

Remodeling of tumor microenvironments enables the enhanced delivery of nanoparticles (NPs). Here we show that the direct priming of a tumor tissue using photosensitization rapidly activates neutrophil infiltration that mediates the delivery of nanotherapeutics into the tumor. We develop a drug delivery platform comprised of NPs coated with anti-CD11b antibodies (Abs) that target activated neutrophils. Intravital microscopy demonstrates that the movement of anti-CD11b Abs-decorated NPs (NPs-CD11b) into the tumor is mediated by neutrophil infiltration induced by photosensitization (PS) because the systemic depletion of neutrophils completely abolishes the nanoparticle tumor deposition. The neutrophil uptake of NPs does not alter neutrophil activation and transmigration. For cancer therapy in mice, tumor PS and photothermal therapy of anti-CD11b Abs-linked gold nanorods (GNRs-CD11b) are combined to treat the carcinoma tumor. The result indicates that neutrophil tumor infiltration enhances nanoparticle cancer therapy. The findings reveal that promoting tumor infiltration of neutrophils by manipulating tumor microenvironments could be a novel strategy to actively deliver nanotherapeutics in cancer therapies.
The innovations in synthesis and bioengineering of nanoparticles (NPs) for targeted drug delivery have had an impact in cancer therapies.[1] The basis of nanoparticle cancer therapies is mainly that tumor vasculature is more permeable than healthy tissues, thus increasing the nanoparticle accumulation in tumors.[2] To further improve the delivery of NPs, priming tumor microenvironments has been exploited, such as normalization of tumor vasculature for enhanced tumor penetration of NPs[3] and upregulating of the P-selectin in a tumor to increase the targeting of NPs.[4] Here, we report a strategy for the direct priming of the tumor tissue using photosensitization that activates neutrophil infiltration to mediate the transport of NPs across the tumor vessel barrier.
Neutrophils are the most abundant circulating leukocytes and are refreshed from the bone marrow. In humans, daily production of neutrophils is 2 × 1011 and they comprise 50 ~ 70% of circulating leukocytes.[5] Importantly, neutrophils play a central role in innate immune responses to infections (bacteria or viruses) or tissue damage to protect homeostasis. Neutrophils are typically the first leukocytes to be recruited to an inflammatory site via the migration across the blood vessel barrier.[6] During acute inflammation, neutrophils in the peripheral blood highly express CD11b (integrin αM, called Mac-1), a marker of neutrophil activation.[6]
Photosensitization (PS) is a process in which a photosensitizer absorbs the visible light and then converts molecular oxygen to a range of reactive oxygen species (ROS).[7] ROS can induce acute inflammation that rapidly activates neutrophil tissue transmigration.[8] We therefore reasoned that creating acute inflammation in tumor tissues by PS could enable the tumor infiltration of neutrophils that transport anti-CD11b antibodies (Abs) coated NPs (NPs-CD11b) into the tumor after the internalization of the NPs by neutrophils. We evaluated this hypothesis using intravital microscopy of tumors, design of NPs and photothermal cancer therapy.
