied by GR wild type or pBox mutant in MCF7 cells treated with E2+Dex.
(E) The interaction of GR with ERα is dependent on its DNA-binding domain (DBD), as shown by coimmunoprecipitation using HA-tagged WT or DBD deleted-GR.
(F) ChIP-qPCR showing GR, GATA3 and RARα binding on ERα-activated enhancers (TFF1 and FOXC1 enhancers) in GR wild type and GR DBD deletion stable MCF7 cells upon E2+Dex treatment. Data are presented as mean ± SD. N≥3, two-tailed Student’s t test. See also Figure S2
To formally test this possibility, we generated engineered MCF7 cells that express either wide-type GR or its DNA-binding domain mutant (pBox mutant) (Uhlenhaut et al., 2013), which is unable to recognize its cognate motifs (Figure S2B). Because GR often functions as a dimer, we knocked down endogenous GR to avoid any potential confounding results. To this aim we used specific shRNAs targeting the 3′UTR region of GR mRNA. We simultaneously induced the expression of the pBox mutant or wild-type GR to enable a ChIP-seq analysis (Figure S2A). The mutation of the DNA binding domain of GR (pBox mutant) did not change the recruitment of GR on the 423 enhancers in response to E2+Dex treatment (Figure 2D). This result strongly supports the hypothesis that the recruitment of GR on these enhancers occurs in trans. In contrast, we found that for half of peaks occupied by wild-type GR (20,830 of the 39,405), the mutation of the pBox domain significantly decrease GR binding, indicating that they correspond to GR direct targets in cis (Figure S2C). These trans or cis binding events was further confirmed by the motif analysis (Figure 2C, S2D). Two representative genomic loci showing GR binding to ERα enhancers in the presence of E2+Dex are shown in Figure S2E. The correlations between ERα and GR peaks identified by ChIP-seq under different stimuli are summarized in Figure S2I.
To further investigate the mechanisms by which GR is recruited in trans on E2-activated enhancers, we generated a stable cell line expressing a deletion mutant of GR lacking the DNA binding domain (DBD). This domain was reported to directly contribute to the interaction with ERα (Karmakar et al., 2013), a finding that has been verified in our lab (Figure 2E). Deletion of the DBD of GR indeed largely abolished the GR binding on the E2-activated enhancers, suggesting that the interaction with ERα is required for GR trans binding. In contrast, the binding of MegaTrans components GATA3 and RARα was significantly increased, as exemplified by the analysis of the TFF1 and FOXC1 enhancers (Figure 2F). The protein expression levels of wild-type and mutant GR were equivalent, as shown in Figure S2F. In addition, when we abolished the expression of ERα by using shRNAs (Figure S2G), this also abolished the binding of GR on those enhancers (Figure S2H). These data strongly support our conclusion that liganded GR bind in trans on the E2-activaed enhancers.
Dex Inhibits E2-Activated Gene Expression by Disassembling the MegaTrans Complex on the ERα-Bound Sites
The MegaTrans complex provides a signature of the most potent functional ERα-regulated enhancers, and is required for transcription of eRNAs or target genes and recruitment of coactivators, including p300 (Liu et al., 2014). To explore the mechanisms involved in Dex-dependent down-regulation of E2-activated genes and eRNAs, we assessed binding of the MegaTrans complex on E2-regulated gene enhancers by ChIP-qPCR. We found that GATA3 and RARα, which function as the “nucleating” components of the MegaTrans complex formation (Liu et al., 2014), but not ERα exhibited significantly reduced binding on E2-activated enhancers, exemplified by TFF1 and FOXC1 enhancers, in MCF7 cells treated with E2+Dex (Figure 3A). AP2ɣ, another component of the MegaTrans complex, also showed a significantly decreased recruitment on the E2-activated enhancers (Figure 3A). As expected, the binding of the pioneer transcription factor FOXA1 on these enhancers was virtually unchanged (Figure 3B). This result is consistent with the reports that FOXA1 is required for ERα and GR binding on the enhancers (Belikov et al., 2009; Belikov et al., 2012; Carroll et al., 2005; Hurtado et al., 2011). Not surprisingly, the coactivator p300 also exhibited diminished binding on these enhancers, consistent with previous observations that MegaTrans is required for coactivator recruitment (Figure 3B) (Liu et al., 2014). ChIP-seq experiments for GATA3 confirmed a significantly decreased binding of this factor on the 423 E2-activated enhancers in proximity of the genes dramatically down-regulated by E2+Dex (Figure 3C). Accordingly, GATA3 binding was not robustly affected on the ERα-dependent enhancers that were not responsive to Dex treatment (Figure S3A). Two representative genomic loci exhibiting GATA3 decreased binding under E2+Dex treatment are shown in Figure S3B. These data suggest a model of competition between GR and MegaTrans components. To test the validity of this model, we performed double-ChIP assays using the stable cell line expressing BLRP-tagged-GR, which clearly demonstrated that the occupancy of GR and MegaTrans components (i.e. GATA3, RARα or AP2ɣ) is mutually exclusive and they cannot be co-recruited by ERα on these enhancers (Figure 3D). After E2 treatment, the MegaTrans complex, rather than GR, was recruited to E2-activated enhancers by ERα. In contrast, upon E2+Dex treatment, ERα recruits GR, but not the MegaTrans complex (Figure 3D). Together, these data demonstrated that GR inhibited MegaTrans binding on the E2-activated enhancers, thereby decreasing eRNA and target gene expression levels.
Figure 3. GR inhibits the assembly of the MegaTrans complex on E2-activated enhancers.
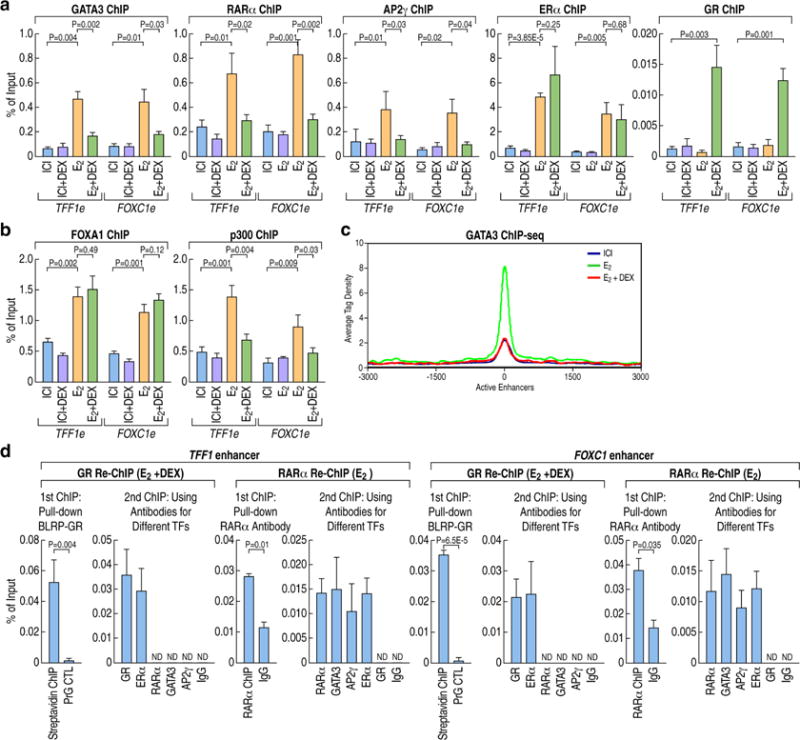
(A) ChIP-qPCR showing the binding of MegaTrans components (GATA3, RARα, AP2ɣ), ERα and GR on E2-activated TFF1 and FOXC1 enhancers in MCF7 cells treated with ICI, ICI+DEX, E2, E2+DEX. Data are presented as mean ± SD. N≥3, two-tailed Student’s t test. TFF1 e: TFF1 enhancer; FOXC1 e: FOXC1 enhancer.