CD11b highly express on the surface of activated neutrophils, so we decorated anti-CD11b Abs to fluorescent polystyrene NPs via biotin-neutravidin binding.[9] (Figure 1A and S1). To address the specificity of anti-CD11b-linked NPs (NPs-CD11b) binding to neutrophils, we investigated a wide range of nanoparticle surface modifications, finding that their sizes were similar and the size range was 200 ~ 250 nm in diameter (Figure S1). We hypothesize that neutrophils are activated after tumor PS produced by the combination of light (660 nm) and pyropheophorbide-a (Ppa).[10] The NPs-CD11b are then internalized by activated neutrophils in blood after intravenously (i.v.) injected. Finally, nanoparticle-laden neutrophils will infiltrate the tumor (Figure 1B). To illustrate this concept, we have established intravital microscopy of a mouse flank tumor model using a laser resonance scanning confocal microscope.[11, 12] 1 h after i.v. injection of Ppa in a tumor bearing mouse, the tumor was exposed to the laser at 660 nm for 45 seconds to generate acute inflammation. 0.75 h later, yellow-green fluorescent NPs-CD11b or PEG coated NPs (NPs-PEG) (Figure S1) were i.v. injected. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (a specific marker of mouse neutrophils) was subcutaneously (s.c.) injected around the tumor to stain neutrophils, and Cy3-BSA was i.v. administered to visualize blood vessels. We found that some neutrophils containing NPs-CD11b accumulated outside of blood vessels, suggesting that neutrophils were able to transport the NPs across blood vessel wall (Figure 1C and Movie 1) into tumor tissues. Some extracellular NPs might be due to the release from neutrophils (Figure 1C and Movie 1). In contrast, we did not observe the existence of NPs-PEG in the inflamed tumor (Figure 1D and Movie 2). Without PS, we found neither NPs-CD11b nor neutrophils in the tumor, indicating that the transport of the NPs is associated with PS-induced neutrophil infiltration (Figure 1E and Movie 3). To further investigate this, we specifically depleted neutrophils in the mouse using anti-LY-6G (Figure S2) before both the treatments of PS and injection of NPs-CD11b (Figure 1F and Movie 4). The result indicated that the depletion of neutrophils completely abolished the transport of NPs into the tumor site, clearly illustrating that the delivery of NPs into the tumor is mediated by neutrophil infiltration induced by PS. Furthermore, neutrophils were isolated from the tumors for confocal imaging. The confocal images showed that NPs-CD11b were internalized in neutrophils (Figure 1G), but no NPs-PEG were seen in neutrophils (Figure S3).
Figure 1.




Neutrophils mediate delivery of nanoparticles (NPs) across blood vessel barrier into the tumor tissues after acute inflammation induced by photosensitization (PS). (a) Scheme of surface modifications of NPs with PEG and antibodies (Abs). (b) The concept of neutrophil-mediated delivery of NPs to tumor tissues inflamed by PS. Intravital microscopic 3D images of mouse tumor treated with (c) both PS and NPs-CD11b (green), (d) PS and NPs-PEG (green), (e) NPs-CD11b only and (f) PS and NPs-CD11b after neutrophil depletion. The experimental protocol of intravital microcopy of tumors is illustrated above figure c. The tumor was treated with PS (intravenous (i.v.) injection of Pyropheophorbide-a (Ppa) and 660 nm laser). 0.75 h later, 1 mg/kg of NPs were i.v. administrated. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was subcutaneously (s.c.) injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel. To deplete neutrophils, anti-LY-6G Abs were intraperitoneally (i.p.) injected 24 h before the administration of Ppa. (g) Fluorescence confocal microscopy of neutrophils purified from tumors after treated with PS and NPs-CD11b (green). Neutrophils were purified from tumor 3 h after the injection of NPs and labeled by Alexa Fluor 647-labeled anti-mouse LY-6G Abs (pink). Nucleus were stained by DAPI (blue). (h) Accumulation of NPs in tumor tissues after mice were treated with 1 mg/kg of NPs-CD11b, PS/NPs-PEG, PS/NPs-CD11b, and PS/NPs-CD11b after neutrophil depletion using anti-LY-6G Abs. The data represent mean ± SD (n = 6 mice per group). The tumor was removed from the mice 24 h after the administration of NPs. The tumor was homogenized and the concentrations of fluorescent NPs in the homogenate were measured by fluorescence of NPs.
To quantitatively analyze the neutrophil infiltration-mediated delivery of NPs in tumor, we measured the concentrations of NPs in tumor after they were resected and homogenized. It was found that NPs-CD11b accumulated in inflamed tumors by 35 times greater than NPs-PEG (Figure 1H). The tumor accumulation of NPs-CD11b in both the normal tumor and the inflamed tumor after systemic neutrophil depletion (Figure 1H) is similar to that of NPs-PEG in inflamed tumors. The results showed that the neutrophil infiltration was responsible for actively moving NPs-CD11b into tumor sites.
To address the molecular mechanism of neutrophil infiltration to transport NPs, we isolated neutrophils in the tumor and quantified nanoparticle uptake in neutrophils. The neutravidin labeled NPs were successfully decorated with biotinylated mouse IgG, rat IgG and rat anti-CD11b Abs and NPs were coated with PEG via carboxylic acid (Figure S1). Neutrophils isolated from tumor tissues were stained with anti-LY-6G Abs and analyzed by flow cytometry. In the acutely inflamed tumor model, nearly 50% of neutrophils contained NPs-CD11b, but it was less than 5% of neutrophils internalizing PEG, mouse IgG or rat IgG coated NPs (Figures 2A, 2B and S4). The mean fluorescence index (MFI) per neutrophil is an indicator of nanoparticle uptake efficiency. The result indicated that MFI for NPs-CD11b increased ~17–130 folds compared with those of rat IgG and mouse IgG coated NPs and NPs-PEG, showing that anti-CD11b Abs are required to mediate the nanoparticle uptake by neutrophils (Figure 2C).
Figure 2.


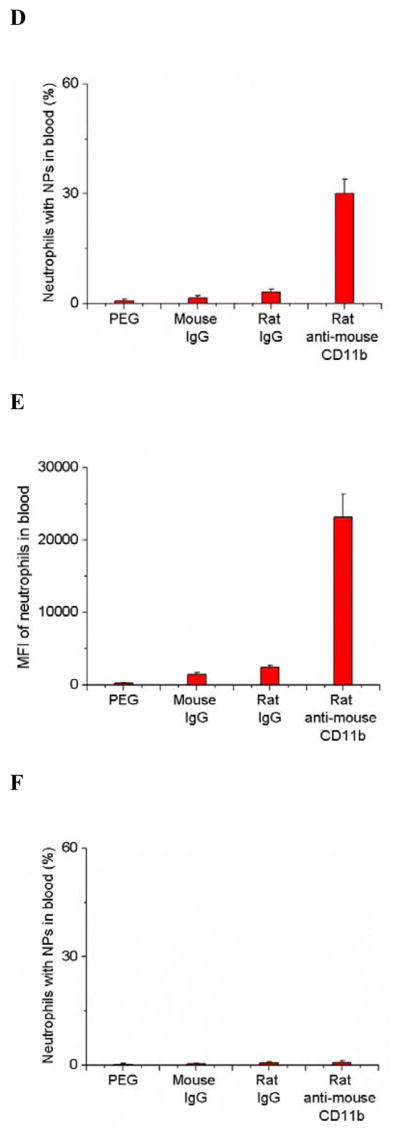
Anti-CD11b coating of NPs mediates their uptake by activated neutrophils. (a) Flow cytometry patterns, (b) percentage and (c) mean fluorescence index (MFI) of neutrophils internalizing NPs in mouse tumor treated with PS after the i.v. injection of PEG, mouse IgG, rat IgG and rat anti-mouse CD11b Abs coated NPs, respectively. (d) Percentage and (e) MFI of neutrophils internalizing NPs in mouse blood after tumor was treated with PS and various NPs. (f) Percentage of neutrophils internalizing NPs in mouse blood without tumor PS. All experiments were performed as below. 1 h after the administration of Ppa, the tumor was exposed to 660 nm laser for 45 seconds. 0.75 h after the laser irradiation, 1 mg/kg of the NPs were i.v. injected. Neutrophils were purified from tumors and blood 3 h after the injection of NPs and were labeled by Alexa Fluor 647-labeled anti-mouse LY-6G Abs for flow cytometry. All data represent mean ± SD (at least 3 mice per group).