(B) ChIP-qPCR showing FoxA1 and P300 binding on ERα-activated enhancers in MCF7 cells treated with ICI, ICI+DEX, E2, E2+DEX. Data are presented as mean ± SD. N≥3, two-tailed Student’s t test.
(C) ChIP-seq tag density profile plot (centered on ERα binding peaks in E2 condition) showing the binding of GATA3 on 423 ERα-activated enhancers under different treatment conditions (ICI, E2 or E2+Dex).
(D) ChIP-reChIP qPCR analysis showing that GR and MegaTrans complex (exemplified by GATA3, RARα, AP2ɣ) could not co-exist on the ERα-activated enhancers in MCF7 cells treated with E2 or E2+Dex. The GR Re-ChIP was done after E2+Dex treatment, the RARα Re-ChIP was done after E2 treatment. ChIP signals are presented as percentage of input. Data are shown as mean ± SD. N≥3, two-tailed Student’s t test. N.D, not detectable. See also Figure S3
GR Trans-Binding to the ERα-Bound Sites Depends on Its SUMOylation Status
SUMOylation of transcription factors has previously been correlated with impaired transcriptional activation and/or transcriptional repression (Hua et al., 2016a; Hua et al., 2016b; Pascual et al., 2005; Perdomo et al., 2005; Yang and Sharrocks, 2004). GR has been reported to harbor three SUMOylation sites, two of which are located in the N- terminal domain (K277 and K293 in human GR), and one in the C-terminal ligand-binding domain (K703 in human GR) (Druker et al., 2013; Paakinaho et al., 2014). The two N-terminal sites have been reported to inhibit GR synergistic activity when there were multiple GREs around the promoter region (Holmstrom et al., 2003). Recently, Hua and colleagues reported that SUMOylation of the GR in the N-terminal domain is required for the glucocorticoid-induced gene repression (Hua et al., 2016a; Hua et al., 2016b). The C-terminal SUMOylation site is reported to exert a positive action on GR’s activity (Druker et al., 2013).
We confirmed that GR is covalently modified by the small ubiquitin-related modifier-1 (SUMO-1) peptide when treated with E2+Dex with the endogenous SUMO in MCF7 cells (Figure 4A). Similar results were obtained by co-transfection of GR, HA-tagged SUMO1 and Ubc9 into HEK293 cells. (Figure S4A). Further, we found that only when all three SUMOylation sites were mutated (GR-3KR), the SUMOylation of GR was completely abolished, compared to either mutation of the C-terminal ligand-binding domain SUMOylation site alone (GR-1KR), or the mutation of the two N-terminal domain SUMOylation sites (GR-2KR) (Figure 4A, S4A). To investigate any functional correlation between GR SUMOylation status and its effects on gene expression, we generated GR knock-out cells via CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 method based on the design of guide RNAs targeting the GR coding region in order to disrupt its open reading frame (ORF) (Figure S4B). We randomly selected 3 independent colonies for further experiments, none of which exhibited any detectable GR expression (Figure S4C). Using these GR knock-out cell lines, we performed rescue experiments by overexpressing either GR-WT and GR SUMOylation sites mutants, achieving an effective lentiviral infection (Figure S4D). Our data demonstrated that the mutation did not alter the subcellular localization of GR (Figure S4E). Based on the GRO-seq and ChIP-seq data, we selected two representative pairs of coding target genes for further analysis. The transcription of the first pair of genes (KLF4, GADD45G) was up-regulated by Dex treatment and GR directly binds in cis to their proximal enhancers (Figure S4F). In contrast, the transcription of the second pair of genes (TFF1, FOXC1) was induced by estrogen and attenuated by the addition of Dex, which induces the binding of GR in trans. (Figure 1D, S2E). The GR SUMOylation mutant rescued KLF4, GADD45G expression similarly to the wild-type GR, indicating that SUMOylation was not required for these GR-mediated activation events. Contrary, the GR-3KR mutant failed to effectively rescue the Dex repression of TFF1, FOXC1 genes on which GR was recruited in trans, indicating that SUMOylation was required for the function of trans bound GR. (Figure 4B).
Figure 4. The ability of GR to bind in trans on E2-activated enhancers depends on its SUMOylation status.
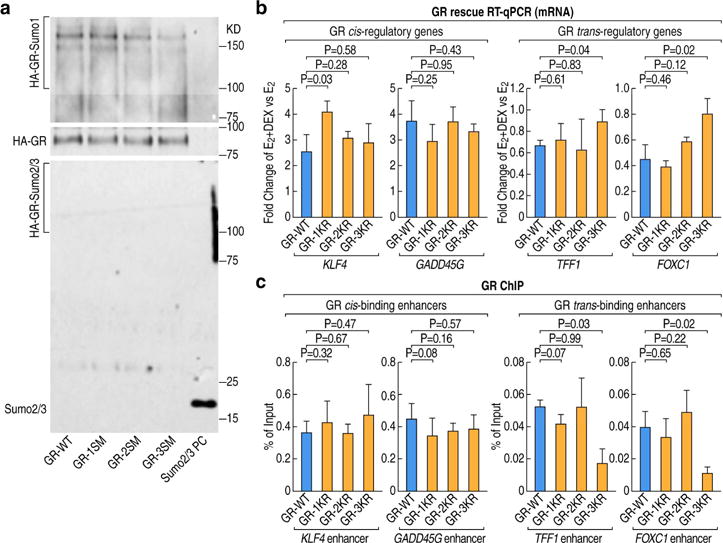
(A) Western blot analysis showing immunoprecipitated wild type GR or SUMOylation mutants in MCF7 cells using different SUMO-specific antibodies upon treatment with E2+Dex. (GR-WT:GR wild type; GR-1KR: GR C-terminal K703R mutation; GR-2KR: GR N-terminal K277R and K293R two sites mutation; GR-3KR: GR K277R, K293R and K703R all three sites mutation, SUMO2/3 PC: HA-SUMO2/3 protein as positive control)
(B) RT-qPCR of GR target genes in GR knockout MCF7 cells upon over-expression of GR wild type or SUMOylation mutants. Fold change of gene expression is presented as comparison of E2+Dex versus E2 treatments. Data are presented as mean ± SD. N≥3, two-tailed Student’s t test.