Since NPs were systemically administrated to a mouse, we investigated the neutrophil uptake of NPs in the blood circulation. The data (Figures 2D, 2E and S5) suggested that NPs-CD11b were internalized by activated neutrophils in blood. To address whether the activation of neutrophils is required to internalize NPs-CD11b in blood, we administrated various NPs in the mice without tumor PS. We found that < 1% of resting neutrophils internalized NPs-CD11b, and this uptake was independent of anti-CD11b Abs as it was similar to the uptake of other surface modified NPs (Figure 2F). The results (Figure 2) suggest that the tumor PS promoted the activation of neutrophils in blood that induced NPs-CD11b uptake in neutrophils and actively delivered NPs into tumor sites. This is consistent with the results of intravital microscopy of tumors (Figure 1C–1F). Furthermore, we addressed whether monocytes, natural killer (NK) cells and T cells also internalized NPs-CD11b since CD11b protein is expressed on those cells.[13] Interestingly, it is unlikely that the immune cells took up NPs-CD11b because we observed 1–3% of the immune cells contained NPs compared to efficient nanoparticle uptake by activated neutrophils (Figure S6). The specificity of neutrophil uptake of NPs might be associated with the activation of neutrophils earlier than other immune cells in acute inflammation. When neutrophils were depleted by antibodies, the tumor deposition of NPs-CD11b is abolished (Figure 1H). This supports that other immune cells unlikely take up NPs-CD11b observed in Figure S6.
To address whether nanoparticle uptake could change the movement and activation of neutrophils, we measured neutrophil transmigration and cytokine release in two animal models. In the tumor model, we found that nanoparticle uptake (NPs-CD11b) did not alter the ratio of neutrophil/tumor cells in inflamed tumors compared with the administration of NPs-PEG that were not internalized by neutrophils (Figure 3A). The results were also comparable with the control (PBS treatment). Uptake of NPs did not affect the cytokine levels either in the tumor or blood, such as IL-6 and TNF-α (Figures 3B to 3E).
Figure 3.

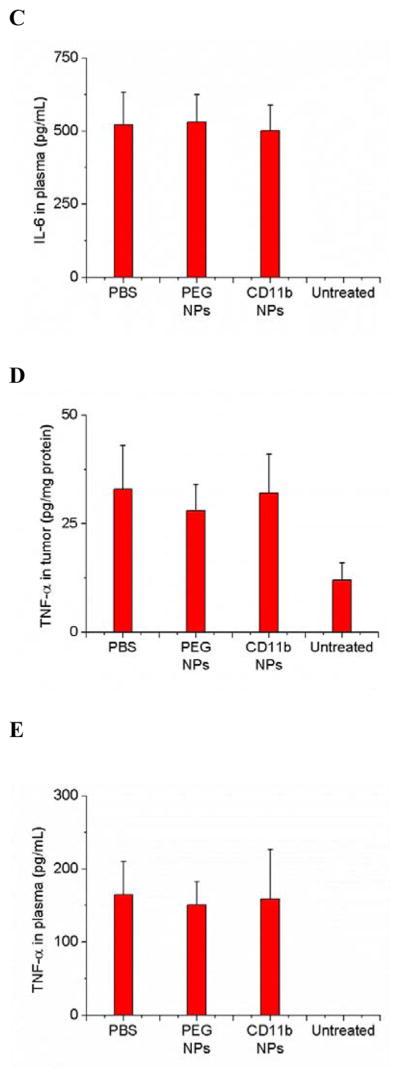
Neutrophil uptake of NPs does not alter neutrophil infiltration into tumors and does not promote neutrophil activation. (a) Ratio of neutrophils/tumor cells, concentrations of IL-6 in (b) tumor and (c) plasma and concentrations of TNF- α in (d) tumor and (e) plasma 3 h after the injection of PBS (pH 7.4), NPs-PEG and NPs-CD11b, respectively. All experiments were conducted as below. 1 h after the administration of Ppa, the tumor was exposed to 660 nm laser for 45 seconds. 0.75 h after the laser irradiation, PBS (pH 7.4) or 1 mg/kg of NPs were i.v. injected. For untreated group, only PBS (pH 7.4) was i.v. injected without the treatment of PS. All data represent mean ± SD (at least 3 mice per group).
It is difficult to quantitatively analyze the transmigration ability of neutrophils after NPs are internalized in neutrophils using the tumor model, so we developed LPS-induced acute lung inflammation mainly regulated by neutrophils.[11] The lung has the unique features with many tiny air sacs (called alveoli) surrounded by blood capillaries to form an interface between circulation and airspace.[14] When acute inflammation occurs in the airspace, neutrophils are capable of migrating from bloodstream to alveoli, where extravascular neutrophils can be easily harvested and investigated.[15] Therefore, the mouse model of acute lung inflammation is a unique system to quantitatively address whether neutrophil uptake of NPs changes neutrophil transmigration (Figure S7A). 4 h after intra-tracheal administration of lipopolysaccharide (LPS) to the lung, we i.v. injected NPs-PEG and NPs-CD11b to the mice, respectively. 20 h after injection of NPs, we collected lung bronchoalveolar lavage fluid (BALF). The cells were isolated and stained with anti-LY-6G Abs to mark neutrophils. To determine the percentage of lung infiltrated neutrophils which have internalized NPs and their MFI, we performed the flow cytometry to analyze the neutrophils in BALF. ~56% of neutrophils in BALF was detected containing NPs-CD11b while only ~1.5% of neutrophils with NPs-PEG (Figures S7B and S7C). The MFI of NPs-CD11b group increased by about 120 times compared with that of NPs-PEG group (Figure S7D). The confocal imaging further confirmed that transmigrated neutrophils internalized NPs-CD11b but not NPs-PEG (Figures S8 and S9). Furthermore, NPs-CD11b did not affect the number of transmigrated neutrophils, cytokine release and vascular permeability (protein concentrations) in BALF compared with PBS and NPs-PEG groups after the LPS challenge (Figure S10). The data indicated that neutrophils containing NPs can biologically function to transmigrate in a deep tissue, and specifically deliver NPs-CD11b into a target.
To demonstrate the usefulness of CD11b-decorated NPs for cancer treatment, we chose gold nanorods (GNRs) as nanotherapeutics. GNRs have been widely investigated for the laser-induced photothermal therapy of cancers.[16] The CD11b-decorated GNRs (GNRs-CD11b) were similarly prepared using biotinylated CD11b Abs and neutravidin coated GNRs as mentioned above for those Abs coated fluorescent NPs (Figure S11A–C). We investigated the biodistribution of GNRs in tumor mice. 0.75 h after the tumor PS, commercially available PEG coated GNRs (GNRs-PEG) or GNRs-CD11b were i.v. injected. The tumor accumulation of GNRs-CD11b was about 20 times higher than that of GNRs-PEG (Figure 4A). When the neutrophils were depleted in mice using anti-LY-6G Abs, the GNRs-CD11b in tumor went down to the level of GNRs-PEG. The similar low tumor deposition of GNRs-CD11b or GNRs-PEG was observed when the mouse tumors were not photosensitized (Figure 4A). Collectively, the results indicated that the accumulation of GNRs-CD11b in tumors was mediated by neutrophils after PS, consistent with the results demonstrated in Figures 1 and 2.
Figure 4.