(C) ChIP-qPCR showing GR wild type and SUMOylation mutants binding on GR cis or trans-binding enhancers in MCF7 cells upon E2+Dex treatment. Data are presented as mean ± SD. N≥3, two-tailed Student’s t test. See also Figure S4
To better elucidate the mechanisms underlying the different effects caused by GR mutants, we performed ChIP assays to compare the binding of GR mutants to the wild-type GR. We found that wild-type GR, GR-1KR, GR-2KR could bind all the interrogated enhancers when treated with E2+Dex (Figure 4C); however, the GR-3KR mutant exhibited a dramatically decreased binding to the TFF1 and FOXC1 ERE-containing enhancers, while still effectively binding KLF4 and GADD45G enhancers (Figure 4C). Together, these data indicate that the SUMOylation of GR is a prerequisite for the trans-recruitment of GR to the ERα-bound activated enhancers; whereas SUMOylation is not necessary for the binding of GR to the cis-bound, activated enhancers we analyzed.
NCoR/SMRT-HDAC3 Complex Stabilizes GR Binding on ERα-Regulated Enhancers
We next wished to investigate whether cofactors brought by GR might be involved in the mechanisms underlying GR-mediated MegaTrans disassembly. It has been reported that cis-bound GR-NCoR/SMRT repressor complexes are required in fluocinolone acetonide (FA) treated epidermis (Surjit et al., 2011) and recently the same group showed that SUMOylation of GR was indispensable for the formation of a GR-small, ubiquitin-related modifiers (SUMOs)-NCoR1/SMRT-HDAC3 repressive complex (Hua et al., 2016a; Hua et al., 2016b, Ki et al., 2005). This would be consistent with our results that GR recruitment to the ERα-bound sites depends on its SUMOylation (Figure 4C). Based on these reports and our data, we tested the possibility that NCoR/SMRT-HDAC3 complex might be required for GR to stably trans-bind ERα-activated enhancers. We first tested the ability of GR to occupy these enhancers after knock-down of the NCoR/SMRT-HDAC3 complex by siRNAs transfection. Indeed, NCoR or SMRT knockdown resulted in a significant decrease of GR binding to these enhancers when compared to control siRNAs in cells treated with E2+Dex (Figure 5A). Concurrently, the MegaTrans complex binding was found to be significantly increased on these gene enhancers, as determined by Chip-qPCR for GATA3, or RARα (Figure 5A). The high knockdown efficiency for each repressive component is shown in Figure S5A. Accordingly, the repressive effects of Dex on E2-activated genes were abolished by knocking down the repressive complex (Figure 5B). Binding of the NCoR/SMRT complex to ERα-activated enhancers showed a significant increase in cells treated with E2+Dex compared to E2 alone (Figure 5C), accompanied by a decrease of H3K9 acetylation (Figures S5B). Additionally, in GR knock-out cells the binding of NCoR/SMRT complex was significantly decreased on these enhancers when treated with E2+Dex (Figure S5C), suggesting that Dex-induced NCoR/SMRT binding to ERα-activated enhancers is GR dependent.
Figure 5. The recruitment of the NCoR/SMRT complex is required for GR-mediated repression on E2-activated genes.
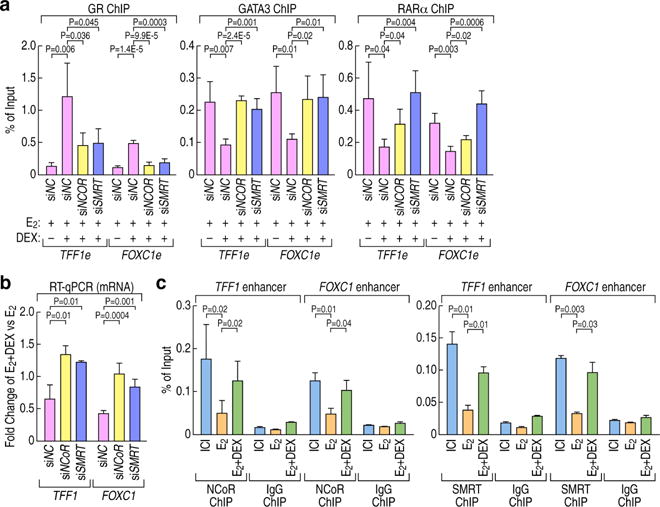
(A) ChIP-qPCR showing GR, GATA3 and RARα binding on ERα-activated enhancers (TFF1 and FOXC1 enhancers) following siRNA-mediated knock-down of NCoR/SMRT complex in MCF7 cells treated with E2 or E2+Dex. ChIP signals are presented as percentage of input. Data are represented as mean ± SD. N≥3, two-tailed Student’s t test. TFF1 e: TFF1 enhancer; FOXC1 e: FOXC1 enhancer.
(B) RT-qPCR showing the effect of DEX treatment on ERα-activated genes (TFF1, FOXC1) following siRNA-mediated knock down of NCoR/SMRT complex in MCF7 cells upon E2 or E2+Dex treatment. The gene expression changes are shown as fold change upon E2+Dex treatment vs E2 stimulation. Data are represented as mean ± SD. N≥3, two-tailed Student’s t test.
(C) ChIP-qPCR showing NCoR/SMRT complex binding on ERα-activated enhancers upon E2+Dex treatment compared with E2 treatment in MCF7 cells. ChIP signals are presented as percentage of input. Data are represented as mean ± SD. N≥3, two-tailed Student’s t test. See also Figure S5
Together, these data support the conclusion that the molecular basis for the action of trans-recruited GR at ERα-activated enhancers is that it competes with the zinc finger components that nucleate formation of the MegaTrans complex, specifically GATA3 and RARα/γ. This process involves GR association with NCoR/SMRT repressive complex that is dependent on GR SUMOylation, permitting effective binding of GR to these enhancers to mediate effective gene and enhancer repression.