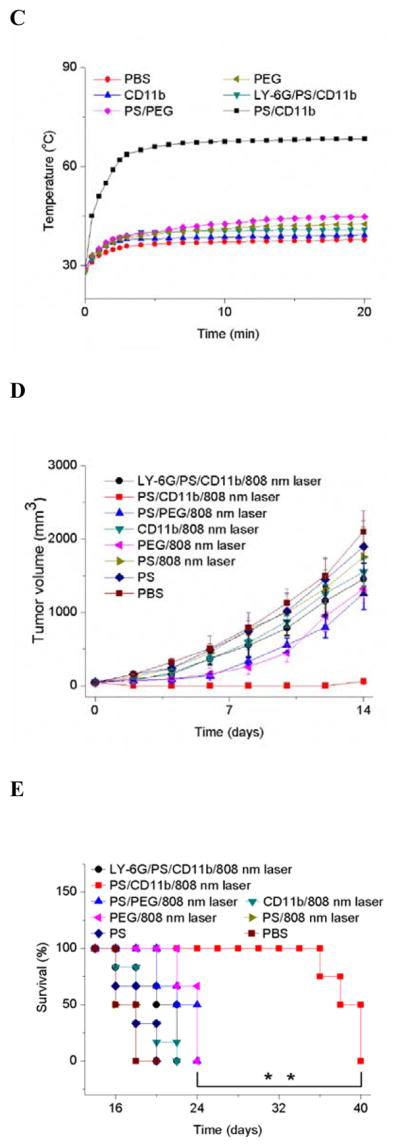
PS mediates the tumor accumulation of anti-CD11b-linked GNRs that significantly diminish the tumor growth and increase the mouse survival after photothermal therapy. The diagram shows the experimental protocol of biodistribution of NPs and photothermal therapies using a laser at 808 nm. Neutrophil depletion with Ly-6G Abs was conducted 24 h before the administration of Ppa. (a) Biodistribution of GNRs in mice bearing tumor treated with PS and NPs-CD11b, PS and NPs-PEG, NPs-PEG only, NPs-CD11b only, PS and NPs-CD11b after neutrophil depletion. Biological samples were collected 24 h after the administration of GNRs. Data represent mean ± SD (n = 6 mice per group). Thermal graphic imaging of mouse tumors (b) and their highest temperature (c) after the tumors were irradiated at 808 nm for 20 min. The mice were i.v. administrated with GNRs-PEG and GNRs-CD11b under either tumor PS or without PS (denoted as PS/PEG, PS/CD11b, PEG and CD11b, respectively), or with both PS and GNRs-CD11b after neutrophil depletion using anti-LY-6G (denoted as LY-6G/PS/CD11b). For the PBS group, the tumor was not treated with PS and only PBS (pH 7.4) was injected with the irradiation at 808 nm. (d) Tumor size and (e) survival rate of the mice bearing tumor irradiated with 808 nm laser after the injection of GNRs-PEG and GNRs-CD11b with or without PS (denoted as PS/PEG, PS/CD11b, PEG and CD11b, respectively), or irradiated with 808 nm laser after the treatment of both PS and GNRs-CD11b after neutrophil depletion (denoted as LY-6G/PS/CD11b). For PBS (without PS and irradiation of laser at 808 nm), PS (with PS but without irradiation of laser at 808 nm) and PS/808 nm laser (with both PS and irradiation of laser at 808nm), only PBS (pH 7.4) was injected. The dose was 4 mg/kg for GNRs-PEG and GNRs-CD11b. Data represent mean ± SD (n = 6 mice per group). Significance of survival rates between treatment groups (**p < 0.01) was assessed by a log-rank (Mantel–Cox) survival analysis test with 95% CIs using GraphPad Prism software (GraphPad Software, Inc., CA).
24 h after i.v. administration of GNRs-PEG or GNRs-CD11b to the tumor bearing mice which have been treated with PS, the tumor was irradiated with 808 nm laser for 20 min. The temperature in the tumor quickly increased to 68 °C when GNRs-CD11b were administrated while it was 45 °C for GNRs-PEG (Figure 4B and 4C). However, when the neutrophils were depleted using anti-LY-6G Abs, the temperature was 41 °C. In the mice without PS, the temperatures on tumor were similar for PBS, GNRs-CD11b and GNRs-PEG after the irradiation with 808 nm laser. The marked temperature increases of the tumor treated with both PS and GNRs-CD11b indicated the GNRs tumor deposition was mediated by neutrophil infiltration induced by PS. The rapid increase of local heat in tumor induced by GNRs would destroy the tumor, so we investigated the therapeutic effect of GNRs in a mouse flank tumor model. We performed a range of experiments, such as, two types of GNRs (NPs-PEG and GNRs-CD11b), tumor manipulation (with or without PS) and neutrophil depletion. After the mice were treated with both tumor PS and GNRs-CD11b, tumor was unmeasurable from day 2 to 12 (Figure 4D) and no mice died within 36 days (Figure 4E). However, in all other control groups, the growth of tumor was not suppressed and all the mice died within 24 days after the various treatments (Figure 4D and 4E). Most importantly, when neutrophils were systemically depleted using anti-LY-6G Abs, the anti-cancer effect using both tumor PS and GNRs-CD11b disappeared. The result further confirmed our hypothesis that tumor PS increases tumor deposition of GNRs mediated by neutrophil infiltration, thus improving photothermal therapy. Moreover, the combination treatments with PS, neutrophil depletion, GNRs and photothermal therapy do not affect mouse weights over the whole period of experiments (Figure S12), indicating mice are tolerant to the therapy of neutrophil-mediated delivery of NPs developed here.
We have demonstrated a new strategy to actively promote the neutrophil movement to tumor sites by PS to deliver nanotherapeutics for cancer therapies. Our versatile design on nanoparticle bio-conjugation and the abilities to manipulate tumor microenvironments[17] allow the rational designs of drug delivery platforms to advance current cancer therapies. Importantly, our studies reveal targeting neutrophils in vivo to actively deliver nanotherapeutics might be a novel approach in cancer therapies.
Supplementary Material
Move 1. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-CD11b (green). Alexa Fluor-647-labeled anti-LY-6G Abs (pink) was subcutaneously (s.c.) injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 2. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-PEG (green). Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 3. Reconstruction of intravital microscopic 3D images of mouse tumor treated with NPs-CD11b (green) only. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 4. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-CD11b after neutrophil depletion using anti-LY-6G Abs. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Acknowledgments
This work was supported by National Institute of Health grants K25HL111157 and RO1GM116823 to Z.W.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Dafeng Chu, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States
Xinyue Dong, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
Prof. Qi Zhao, Faculty of Health Sciences, University of Macau, Taipa, Macau, China
Prof. Jingkai Gu, Research Center for Drug Metabolism, Jilin University, Changchun, Jilin 130021, China
Prof. Zhenjia Wang, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States
References
- 1.Davis ME, Chen ZG, Shin DM. Nature reviews Drug discovery. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]; Petros RA, DeSimone JM. Nature reviews Drug discovery. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]; Shi J, Kantoff PW, Wooster R, Farokhzad OC. Nature reviews Cancer. 2017;17:20. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]; Torchilin VP. Nature reviews Drug discovery. 2014;13:813. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Cancer research. 2013;73:2412. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Nat Nanotechnol. 2012;7:383. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang W, Huang YH, An Y, Kim BYS. Acs Nano. 2015;9:8689. doi: 10.1021/acsnano.5b02028. [DOI] [PubMed] [Google Scholar]; Khawar IA, Kim JH, Kuh HJ. Journal of controlled release: official journal of the Controlled Release Society. 