DISCUSSION
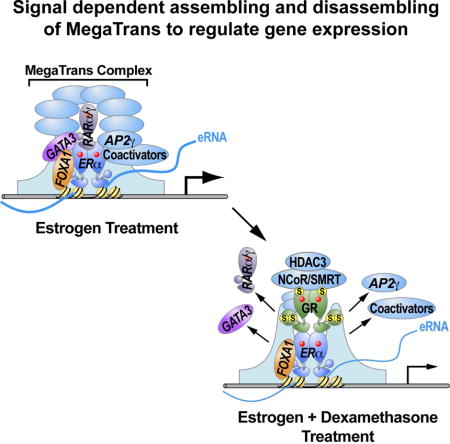
Increasing evidence supports the proposal that GR plays an important role in ERα positive breast cancer and associates with more favorable clinical outcomes (Abduljabbar et al., 2015; Kach et al., 2015; Karmakar et al., 2013; Pan et al., 2011). Upon ligand induction, GR modulates specific genomic sites that are occupied by ERα, either by direct recognition of EREs or through indirect interaction with other factors (Miranda et al., 2013). However, the mechanisms underlying ERα/GR crosstalk at the genomic level have been poorly understood. In this study, we have elucidated a previously unsuspected mechanism by which ligand-bound GR regulates a large E2 transcriptional program, resulting in repression of cell growth. We have found that treatment with E2+Dex results in binding of GR to ERα-regulated enhancers in trans, which diminished the effective assembly of the MegaTrans complex. This reflects the ability of GR to compete with GATA3 and RARα/γ for the fine-tuning regulation of ERα-regulated enhancers. In accord with our previous study (Liu et al., 2014), these ERα-tethered MegaTrans transcription factors work as a new category of ERα “co-activators”, and decommissioning of these “co-activators” causes the failure of activation of the ERα-bound enhancers, as well as their coding target genes. Further, binding of GR to these enhancers requires its SUMOylation-dependent association with the NCoR/SMRT-HDAC3 complex (Figure 6). Here, we have found that GR SUMOylation, while required for its trans-binding to ERα-bound enhancers, is apparently not needed for its cis binding to the enhancers evaluated - the KLF4 and GADD45G enhancers- presumably reflecting the role of SUMOylation in determining the interaction of trans-bound GR with corepressor complexes. The finding that SUMOylation is required for the actions of GR is consistent with the trans recruitment of PPARγ, LXRα/β and NURR1 to the gene targets they repress; in those cases, recruitment seems to depend on interactions with previously-bound corepressors (Ghisletti et al., 2007; Glass and Saijo, 2010; Pascual et al., 2005; Venteclef et al., 2010).
Figure 6. Working model for Dex repressing ERα-activated genes expression.
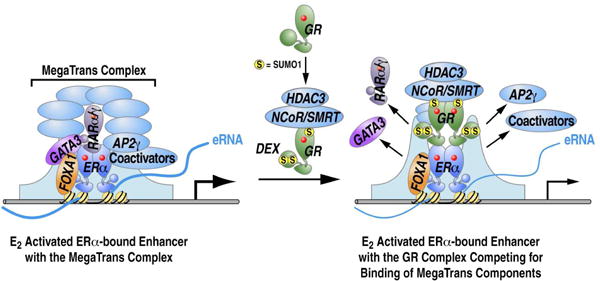
Previous studies have documented repressive actions of GR on specific ERα target genes. For example, liganded GR can induce estrogen sulfotransferase expression and activity to sulfonate estrogens, which leads to the inability of estrogens to activate ERα (Gong et al., 2008). On some specific gene targets, it was noted that GR could result in displacement of ERα and the coactivator SRC3 (Karmakar et al., 2013), but a full molecular explanation was not provided. By using genome wide approaches, including GRO-seq for transcriptomic analysis and ChIP-seq for ERα and GR binding, we found that after treatment with E2+Dex versus E2 only, GR binding on E2-activated enhancers dramatically increased, while ERα binding did not show significant alteration. Thus, globally, altered ERα binding to enhancers does not appear to provide a more generally- applicable explanation for Dex-induced repression events. Instead we have found that Dex treatment inhibits MegaTrans complex recruitment on these enhancers, which has been shown to serve as a functional signature and a new category of “co-activators” for ERα-activated enhancers. This complex is suggested to be sequentially recruited based on initial recruitment of GATA3 and RARα/γ(Liu et al., 2014). As would be predicted with the failure to recruit the MegaTrans complex, well-established coactivators, such as p300, also exhibited impaired recruitment to these enhancers, and eRNA induction was inhibited. We can therefore propose that inhibition of the MegaTrans complex formation is a primary mechanism underlying Dex repression effects on the E2 activated gene transcriptional program. Our ChIP-qPCR and sequential ChIP data indicate that the GR and MegaTrans complex do not co-exist on the regulated ERα-bound enhancers, consistent with a competition model between the “nucleating” components of the MegaTrans complex (GATA3 and RARα/γ) and liganded GR for binding to ERα-bound enhancers.
Collectively, our data demonstrate a previously unsuspected model of repression of a large, biologically important estrogen-regulated transcriptional program based on the competition between zinc finger transcription factors and GR for trans-recruitment by ERα to regulatory enhancers, providing a potentially generalizable insight into understanding transcription factor cross-talk at enhancers. .
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
METHOD DETAILS
shRNA Lentivirus Package, and infection
qPCR
GR Knockout Cell Lines Construction and GR Rescue Experiment
ChIP, ChIP-Seq, Biotin ChIP, Biotin ChIP-Seq, and ChIP-ReChIP
GRO-Seq
SUMOylation Experiments
Cloning, Mutagenesis, and Generation of Biotin-Tagged Inducible MCF7 Stable Cell
Lines
Soft Agar Colony Formation Assay
Kaplan-Meier Analyses
Immunofluorescence
QUANTIFICATION AND STATISTICAL ANALYSIS
Primary Analysis of ChIP-Seq Data Sets
Identification of ChIP-Seq Peaks, Heatmap and Tag Density Analyses
Motif Analysis
GRO-seq Analysis
Data Visualization
Statistical Analysis
DATA AND SOFTWARE AVAILABILITY
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the lead contact, Professor Michael. G. Rosenfeld (mrosenfeld@ucsd.edu), School of Medicine, University of California, San Diego, LA JOLLA, CA 92093, USA.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MCF7 and HEK293T cells obtained from ATCC were cultured in DMEM (GIBCO #10566) media supplemented with 10% FBS in a 5% CO2 humidified incubator at 37 °C. When cells reached 70%–80% confluency, the culture media was replace with phenol red free DMEM media plus 5% charcoal-depleted FBS for at least 72h. For most of the experiments, cells were treated with 100nM 17β-estradiol (E2) (Sigma) or Dexamethasone (DEX) (Sigma) for 1 hr, unless otherwise stated. To induce the knockdown of ERα protein, we treated MCF7 with100nM ICI 182780 (Sigma) for 3 hr, as previously reported (Ross-Innes et al., 2010; Wakeling et al., 1991). Transfection of siRNAs into MCF7 cells was performed using Lipofectamine 2000 (Life Technologies), following manufacturer’s instructions and 50nM final concentration of specific siRNAs. The sequences of siRNAs are listed in Table S2.
METHOD DETAILS
shRNA Lentivirus Package, and infection
pLKO lentiviral shRNA targeting GR 3′UTR region and control shRNA vectors were purchased from Sigma (See Table S2). shRNAs-mediated knockdown assays were conducted according to the standard protocols from Addgene. Briefly, pLKO-based lentiviral shRNA plasmids were co-transfected with packaging plasmids (psPAX2 and pMD2.G) into human 293T cells. Culture medium containing lentiviruse particles was harvested, filtered, and used to infect MCF7 cells. For stable knockdown of MCF7 cells, 1 mg/ml puromycin was used for selection and cells were collected for experiments within 5 days.
qPCR
For qRT-PCR experiments, the MCF7 cells were treated with E2 or DEX for 4 hr before collection. RNA was isolated with RNeasy column (QIAGEN) and reverse-transcribed using SuperScript III Reverse Transcriptase (Life Technologies) or iScript Select cDNA Synthesis Kit (Bio-Rad) following manufacturer’s instructions. qPCRs were performed in StepOne™ Real-Time PCR Systems (Applied Biosystems) using 2X qPCR master mix from Affymetrix. Relative quantities (RQ) of gene expression levels were normalized to β-actin. A list of primers used for qPCR is provided (Table S1).
GR Knockout Cell Lines Construction and GR Rescue Experiment
We generated GR knockout cells using CRISPR/Cas9 technology. The sgRNAs targeting GR mRNA coding region were predicted by an online software (http://crispr.mit.edu/). The sgRNA target sites are ACTACGCTCAACATGTTAGG AGG and CGCTCAACATGTTAGGAGGGCGG (PAM sequence are underlined). Cloning strategy using the PX459 vector (Addgene, #48139) has been previously reported (Ran et al., 2013). The px459 vectors containing sgRNA and spCas9 were transfected into MCF-7 cell using Lipofectamine 2000(invitrogen). Two days later puromycin was added (0.4ug/ml) to the medium for selection. The positive clones were confirmed by western blotting assay.
For the rescue experiments, we used the FM5 lentiviral vector to overexpress HA-tagged GR wild type or SUMOylation sites mutants or DNA binding domain deletion GR in GR knockout cells.
ChIP, ChIP-Seq, Biotin ChIP, Biotin ChIP-Seq, and ChIP-ReChIP
ChIP or ChIP-Seq experiments were performed as previously described (Furlan-Magaril et al., 2009; Liu et al., 2014). Briefly, cells were cross-linked with 1% formaldehyde at room temperature for 10 min. For selected experiments (e.g., ChIP for NCoR, SMRT), cells were double cross-linked with 2mM DSG (ProteoChem) for 1 hr and then with1% formaldehyde for 10 min. Cross-linking was quenched with 0.125M glycine for 5 min at RT. Chromatin was fragmented using a tip sonicator or Bioruptor to get the fragments in the range of 200–500bp and precleared using 15ul Protein G Dynabeads (Life Technologies). Subsequently, the soluble chromatin was incubated with 2–5 ug antibodies at 4°C overnight. Immunoprecipitated complexes were collected using 20ul Protein G Dynabeads (Life Technologies) per reaction. After washing, the protein-DNA complexes were eluted and de-crosslinked overnight at 65°C.
To perform biotin ChIP or biotin ChIP-seq experiments using BLRP-tagged GR and GATA3, we followed previously described protocols (Liu et al., 2014). Briefly, cross-linked protein-DNA complexes were pulled down with Nanolink Streptavidin Magnetic beads (Solulink). The beads were washed twice with 1% SDS in TE and twice with 1% Triton X-100 in TE (20 min each). The streptavidin beads were then subjected to TEV protease (Life Technologies) digestion to elute tagged protein and DNA complex before de-crosslinking at 65°C overnight.
To perform the HA-tag ChIP, we used infected GR knockout MCF7 cells with lentivirus expressing HA-tagged GR carrying mutation for different SUMOylation sites or DNA binding domain deletion.
For ChIP-reChIPs of biotin-tagged GR we followed a previously described protocol (Liu et al., 2014). Briefly, the first biotin ChIP was performed as described above with protein-DNA complexes eluted through TEV protease (Life Technologies) digestion. Protein G Dynabeads (Life Technologies) were used as a negative control for the first biotin ChIPs. The first biotin ChIP elution was diluted at least 10 times with dilution buffer (20mM Tris-HCl pH7.4, 100mM NaCl, 0.5% Triton X-100, 2mM EDTA) and then incubated with the second ChIP antibody or IgG (as control). The second ChIP procedure was performed the same way as described above for regular ChIP.
For ChIP-reChIP using specific antibodies we used a previously reported protocol with some modifications (Furlan-Magaril et al., 2009). Briefly, the first ChIP beads were resuspended in 75 m L TE/10 mM DTT and the immunocomplexes were eluted by incubating 30 min at 37°C. After centrifugation, the samples were diluted 20 times (to a final volume of 1.5 mL) with dilution buffer and then incubated with the second ChIP antibody or IgG (as control). The second ChIP procedure was performed the same way as described above for regular ChIP.
For all ChIPs, final ChIP DNA was extracted and purified using QIAquick spin columns (QIAGEN). The ChIP-seq libraries were constructed following Illumina’s ChIP-seq Sample prep kit.
GRO-Seq
GRO-seq experiments were performed as previously reported (Core et al., 2008; Li et al., 2013; Wang et al., 2011). Briefly, 10–20 millions of MCF7 cells treated with E2, DEX or E2+DEX for 1 hr were washed 3 times with cold PBS and then sequentially swelled in swelling buffer (10mM Tris-HCl pH7.5, 2mM MgCl2, 3mM CaCl2) for 5 min on ice, harvested, and lysed in lysis buffer (swelling buffer plus 0.5% NP-40, 20 units of SUPERase-In, and 10% glycerol). The resultant nuclei were washed two more times with 10ml lysis buffer. For the run-on assay, resuspended nuclei were mixed with an equal volume of reaction buffer (10mM Tris-HCl pH 8.0, 5mM MgCl2, 1mM DTT, 300mM KCl, 20 units of SUPERase-In, 1% sarkosyl, 500 mM ATP, GTP, and Br-UTP, 2 mM CTP) and incubated for 5 min at 30°C. The resultant nuclear-run-on RNA (NRO-RNA) was then extracted with TRIzol LS reagent (Life Technologies) following manufacturer’s instructions. NRO-RNA was fragmented to 300–500nt by alkaline base hydrolysis on ice for 30 min and followed by treatment with DNase I and antarctic phosphatase. At this step, only a small portion of all the RNA species are BrU-labeled. To purify the Br-UTP labeled nascent RNA, the fragmented NRO-RNA was immunoprecipitated with anti-BrdU argarose beads (Santa Cruz Biotechnology) in binding buffer (0.5XSSPE, 1mM EDTA, 0.05% tween) for 1–3 hr at 4°C with rotation. Subsequently, T4 PNK was used to repair the ends of the immunoprecipitated Br-UTP labeled nascent RNA at 37°C for 1 hr. The RNA was extracted and precipitated using acidic phenol-chloroform. cDNA synthesis was performed as per a published method (Ingolia et al., 2009) with few modifications. The RNA fragments were subjected to poly-A tailing reaction by poly-A polymerase (NEB) for 30 min at 37°C. Subsequently, reverse transcription was performed using oNTI223 primer and superscript III RT kit (Life Technologies). The cDNA products were separated on a 10% polyacrylamide TBE-urea gel and only those fragments migrating between 100–500bp were excised and recovered by gel extraction. Next, the first-strand cDNA was circularized by CircLigase (Epicenter) and relinearized by APE1 (NEB). Relinearized single strand cDNA (sscDNA) was separated on a 10% polyacrylamide TBE gel and the appropriately sized product (120–320bp) was excised and gel-extracted. Finally, sscDNA template was amplified by PCR using the Phusion High-Fidelity enzyme (NEB) according to the manufacturer’s instructions. The oligonucleotide primers oNTI200 and oNTI201 were used to generate DNA for deep sequencing (see Table S3 for all GRO-seq primer sequences).
SUMOylation Experiments
HA-GR expression human MCF7 stable cells were seeded onto 10cm plates. After stripping for 72 hours, cells were treated with indicated ligands for 1 hour, and harvested in NP40 lysis buffer with protease inhibitor cocktail (PIC)(Roche) and 20 mM N-ethylmaleimide (Sigma). Endogenous GR was immunoprecipitated with a HA antibody and SUMOylation was detected using SUMO1 and SUMO2/3 antibodies. For in vitro SUMOylation assay, human 293T cells were seeded onto 10cm plates and co-transfected with GR wild type or SUMOlytion mutants, Ubc9 and HA-tagged SUMO-1. After co-treatment with E2+Dex, cells were harvested in NP40 lysis buffer supplemented with protease inhibitor cocktail (PIC) (Roche) and 20 mM N-ethylmaleimide (Sigma), then were analyzed by SDS-PAGE and Western blotting.
Cloning, Mutagenesis, and Generation of Biotin-Tagged Inducible MCF7 Stable Cell Lines
Inducible MCF7 stable cell lines expressing biotin-tagged proteins were generated as previously described (Liu et al., 2014). Briefly, GR wild type and pBox mutant were fused in-frame with the C terminus of the peptide MAGGLNDIFEAQKIEWHEDTGGGGSGGGGSGENLYFQSDYKDDDDK in the BLRP expression retrovirus construct. Then, the retrovirus was transduced into a parental MCF7 stable line that was engineered to stably express BirA and a Tet Repressor. G418 (500 mg/ml), hygromycin (200 mg/ml), and puromycin (0.3 mg/ml) were used for stable selection. Multiple stable cell lines were isolated, treated with doxycycline, and screened for BLRP-tagged protein expression that was similar to the levels of the respective endogenous genes as revealed by immunoblotting with specific antibodies. To induce BLRPtagged protein expression, 2 mg/ml doxycycline was added into culture media approximately 24 hr before hormone treatment and collection. The following primers were used for PCR cloning the full-length GR wild type into the NotI and XbaI sites (underlined) of the pcDNA3.1 expression construct. GR-WT-insertion-F: AAAGCGGCCGCATGGACTCCAAAGAATCATTAAC GR-WT-insertion-R: AAATCTAGATCACTTTTGATGAAACAGAAG The following primers were used for cloning full-length GR at the NotI and MluI sites (underlined) in the BLRP-Retroviral Tet-On vector. GR-BLRP-insertion-F: AAAGCGGCCGCATGGACTCCAAAGAATCATTAAC GR-BLRP-insertion-R: AAAACGCGTTCACTTTTGATGAAACAGAAG.
Mutations of the DNA-binding domains and SUMOylation sites of GR were generated by QuickChange II site-directed mutagenesis(Stratagene) using the following oligonucleotides, with mutated sequence in caps. For the pBox mutation at GR, three amino acids (EGG) in the pBox region (GR, amino acid 439, 440 and 443) were mutated to tryptophans, which blocks nuclear receptor binding to its DNA recognition motif. For sumoylation sites mutation, we mutated N-terminal two sites (Lysine 277, 293), C-terminal 1 site (Lysine 703) to arginines. GR-pBox mutant-F: GTGCTGTCCTTCCACTGCTCTTTTGAAGAACCATTTACACCACCAACAAGTTAAGACTCCATAATGACATCCTGA GR-pBox mutant-R: TCAGGATGTCATTATGGAGTCTTAACTTGTTGGTGGTGTAAATGGTTCTTCAAAAGAGCAGTGGAAGGACAGCAC The GR SUMOlytion mutant fragments finally were cloned into XbaI and NotI sites of FM5 lentivirus expression vector with HA-tag in the N terminal using the following primers. GR -1st SUMOmutant-F: GATGAAATCTTCTTTTTCTGTTCTCACTTGGGGCAGTGTTACATT GR -1st SUMOmutant-R: AATGTAACACTGCCCCAAGTGAGAACAGAAAAAGAAGATTTCATC GR -2ndSUMOmutant-F: CAGTTTCTCTTGCCTAATTACCCCAGGGGTGCAG GR -2ndSUMOmutant-R: CTGCACCCCTGGGGTAATTAGGCAAGAGAAACTG GR -3rdSUMOmutant-F: TGGAGTTTCCTTCCCTCCTGACAATGGCTTTTCCT GR -3rdSUMOmutant-R: AGGAAAAGCCATTGTCAGGAGGGAAGGAAACTCCA The GR DNA binding domain deletion mutant construct was based on GR wild type with the following primers: GR-DBD deletion-F: CTTCAGGATGTCATTATGGAGTCTCTGAAAATCCTGGTAACAAAAC GR-DBD deletion-R: GTTTTGTTACCAGGATTTTCAGAGACTCCATAATGACATCCTGAAG.
Soft Agar Colony Formation Assay
MCF7 cells were suspended in medium containing 1% agar and overlaid on 2% agar in 12-well plates (1000 cells/well), respectively. After 10 days, colonies were counted (size >=50μm) and photographed.
Kaplan-Meier Analyses
Kaplan-Meier estimators (Dinse and Lagakos, 1982)were generated using the online GOBO tool (http://co.bmc.lu.se/gobo) (Ringner et al., 2011). The 465 E2+Dex significantly down-regulated gene set determined by GRO-seq was provided as inputs to assess patient outcomes in ER-positive breast cancer subtypes, and the randomly picked up same size of E2-upregulated genes as control.
Immunofluorescence
After ICI, E2, or E2+Dex treatment for 1h, MCF7 cells grown on cover slip were washed by PBS twice, fixed with 4% paraformaldehyde for 15min and permeabilized by 0.1% Triton X-100 in PBS for 20min at room temperature. Then the cells were blocked by 3% BSA and sequentially incubated with the primary antibody for 1.5hr and the secondary antibody for 1hr at room temperature. Cover slips were mounted by mounting medium with DAPI (Vector Laboratories) and sealed with nail polish before analysis by microscopy.
QUANTIFICATION AND STATISTICAL ANALYSIS
Primary Analysis of ChIP-Seq Data Sets
The sequencing data sample image analysis and base calling were analyzed by Illumina’s Genome Analysis pipeline (http://www.illumina.com/documents/products/datasheets/datasheet_genome_analyzer_software.pdf). The sequencing reads alignment to the human genome (hg18 version) was performed by using Bowtie2 short reads alignment programs (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Only uniquely aligned reads were kept for downstream analysis (if a read aligns to multiple genome locations, only one location is arbitrarily chosen). The multiple reads were collapsed into single read per genomic location only in order to reduce the PCR biases. The aligned reads were used for down-stream peak finding with HOMER (http://homer.salk.edu/homer/motif/).
Identification of ChIP-Seq Peaks, Heatmap and Tag Density Analyses
The identification of ChIP-seq peaks (bound & enriched regions) was performed by using HOMER as previously described ((Li et al., 2015; Liu et al., 2013; Ma and Telese, 2015; Telese et al., 2015). For all the ChIP-seq reads we kept only one tag for each unique genomic position to minimize artifacts due to clonal amplification. Peak finding was conducted by calling the regions that showed enriched tag density (> 4 Folds) compared to the surrounding 10kb regions. This strategy is adopted to minimize duplication or non-localized bindings. Meanwhile, a cumulative p-value of 0.0001 calculated by Poisson distribution was applied to determine statistically significant tag enrichment. All the ChIP-seq samples have a minimum of 10 million uniquely mappable reads. This greatly helped to filter out false positive regions with low tag counts. Significant peaks have a false discovery rate of 0.001 that was calculated using randomized tag positions in a genome with an effective size of 2×109 bp. We customized different parameters to identify TFs or histone marks enriched peaks due to distinct characteristics of tag distribution. For transcription factor/cofactor binding, we applied 1% cutoff of tags in peaks versus total tags as the threshold to determine TFs ChIP-seq samples’ quality. Only peaks passing over this cutoff could to be considered as valid peaks to be used in following steps. For histone marks, we used an initial seed region of 500bp and a false discovery rate of 0.001, a strategy that is more feasible to capture broad region of enrichment characteristic of histone marks. For tag density analyses read counts were collected within a ±500bp window apart from the center of the identified peaks. And ±3000bp windows apart from the center of the identified peaks (±3kb of the peak center) was used for generating read density heatmap and average tag density profile plot analysis, which is visualized by using Java TreeView (http://jtreeview.sourceforge.net/)as described as previous(Wang et al., 2015). The co-bound peaks were identified as those in which the distance between two peaks’ is less than 200bp, based on center of the peaks’ positions. All identified peaks were then associated and annotated using the NCBI Reference Sequence Database (RefSeq). All the annotation information for position of promoters, exons, introns and other regions were described from transcripts and repeat information included in University of California, Santa Cruz database. The NSC and RSC are calculated according to the literatures (Kharchenko et al., 2008; Landt et al., 2012).
Motif Analysis
Motif finding was performed by using algorithms described in HOMER (Heinz et al., 2010). The detection region for TFs motif finding was performed on sequence from ±100 bp relative to the peak center. Sequence logos were plotted by using WebLOGO (http://weblogo.berkeley.edu).
GRO-seq Analysis
For GRO-Seq, the sequencing tags were aligned to hg18 Refseq database by using Bowtie2 and only three tag per genomic location at most were applied as cutoff to get rid of spike enriched regions and clonal amplification. The gene transcription was calculated over the entire gene body by Homer. The tag counts were calculated strand specifically and then plotted as described previously (Li et al., 2013; Skowronska-Krawczyk et al., 2014). The differential gene expression level were calculated by using EdgR (http://www.bioconductor.org/packages/release/bioc/html/edgeR.html) with FDR <0.001. The eRNAs expression were measured by counting tags based on regions that are ±500bp window apart from the center of enhancers binding sites.
Data Visualization
Visualization of the data for ChIP-seq and GRO-seq was performed by organizing custom tracks onto the University of California, Santa Cruz, (UCSC) genome browser using HOMER software package. The total mappable reads for each experiment were normalized to 107 bp to facilitate the comparison between different tracks.
Statistical Analysis
For all qRT-PCRs, data were analyzed and statistics were performed using two-tailed Student’s t test. The results were shown as mean ± SD. Results are representative of at least two independent experiments.
Data and Software Availability
Software
See Key Resources Table.
Data Resources
All the GRO-seq and ChIP-seq data generated in this study have been deposited in the NCBI Gene Expression Omnibus. The accession GEO number is GSE81512. The MegaTrans ChIP-seq data used in this study have been previously deposited under accession numbers GEO: GSE60270.
The Raw image data for all the figures have been deposited at Mendeley with the DOI is 10.17632/p8jjsj4c5w.1.
Supplementary Material
Highlights.
Dex liganded GR inhibits the E2-activated transcriptome.
GR binds ERα-activated enhancers in trans, and is dependent on its SUMOylation.
Liganded GR disassembles the E2-induced MegaTrans complex at ERα-activated enhancers.
The NCoR/SMRT-HDAC3 complex is required for GR binding on ERα-activated enhancers
Acknowledgments
The authors are grateful to Janet Hightower for assistance with figure preparation. M.G.R. is an investigator with the Howard Hughes Medical Institute. This work was supported by NIH grants (DK018477, HL065445, NS034934,DK039949, and CA173903)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
M.G.R. and F.Y., with input from Z. L, conceived the original ideas, designed the project and wrote the manuscript. F.Y. performed the majority of the experiments with participation from Y.T, Y.H. and K.A.O. W.L. performed the GRO-seq experiment. C.J performed all the immunofluorescence assays. J.S performed the soft agar colony formation assay. Q.M. performed all of the bioinformatic analyses. F.T helped to edit the manuscript.
References
- Abduljabbar R, Negm OH, Lai CF, Jerjees DA, Al-Kaabi M, Hamed MR, Tighe PJ, Buluwela L, Mukherjee A, Green AR, et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res Treat. 2015;150:335–346. doi: 10.1007/s10549-015-3335-1. [DOI] [PubMed] [Google Scholar]
- Belikov S, Astrand C, Wrange O. FoxA1 binding directs chromatin structure and the functional response of a glucocorticoid receptor-regulated promoter. Mol Cell Biol. 2009;29:5413–5425. doi: 10.1128/MCB.00368-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belikov S, Holmqvist PH, Astrand C, Wrange O. FoxA1 and glucocorticoid receptor crosstalk via histone H4K16 acetylation at a hormone regulated enhancer. Exp Cell Res. 2012;318:61–74. doi: 10.1016/j.yexcr.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, Rogatsky I. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012;109:11776–11781. doi: 10.1073/pnas.1206059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- Druker J, Liberman AC, Antunica-Noguerol M, Gerez J, Paez-Pereda M, Rein T, Iniguez-Lluhi JA, Holsboer F, Arzt E. RSUME enhances glucocorticoid receptor SUMOylation and transcriptional activity. Mol Cell Biol. 2013;33:2116–2127. doi: 10.1128/MCB.01470-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene-and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song WC, DeFranco DB, Cheng SY, et al. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386–7393. doi: 10.1158/0008-5472.CAN-08-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R, Muse GW, Chinenov Y, Adelman K, Rogatsky I. Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proc Natl Acad Sci U S A. 2013;110:14616–14621. doi: 10.1073/pnas.1309898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Holmstrom S, Van Antwerp ME, Iniguez-Lluhi JA. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc Natl Acad Sci U S A. 2003;100:15758–15763. doi: 10.1073/pnas.2136933100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Ganti KP, Chambon P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc Natl Acad Sci U S A. 2016a;113:E635–643. doi: 10.1073/pnas.1522826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Paulen L, Chambon P. GR SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci U S A. 2016b;113:E626–634. doi: 10.1073/pnas.1522821113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kach J, Conzen SD, Szmulewitz RZ. Targeting the glucocorticoid receptor in breast and prostate cancers. Sci Transl Med. 2015;7:305ps319. doi: 10.1126/scitranslmed.aac7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S, Jin Y, Nagaich AK. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J Biol Chem. 2013;288:24020–24034. doi: 10.1074/jbc.M113.473819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki SH, Cho IJ, Choi DW, Kim SG. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol. 2005;25:4150–4165. doi: 10.1128/MCB.25.10.4150-4165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hu Y, Oh S, Ma Q, Merkurjev D, Song X, Zhou X, Liu Z, Tanasa B, He X, et al. Condensin I and II Complexes License Full Estrogen Receptor alpha-Dependent Enhancer Activation. Mol Cell. 2015;59:188–202. doi: 10.1016/j.molcel.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225–240. doi: 10.1146/annurev-physiol-030212-183708. [DOI] [PubMed] [Google Scholar]
- Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014;159:358–373. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grontved L, Schiltz RL, Hager GL. Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res. 2013;73:5130–5139. doi: 10.1158/0008-5472.CAN-13-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, Taipaleenmaki H, Ben-Batalla I, Scheller M, Loges S, et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014;8:460–469. doi: 10.1016/j.celrep.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Szanto A, Szatmari I, Szeles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–789. doi: 10.1152/physrev.00004.2011. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakinaho V, Kaikkonen S, Makkonen H, Benes V, Palvimo JJ. SUMOylation regulates the chromatin occupancy and anti-proliferative gene programs of glucocorticoid receptor. Nucleic Acids Res. 2014;42:1575–1592. doi: 10.1093/nar/gkt1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71:6360–6370. doi: 10.1158/0008-5472.CAN-11-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo J, Verger A, Turner J, Crossley M. Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol Cell Biol. 2005;25:1549–1559. doi: 10.1128/MCB.25.4.1549-1559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci U S A. 2002;99:16701–16706. doi: 10.1073/pnas.262671599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2003;100:13845–13850. doi: 10.1073/pnas.2336092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever R, Glass CK. Signaling by nuclear receptors. Cold Spring Harb Perspect Biol. 2013;5:a016709. doi: 10.1101/cshperspect.a016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hubner N, Evans RM. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49:158–171. doi: 10.1016/j.molcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson LM, Parini P, Janne OA, Gustafsson JA, et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010;24:381–395. doi: 10.1101/gad.545110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell. 2004;13:611–617. doi: 10.1016/s1097-2765(04)00060-7. [DOI] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. doi: 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinse GE, Lagakos SW. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics. 1982;38:921–932. [PubMed] [Google Scholar]
- Furlan-Magaril M, Rincon-Arano H, Recillas-Targa F. Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. Methods Mol Biol. 2009;543:253–266. doi: 10.1007/978-1-60327-015-1_17. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hu Y, Oh S, Ma Q, Merkurjev D, Song X, Zhou X, Liu Z, Tanasa B, He X, et al. Condensin I and II Complexes License Full Estrogen Receptor alpha-Dependent Enhancer Activation. Mol Cell. 2015;59:188–202. doi: 10.1016/j.molcel.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal AK, Rosenfeld MG. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155:1581–1595. doi: 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014;159:358–373. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Telese F. Genome-wide epigenetic analysis of MEF2A and MEF2C transcription factors in mouse cortical neurons. Commun Integr Biol. 2015;8:e1087624. doi: 10.1080/19420889.2015.1087624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 2011;6:e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, Carroll JS. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010;24:171–182. doi: 10.1101/gad.552910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronska-Krawczyk D, Ma Q, Schwartz M, Scully K, Li W, Liu Z, Taylor H, Tollkuhn J, Ohgi KA, Notani D, et al. Required enhancer-matrin-3 network interactions for a homeodomain transcription program. Nature. 2014;514:257–261. doi: 10.1038/nature13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telese F, Ma Q, Perez PM, Notani D, Oh S, Li W, Comoletti D, Ohgi KA, Taylor H, Rosenfeld MG. LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation. Neuron. 2015;86:696–710. doi: 10.1016/j.neuron.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DM, Won KJ, Nguyen N, Lazar MA, Chen CS, Steger DJ. ATF4 licenses C/EBPbeta activity in human mesenchymal stem cells primed for adipogenesis. Elife. 2015;4:e06821. doi: 10.7554/eLife.06821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing D, Bhadri VA, Beck D, Thoms JA, Yakob NA, Wong JW, Knezevic K, Pimanda JE, Lock RB. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood. 2015;125:273–283. doi: 10.1182/blood-2014-05-576470. [DOI] [PubMed] [Google Scholar]
- Luca F, Maranville JC, Richards AL, Witonsky DB, Stephens M, Di Rienzo A. Genetic, functional and molecular features of glucocorticoid receptor binding. PLoS One. 2013;8:e61654. doi: 10.1371/journal.pone.0061654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao NA, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, Kolisis FN, Alexis MN, Mitsiou DJ, Stunnenberg HG. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011;21:1404–1416. doi: 10.1101/gr.118042.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, Janne OA. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–1580. doi: 10.1158/0008-5472.CAN-12-2350. [DOI] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell. 2016;165:593–605. doi: 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AL, Coarfa C, Qian J, Wilkerson JJ, Rajapakshe K, Krett NL, Gunaratne PH, Rosen ST. Identification of potential glucocorticoid receptor therapeutic targets in multiple myeloma. Nucl Recept Signal. 2015;13:e006. doi: 10.1621/nrs.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.