2015;201:78. doi: 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 4.Shamay Y, Elkabets M, Li H, Shah J, Brook S, Wang F, Adler K, Baut E, Scaltriti M, Jena PV, Gardner EE, Poirier JT, Rudin CM, Baselga J, Haimovitz-Friedman A, Heller DA. Science translational medicine. 2016;8:345ra87. doi: 10.1126/scitranslmed.aaf7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borregaard N. Immunity. 2010;33:657. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]; Mestas J, Hughes CC. Journal of immunology. 2004;172:2731. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 6.Kolaczkowska E, Kubes P. Nature reviews Immunology. 2013;13:159. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]; Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Nature reviews Immunology. 2007;7:678. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]; Mantovani A, Cassatella MA, Costantini C, Jaillon S. Nature reviews Immunology. 2011;11:519. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]; Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Trends in immunology. 2011;32:461. doi: 10.1016/j.it.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L, Wang WC, Unger E, Henderson BW. British journal of cancer. 2003;88:1772. doi: 10.1038/sj.bjc.6600864. [DOI] [PMC free article] [PubMed] [Google Scholar]; Firczuk M, Nowis D, Golab J. Photochemical & photobiological sciences: Official journal of the European Photochemistry Association and the European Society for Photobiology. 2011;10:653. doi: 10.1039/c0pp00308e. [DOI] [PubMed] [Google Scholar]; Castano AP, Mroz P, Hamblin MR. Nature reviews Cancer. 2006;6:535. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]; Korbelik M. Lasers in surgery and medicine. 2006;38:500. doi: 10.1002/lsm.20337. [DOI] [PubMed] [Google Scholar]
- 8.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Antioxidants & redox signaling. 2014;20:1126. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sellak H, Franzini E, Hakim J, Pasquier C. Blood. 1994;83:2669. [PubMed] [Google Scholar]; Marriott HM, Jackson LE, Wilkinson TS, Simpson AJ, Mitchell TJ, Buttle DJ, Cross SS, Ince PG, Hellewell PG, Whyte MK, Dockrell DH. American journal of respiratory and critical care medicine. 2008;177:887. doi: 10.1164/rccm.200707-990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi H, Sakahara H, Endo K, Hosono M, Yao ZS, Toyama S, Konishi J. Japanese journal of cancer research: Gann. 1995;86:310. doi: 10.1111/j.1349-7006.1995.tb03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rapozzi V, Zorzet S, Zacchigna M, Drioli S, Xodo LE. Investigational new drugs. 2013;31:192. doi: 10.1007/s10637-012-9844-4. [DOI] [PubMed] [Google Scholar]
- 11.Chu D, Gao J, Wang Z. ACS nano. 2015;9:11800. doi: 10.1021/acsnano.5b05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Li J, Cho J, Malik AB. Nature nanotechnology. 2014;9:204. doi: 10.1038/nnano.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muto S, Vetvicka V, Ross GD. Journal of clinical immunology. 1993;13:175. doi: 10.1007/BF00919970. [DOI] [PubMed] [Google Scholar]
- 14.Ware LB, Matthay MA. N Engl J Med. 2005;353:2788. doi: 10.1056/NEJMcp052699. [DOI] [PubMed] [Google Scholar]
- 15.Matthay MA, Ware LB, Zimmerman GA. The Journal of clinical investigation. 2012;122:2731. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]; Matthay MA, Zemans RL. Annu Rev Pathol. 2011;6:147. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shanmugam V, Selvakumar S, Yeh CS. Chemical Society reviews. 2014;43:6254. doi: 10.1039/c4cs00011k. [DOI] [PubMed] [Google Scholar]; Zhang P, Hu C, Ran W, Meng J, Yin Q, Li Y. Theranostics. 2016;6:948. doi: 10.7150/thno.15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeshima T, Pop LM, Laine A, Iyengar P, Vitetta ES, Hannan R. Proc Natl Acad Sci U S A. 2016;113:11300. doi: 10.1073/pnas.1613187113. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. Nat Nanotechnol. 2016;11:295. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Move 1. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-CD11b (green). Alexa Fluor-647-labeled anti-LY-6G Abs (pink) was subcutaneously (s.c.) injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 2. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-PEG (green). Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 3. Reconstruction of intravital microscopic 3D images of mouse tumor treated with NPs-CD11b (green) only. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.
Movie 4. Reconstruction of intravital microscopic 3D images of mouse tumor treated with both PS and NPs-CD11b after neutrophil depletion using anti-LY-6G Abs. Alexa Fluor-647-labeled anti-mouse LY-6G Abs (pink) was s.c. injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel.