

Abstract
Background
Pemphigus foliaceus (PF) is an autoimmune blistering disease caused by autoantibodies (Abs) against desmoglein 1 (Dsg1). PF sera contain polyclonal Abs which are heterogeneous mixture of both pathogenic and non-pathogenic Abs, as shown by isolation of monoclonal Abs (mAbs).
Objective
To investigate how pathogenic and non-pathogenic anti-Dsg1 Abs contribute to blister formation in PF.
Methods
Using organ-cultured human skin, we compared the effect of a single pathogenic anti-Dsg1 IgG mAb, a single non-pathogenic anti-Dsg1 IgG mAb, and their mixture on blister formation as analyzed by histology, subcellular localization of IgG deposits and desmosomal proteins by confocal microscopy, and desmosomal structure by electron microscopy. In addition, we measured keratinocyte adhesion by an in vitro dissociation assay.
Results
24 h after injection, a single pathogenic anti-Dsg1 IgG caused a subcorneal blister with IgG and Dsg1 localized linearly on the cell surface of keratinocytes. A single non-pathogenic anti-Dsg1 IgG bound linearly on the keratinocytes but did not induce blisters. A pathogenic and a non-pathogenic IgG mAb injected together caused an aberrant granular pattern of IgG and Dsg1 in the lower epidermis with blister formation in the superficial epidermis. Electron microscopy demonstrated that the mixture of mAbs shortened desmosomal lengths more than a single mAb in the basal and spinous layers. Furthermore, although Dsg1 clustering required both cross-linking of Dsg1 molecules by the non-pathogenic IgG plus a pathogenic antibody, the latter could be in the form of a monovalent single chain variable fragment, suggesting that loss of trans-interaction of Dsg1 is required for clustering. Finally, a p38MAPK inhibitor blocked Dsg1 clustering. When pathogenic strength was measured by the dissociation assay, a mixture of pathogenic and non-pathogenic IgG mAbs disrupted keratinocyte adhesion more than a single pathogenic mAb. This pathogenic effect was only partially suppressed by the p38MAPK inhibitor.
Conclusion
These findings indicate that a polyclonal mixture of anti-Dsg1 IgG antibodies enhances pathogenic activity for blister formation associated with p38MAPK-dependent Dsg1 clustering and that not only pathogenic antibodies but also non-pathogenic antibodies coordinately contribute to blister formation in PF.
Keywords: Pemphigus foliaceus, Desmoglein 1, Desmoglein 1 clustering, p38MAPK signaling
1. Introduction
Pemphigus is an autoimmune blistering disease of skin and/or mucous membranes characterized by the loss of intercellular adhesion of keratinocytes, acantholysis, due to the binding of IgG autoantibodies to desmogleins (Dsgs) [1]. Pemphigus consists of two major subtypes, pemphigus foliaceus (PF) and pemphigus vulgaris (PV), which are characterized by autoantibodies against desmoglein 1 (Dsg1) and desmoglein 3 (Dsg3), respectively. PF exhibits superficial blistering in the skin, while PV shows suprabasilar blistering of the skin and mucous membranes. A characteristic clinical finding of pemphigus is Nikolsky’s sign, in which blisters can be induced in normal appearing skin by applying mechanical shear force, reflecting the loss of cell adhesion in the epidermis.
Dsgs and desmocollins (Dscs) are cadherin type adhesion molecules located in desmosomes. Extracellularly, Dsgs comprise four domains (EC1-4) which consist of cadherin repeats of approximately 110 amino acids each, and juxtamembrane anchor (EC5). Dsgs and Dscs are predicted to form homophilic (or perhaps heterotypic with each other) trans-interactions via their EC1 domains or cis-interactions via their EC1 and EC2 domains [2]. Desmosomal cadherins are connected to keratin intermediate filaments by cytoplasmic plaque proteins, including plakoglobin (PG) and desmoplakin [3].
Two major mechanisms have been proposed for blister formation in pemphigus. One is steric hindrance, in which pathogenic autoantibodies induce the loss of cell adhesion by directly interfering with the trans- or cis-interaction of Dsgs [4–8]. The other proposed pathologic mechanism is that autoantibodies cause a cellular response, that includes internalization and degradation of Dsgs, associated with intracellular signaling, such as p38 mitogen-activated protein kinase (p38MAPK), Rho family GTPase, protein kinase C and phospholipase C [9–13]. However, the relative contribution of the two major mechanisms to loss of cell– cell adhesion has not been determined. A recent study on PV showed that polyclonal PV serum IgG induced p38MAPK-dependent Dsg3 clustering on the cell surface that subsequently leads to its endocytosis. On the other hand, a pathogenic monoclonal anti-Dsg3 IgG, AK23, induced the loss of cell adhesion independently of the p38MAPK pathway, possibly functioning through the steric hindrance interfering with Dsg adhesion [14].
The mechanism for blister formation in PF is less well understood. To better understand the pathogenesis of PF, we previously isolated multiple anti-Dsg1 monoclonal antibodies (mAbs) as single-chain variable fragment (scFv) from a PF patient by phage display [15]. Out of 67 generated unique anti-Dsg1 scFv mAbs, only two scFv mAbs showed the ability to induce blister formation when injected into cultured skin and neonatal mice. Therefore this data suggested that PF sera consisted of both pathogenic and non-pathogenic antibodies against Dsg1. Analysis of the strongly pathogenic anti-Dsg1 scFv mAb, 3–30/3 h (PF1-8-15), revealed that it recognized the amino-terminal trans-adhesive region of the EC1 domain of Dsg1, which defined a major pathogenic epitope targeted by most PF sera [16] and caused trans-interaction blocking.
However, several questions are still unsolved. It is unknown how each anti-Dsg1 mAb contributes to blister formation under polyclonal conditions in vivo. For example: Do non-pathogenic anti-Dsg1 Abs have any role in blister formation? Is p38MAPK signaling necessary for blister formation in PF? Do monoclonal and polyclonal anti-Dsg1 Abs induce blisters by different mechanisms?
To assess these questions, we compared the effects of a single anti-Dsg1 mAb and a mixture of anti-Dsg1 mAbs on blister formation, subcellular localization of desmosomal molecules, and desmosomal structural changes in a human skin organ culture and primary keratinocyte. We found that a mixture of a pathogenic and a non-pathogenic anti-Dsg1 IgG mAb induced Dsg1 clustering, which enhanced the loss of cell adhesion compared to the pathogenic antibody alone. Furthermore, a p38MAPK inhibitor prevented Dsg1 clustering. Although the inhibitor did not prevent blistering in the ex vivo injection assay, it did partially block the loss of cell adhesion measured by the in vitro keratinocyte dissociation assay. These findings indicated that a polyclonal mixture of pathogenic and non-pathogenic anti-Dsg1 mAbs caused p38MAPK-dependent Dsg1 clustering that was associated with enhanced pathogenic activity beyond that of the single pathogenic mAb.
2. Materials and methods
2.1. Preparation and purification of anti-Dsg1 scFv mAbs
We used two anti-Dsg1 scFv antibodies isolated from a PF patient by phage display (Table 1). Soluble scFv mAbs were produced in the Top10 F’ strain of E. coli (Invitrogen, Carlsbad, CA) and purified by Talon metal affinity resin (Clontech, Mountain View, CA) as previously described [15].
Table 1.
Epitope location, pathogenicity and antibody form of anti-Dsg1 monoclonal antibody.
| Name of antibodya | Epitope location of Dsg1 | Antibody form | |
|---|---|---|---|
| Pathogenic antibody | PF1-8-15 (3-30/3h) | Conformational epitope of AA89-101 of EC1 (Putative trans-adhesive interface) |
IgG and scFv |
| Non-pathogenic antibody | PF1-2-6 (1-18/L12) | Linear epitope of EC3 | IgG and scFv |
| PF1-2-22 (1-08/O12O2) | AA 1-161 of Dsg1 | IgG |
Abbreviations: Dsg1, desmoglein 1; PF, pemphigus foliaceus; EC, extracellular domain; AA, amino acid; scFv, single chain variable fragment.
Antibody name in parenthesis is based on gene usage of heavy and light chain [15].
2.2. Generation of divalent IgG1 mAbs from cDNA encoding anti-Dsg1 scFv mAbs
Divalent IgG1 mAbs were generated from anti-Dsg1 scFv mAbs as described previously [17]. The eukaryotic PIGG expression vector was provided by Carlos Barbas (Scripps Research institute). Primers used to subclone scFv mAbs into the PIGG vector are shown in Supplemental Table 1. The variable region of the heavy chain (VH) was amplified by PCR over PF mAbs, contained in the scFv phagemid expression vector pComb3X. The PCR product was subcloned into the SacI-ApaI site in the PIGG vector. The constant region of the kappa light chain (CK) was amplified over PA-H6-HA-TT vector which was provided by Donald Siegel (University of Pennsylvania) as the PCR template for PF1-2-6 and PF1-2-22. The constant region of the lambda light chain (CL) was amplified over pIgG-E1M2 as the PCR template for PF1-8-15. The variable region of the light chain (VL) was hybridized by overlap PCR using primers shown in Supplemental Table 1 and subcloned in the HindIII-XbaI site of the PIGG vector. All recombinant constructs were verified by sequencing.
2.3. Production of IgG form of anti-Dsg1 mAbs
Transfection of generated anti-Dsg1 IgG mAb in PIGG vectors was performed into 293FT cells (Invitrogen) using FUGENE6 transfection reagent (Promega, Madison, WI) according to the standard protocol, using five 10 cm cell culture plates at approximately 90% cell density and Dulbecco’s Modified Eagle’s Medium (Wako, Osaka, Japan) plus 10% ultralow IgG fetal bovine serum (Invitrogen) as harvest media. Expressed antibody was harvested from the cell culture supernatant at 3 and 6 days. IgG antibody was purified from the harvested media with protein A agarose beads (GE Healthcare, Uppsala, Sweden). Samples were dialyzed into phosphate buffered saline (PBS) (pH 7.4) and concentrated to approximately 1 μg/μl using Amicon Ultra-4 (Merck Mllipore, Co.cork, Ireland). Antibody binding to Dsg1 was confirmed by ELISA (MBL, Aichi, Japan) according to the manufacturer’s protocols.
2.4. SDS-PAGE of anti-Dsg1 IgG mAb
Purified anti-Dsg1 IgG mAb was mixed in Laemmli sample buffer (BIO-RAD, Alfred Nobel Drive Hercules, CA) with or without β-mercaptoethanol (Wako, Osaka, Japan), then separated by SDS-PAGE. Fractionated proteins were detected by coomassie brilliant blue stain (ATTO, Tokyo, Japan). SDS-PAGE under reducing conditions showed that purified IgG mAbs produced from the scFvs exhibit two bands, with molecular weights corresponding to the light and heavy chains of IgG. Under non-reduced conditions, the purified IgG were detected as single band, with molecular weight of the divalent IgG (Supplemental Fig. S1).
2.5. Human skin organ culture injection assay
Specimens were obtained from leftover normal skin after excisional surgery after written informed consent. The specimens were trimmed by removing fat tissue and cut into 5 mm diameter pieces. Individual anti-Dsg1 IgG mAbs (50 μg) or a mixture of two anti-Dsg1 IgG mAbs (50 μg each) or PBS as control were injected from dermal side by using insulin syringe. In some experiments, we used 25 μg of anti-Dsg1 scFv mAb to equalize the valency of antigen-binding site between IgG and scFv. As molecular weight of scFv is 37 kD and that of IgG is 146 kD, we calculated it by following formula: Y(weight of scFv)/37 = [X(weight of IgG)/146] × 2. The injected skin specimens were put on the insert of Transwells (Corning, Tewksbury, MA) with defined keratinocyte-SFM medium (Invitrogen) containing 1.2 mM CaCl2 in the outer compartment and then cultured at 37 °C. After 22–24 h incubation, the skins were harvested for histology after mechanical shear stress by slight friction of epidermis and processed for immunofluorescence staining and electron microscopy without mechanical stress. For p38MAPK inhibition assay, we used SB202190 (Sigma-Aldrich, St. Louis, MO), a highly selective, potent and cell permeable inhibitor of p38MAPK. SB202190 bound to both the inactive and the active forms of p38MAPK. Specimens were pretreated by intradermal injection with 16.7 μg SB202190 or dimethyl sulfoxide (DMSO) as control for 1.5 h at 37 °C. Pretreated skin specimens were injected with a single anti-Dsg1 IgG mAb or a mixture of anti-Dsg1 IgG mAbs together with 16.7 μg of SB202190 or PBS with DMSO as control, then put on the insert of Transwells, and incubated with the medium containing 1.2 mM CaCl2 and 100 μM SB202190 in the outer compartment for 22–24 h at 37 °C. All experiments were performed at least 3 times.
2.6. Immunofluorescence microscopy
Injected skin specimens without mechanical stress area were frozen in OCTcompound (SAKURA, Tokyo, Japan) by liquid nitrogen and kept at −80 °C until use. Skin samples were sliced into 4 μm sections. Skin specimens were blocked with Tris-bufferd saline (TBS) containing 1% albumin from bovine serum and 1 mM CaCl2 for 30 min at room temperature, followed by incubation with primary antibodies in blocking solution for 1 h at room temperature. After three time washes in TBS-Ca, the skin sections were incubated with appropriated conjugated secondary antibodies diluted 1:400 in the blocking solution for 1 h at room temperature. The following mAbs were used for primary antibodies: a mouse anti-Dsg1 mAb (27B2, 1:20 dilution, Abcam, Cambridge, UK), a mouse anti-PG mAb (15F11, 1:1000 dilution, Sigma-Aldrich), a mouse anti-Dsc1 mAb (U100, undiluted, PROGEN Biotechnik GmbH, Heidelberg, Germany), and a mouse anti- Dsg3 mAb (G194, undiluted, Acris Antibodies GmbH, Herford, Germany). Double staining of deposited IgG and adhesion molecules was performed with Alexa Fluor 488 anti-mouse IgG (Invitrogen) and Alexa Fluor 594 anti-human IgG (Invitrogen) as secondary Abs. Immunofluorescence was observed at ×1000 magnification by Nikon laser confocal microscope A1R (Nikon, Tokyo, Japan).
2.7. Cell culture and immunofluorescence studies
Primary keratinocytes (a gift from Ehime University) were cultured on collagen-coated chamber slide (Iwaki Science products, Tokyo, Japan) in medium MCDB153 until 100% confluent. Cells were changed to 0.5 mM calcium-containing medium for 24 h before treatment with anti-Dsg1 mAbs. Individual anti-Dsg1 IgG mAbs (50 μg/ml), individual anti-Dsg1 scFv mAbs (25 μg/ml), a mixture of these mAbs or PBS as control were added to cell culture medium. After 24 h incubation at 37 °C, cells were washed three times with PBS(+), and then fixed in −20 °C methanol for 5 min and processed for immunofluorescence indicated above. For p38MAPK inhibition assay, cells were treated with 25 μM SB202190 or DMSO as control 1.5 h prior to antibody addition. SB202190 remained in the medium for the duration of the experiment. All experiments were performed at least 3 times.
2.8. Electron microscopy
The injected skins without mechanical shear stress area were fixed by 2.5% glutaraldehyde in 0.1 M phosphate buffer and post-fixed in 1% osmiumtetroxide, then dehydrated and embedded in epon resin. Ultrathin sections were cut into 80 nm and stained with uranyl acetate and lead citrate and observed by JEL-1400C transmission electron microscope (JEOL Ltd., Tokyo, Japan). To analyze lengths of desmosomes, image pictures of ultrathin sections were taken at the same magnification (×8000). About one to two hundred of desmosomal length in the each layer was averaged.
2.9. Modified dispase-based dissociation assay
We used a previously described in vitro dissociation assay with some modifications [18]. Primary keratinocytes were cultured on collagen-coated 12-well microplates (Iwaki) in medium MCDB153 until 100% confluent. Culture medium was changed to 0.5 mM calcium-containing medium for 24 h before treatment with anti-Dsg1 mAbs. Individual anti-Dsg1 IgG mAbs (50 μg/ml), individual anti-Dsg1 scFv mAbs (25 μg/ml), a mixture of these mAbs or PBS as control with AK23 (1 μg/ml) were added to each wells. After 24 h incubation with these mAbs, the cells were rinsed with PBS (+) twice and incubated with 300 μl dispase at 37 °C for 15 min. Released cell sheets were rinsed with PBS (+) three times and pipetted five times by using a 1 ml pipette. Fragmented cell sheets were fixed by adding 1 ml of 10% buffered formalin phosphate and stained by crystal violet. Three pictures were taken and used for counting the number of fragments manually. To inhibit p38MAPK signaling, the cells were pretreated with 25 μM SB202190 or DMSO as control for 1.5 h at 37 °C before adding anti-Dsg1 mAbs with AK23. The inhibitor remained in the medium during the experiment. We set the concentration of the inhibitor at which Dsg1 clustering was not detectable by immunofluorescence staining. Dissociation index were calculated from the number of fragments (N) using following formula: N with anti-Dsg1 mAb + AK23/N with PBS + AK23. All experiments were performed at least 3 times.
2.10. Statistical analysis
The data were analyzed by one-way analysis of variance followed by Bonferroni correction (Fig. 2B) or Tukey’s method (Figs. 5 B and 6 B) for multiple-group samples. Statistical significance was presumed for p < 0.05.
Fig. 2.
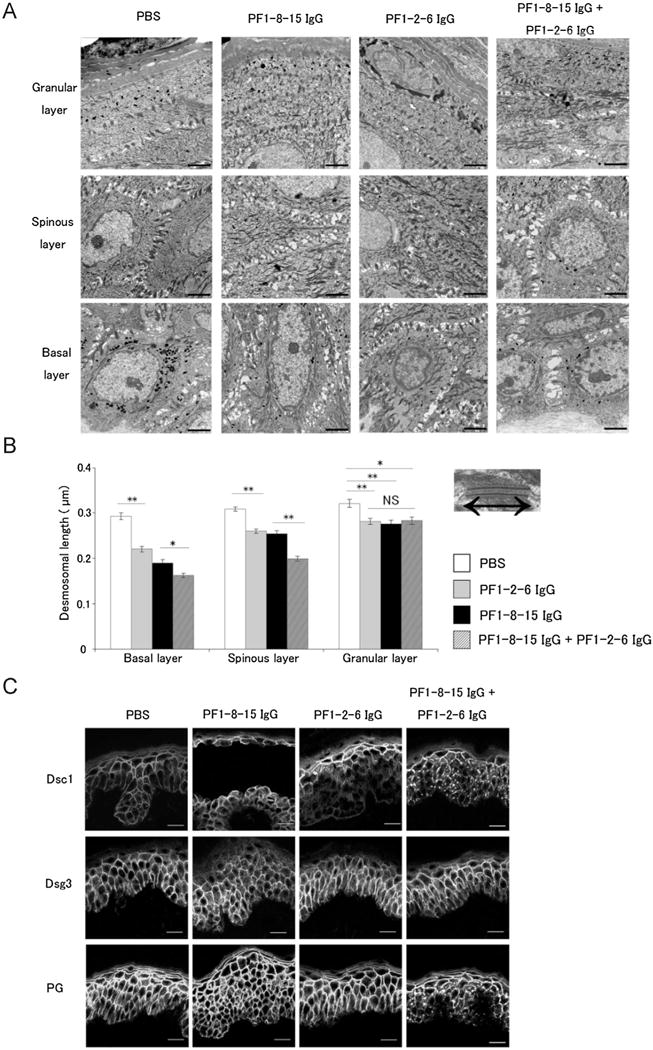
A mixture of anti-desmoglein 1 (Dsg1) IgG monoclonal antibodies (mAbs) accelerated alteration of desmosomal morphology and desmosomal proteins in the lower epidermis. A: Non-blister area of anti-Dsg1 IgG mAb injected human skin was observed by electron microscopy. Note that every mAbs injection pattern caused intercellular space widening in the basal and spinous layers. Bar = 2.0 μm. B: Desmosomal length in the different layers of epidermis. Right and left arrow in the photo of the desmosome represents the length of desmosome in the electron microscopy. The desmosomal lengths in every pattern injection of anti-Dsg1 IgG mAbs were shorter than those in PBS. Especially, desmosomal length of the mixture injection (PF1-8-15 IgG + PF1-2-6 IgG) was shorter than PF1-8-15 IgG alone at the basal and spinous layers. Data are mean SEM. *p < 0.05, **p < 0.01. NS, not significant. C: Immunofluorescence staining of desmocollin 1 (Dsc1), desmoglein 3 (Dsg3) and plakiglobin (PG) in human skin specimens injected with anti-Dsg1 IgG mAbs was captured by confocal microscopy. The mixture injection (PF1-8-15 IgG + PF1-2-6 IgG) induced clustering of Dsc1 and PG in the lower epidermis, but individual anti-Dsg1 IgG mAb injection did not. Anti-Dsg1 IgG mAb injections did not affect Dsg3 distribution. Bar = 20 μm.
Fig. 5.
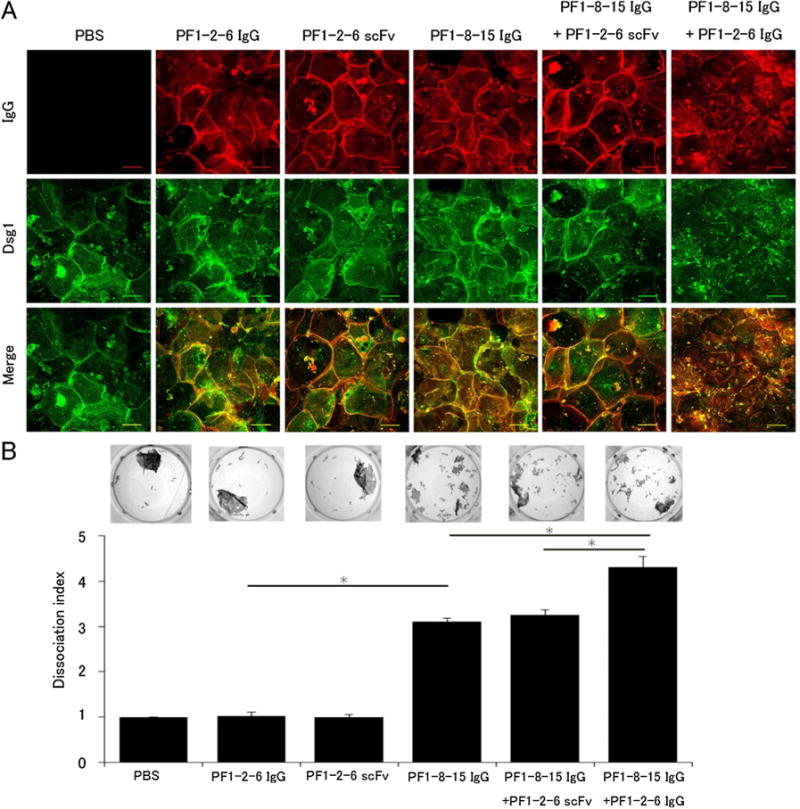
A mixture of pathogenic and non-pathogenic anti-desmoglein 1 (Dsg1) IgG monoclonal antibodies (mAbs) induced Dsg1 clustering which enhanced loss of cell adhesion in primary keratinocytes. A: Immunofluorescence imaging of primary keratinocytes. Keratinocytes were incubated in 0.5 mM calcium-containing medium for 24 h to induce Dsg1 expression and then treated anti-Dsg1 IgG mAbs for 24 h. Keratinocytes treated with the mixture of PF1-8-15 IgG and PF1-2-6 IgG (PF1-8-15 IgG + PF1-2-6 IgG) showed Dsg1 clustering. Bar = 50 μm. B: Dissociation assay corresponding to the immunofluorescence imaging series (n = 6 per group). PF1-8-15 IgG + PF1-2-6 IgG showed higher dissociation index than the mixture of PF1-8-15 IgG and PF1-2-6 scFv (PF1-8-15 IgG + PF1-2-6 scFv) or PF1-8-15 IgG alone. Photos show the fragments condition of each well. Data are mean SEM. *p < 0.05.
Fig. 6.
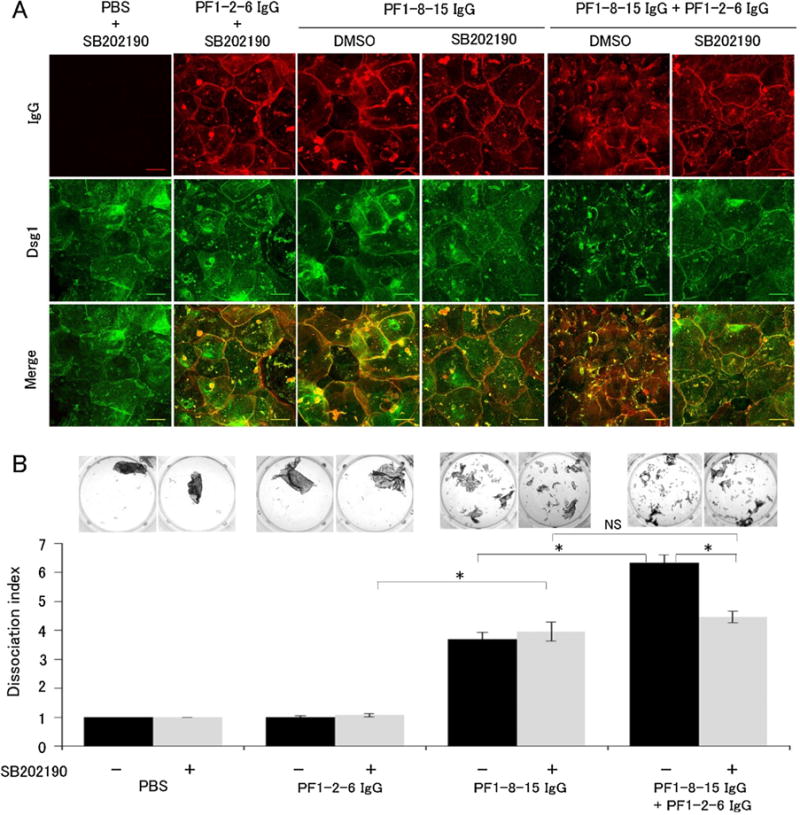
p38 mitogen-activated protein kinase (p38MAPK) inhibition prevented anti-desmoglein 1 (Dsg1) IgG monoclonal antibodies (mAbs) Dsg1 clustering, but not loss of cell adhesion in vitro. A: Immunofluorescence imaging of primary keratinocytes with the p38MAPK inhibitor SB202190. Cells were pre-incubated with SB202190 for 1.5 h followed by co-incubated with anit-Dsg1 IgG mAbs and SB202190 for 24 h. SB202190 prevented Dsg1 clustering induced by the mixture of PF1-8-15 IgG and PF1-2-6 IgG in cultured primary keratinocytes. Bar = 50 μm. B: Dissociation assay with SB202190 corresponding to the immunofluorescence imaging series (n = 6 per group). SB202190 failed to prevent loss of cell adhesion in keratinocytes incubated with the single PF1-8-15 IgG. On the other hand, SB202190 partially suppressed loss of cell adhesion in keratinocytes incubated with the mixture of PF1-8-15 IgG and PF1-2-6 IgG. Dissociation index of the single PF1-8-15 IgG with SB202190 was almost the same as that of the mixture of PF1-8-15 IgG and PF1-2-6 IgG with SB202190. Photos show the fragments condition of each well. Data are mean SEM. *p < 0.05. NS, not significant.
3. Results
3.1. Production of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs
We have previously cloned both pathogenic and non-pathogenic anti-Dsg1 mAbs as scFv from lymphocytes of a PF patient by phage display technique. ScFvs form antibody fragments that express the monovalent antigen-binding site of the native immunoglobulin molecules. In order to analyze the mechanism of blister formation in PF, the anti-Dsg1 scFvs were converted to divalent full length IgG1 molecules using the PIGG vector [19]. Anti-Dsg1 IgG and scFv mAbs used in this study are shown in Table 1. A pathogenic PF1-8-15 IgG mAb recognized a conformational epitope within amino acid 89–101 on the putative trans-adhesive interface of the EC1 domain, which defines a major pathogenic epitope targeted by most PF sera [16]. A non-pathogenic PF1-2-6 IgG mAb recognized a linear epitope on EC3 and another non-pathogenic PF1-2-22 IgG mAb recognized amino acid 1–161 of Dsg1 [15].
3.2. A mixture of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs induced clustering of Dsg1 in the lower epidermis
Previous several studies using PV sera or a mixture of anti-Dsg3 mAbs reported that those Abs induced clustering and endocytosis of Dsg3 [14,20]. In PF, several papers reported that PF patients’ Nikolsky positive skin showed clustered deposition of IgG and Dsg1 in the basal and spinous layers [21,22]. In this study, we first analyzed the effects of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs on alterations of Dsg1 sublocalization and IgG deposits. We used a human skin organ culture system to reflect in vivo conditions. Excised human skin was injected with three different patterns of mAbs; a pathogenic PF1-8-15 IgG (50 μg), a non-pathogenic PF1-2-6 IgG (50 μg), a mixture of pathogenic PF1-8-15 IgG (50 μg) and non-pathogenic PF1-2-6 IgG (50 μg). We analyzed hematoxylin-eosin staining, direct immunofluorescence (DIF) of IgG deposits and staining of Dsg1 captured by confocal microscopy. PF1-8-15 IgG injection caused an acantholytic superficial blister (Fig. 1A). DIF showed PF1-8-15 IgG bound linearly on the cell surface of keratinocytes throughout the epidermis (Fig. 1B) coincident with the Dsg1 distribution (Fig. 1C, D). PF1-2-6 IgG did not cause acantholysis, as expected (Fig. 1E). DIF of PF1-2-6 IgG and Dsg1 distribution also showed coincident linear staining on the cell surface of keratinocytes throughout the epidermis (Fig. 1F–H). On the other hand, the mixture of PF1-8-15 IgG and PF1-2-6 IgG injection caused superficial acantholysis (Fig. 1I) and revealed IgG and Dsg1 staining, mostly merged, with an aberrant granular pattern in the lower epidermis in addition to the usual linear cell surface staining elsewhere (Fig. 1J–L). At a longer incubation time of 36 h, Dsg1 clustering was observed at whole layers of the skin injected the mixture of PF1-8-15 IgG and PF1-2-6 IgG (Supplemental Fig. 2). To exclude the possibility that the Dsg1 clustering was resulted from excessive doses of PF IgG rather than the mixture, we injected double doses of pathogenic PF1-8-15 IgG (100 ug) and non-pathogenic PF1-2-6 IgG (100 ug). The single mAb injection did not induce Dsg1 clustering (Supplemental Fig. 3). These findings suggest that in PF, as has been shown in PV, clustering of Dsg1 requires polyclonal anti-Dsg1 antibodies.
Fig. 1.
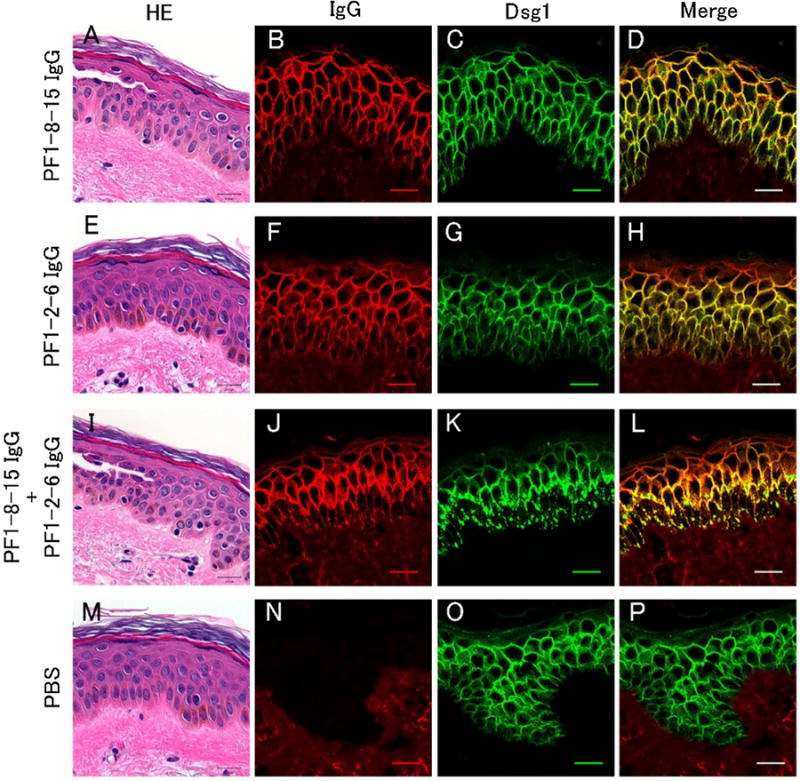
A mixture of pathogenic and non-pathogenic IgG monoclonal antibodies (mAbs) induced clustering of desmoglein 1 (Dsg1) in the organ-cultured human skin. Each individual mAb and a mixture of anti-Dsg1 IgG mAbs were injected into human skin specimens that were then cultured for 22–24 h. The skins were harvested for histology after mechanical shear stress by slight friction of epidermis. Histology and direct immunofluorescence of IgG deposits and staining of Dsg1 were captured by confocal microscopy. PF1-8-15 IgG (50 μg) caused superficial epidermal acantholysis (A), but PF1-2-6 IgG (50 μg) did not (E). Note that an individual injection of PF1-8-15 IgG or PF1-2-6 IgG showed of IgG and Dsg1 linear distribution cell surface of keratinocytes on F–H), while the mixture injection of PF1-8-15 IgG (50 μg) the and PF1-2-6 IgG (50 μg) (B–D, (PF1-8-15 IgG + PF1-2-6 IgG) showed aberrant granular IgG and Dsg1 distribution in the lower epidermis (J–L), in addition to superficial acantholytic blister (I). In PBS, epidermal morphology and Dsg1 distribution were normal (M, O) with no detectable IgG deposition in the epidermis (N). Bar = 20 μm.
3.3. A mixture of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs causes more remarkable ultrastructural changes in the basal and spinous layers than a single anti-Dsg1 IgG mAb injection
To evaluate the ultrastructure of cultured human skin injected with anti-Dsg1 IgG mAbs, the Nikolsky positive non-blister area was observed by electron microscopy. Each individual mAb and the mixture of mAbs caused widening of intercellular space in the basal and spinous layers, but not in the granular layer (Fig. 2A). Fig. 2B shows the average desmosomal length in the different layers of epidermis with the three different mAb conditions as well as PBS as a control. The mixture of PF1-8-15 IgG and PF1-2-6 IgG shortened the desmosomal length much more than PF1-8-15 IgG or PF1-2-6 IgG alone in the basal and spinous layers. In the granular layer, the desmosomal length in the skin injected with anti-Dsg1 mAbs was shorter than that with PBS. These findings suggested that a mixture of anti-Dsg1 mAbs promoted ultrastractural changes of desmosomes.
3.4. A mixture of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs injection induced clustering of Dsc1 and PG
To check the distribution of other desmosomal proteins in the injected skin, we stained Dsc1, Dsg3 and PG (Fig. 2C). In the skin injected with PF1-8-15 IgG or PF1-2-6 IgG alone, Dsc1, Dsg3 and PG were distributed linearly on the cell surface of keratinocyte throughout the epidermis. On the other hand, in the skin injected with the mixture of PF1-8-15 IgG and PF1-2-6 IgG, Dsc1 and PG were clustered in the lower epidermis similar to Dsg1 staining while Dsg3 remained in the normal linear pattern. This data showed that Dsg1 clustering was associated with alterations of the sub-cellular localizations of Dsc1 and PG as well.
3.5. Dsg1 clustering is due to cross-linking of Dsg1 molecules by non-pathogenic anti-Dsg1 IgG antibody only when trans-interaction of Dsg1 molecules is blocked by a pathogenic anti-Dsg1 antibody
To test if Dsg1 clustering is due to cross-linking by IgG mAbs, we compared the effects of the IgG (divalent) and scFv (monovalent) forms of anti-Dsg1 mAbs. The correspondence between mAb injections and Dsg1 clustering is shown in Table 2 and representative microscopic findings are shown in Fig. 3 and Supplemental Fig. 4A. The mixture of PF1-8-15 IgG and PF1-2-6 IgG caused Dsg1 clustering (Fig. 3A). In contrast, when a mixture of PF1-8-15 scFv and PF1-2-6 scFv was injected, Dsg1 clustering was not observed although superficial blistering was induced (Fig. 3B). A mixture of PF1-8-15 IgG with another non-pathogenic PF1-2-22 IgG also caused Dsg1 clustering (data not shown). Therefore, divalent IgG was necessary to induce Dsg1 clustering, but not blister formation. To further test which mAb was necessary for Dsg1 clustering, we injected various combinations of IgG and scFv forms of anti-Dsg1 mAbs. When a mixture of PF1-8-15 IgG and PF1-2-6 scFv was injected, the Dsg1 clustering was not observed (Fig. 3C). On the other hand, when a mixture of PF1-8-15 scFv and PF1-2-6 IgG was injected, the Dsg1 clustering was observed (Fig. 3D). These findings suggest that Dsg1 clustering is due to cross-linking of Dsg1 molecules by non-pathogenic IgG mAbs. When injected with a mixture of two different non-pathogenic anti-Dsg1 IgG mAbs (PF1-2-6 IgG + PF1-2-22 IgG), Dsg1 clustering was not observed (Fig. 3E). This suggests that blocking of trans-interaction of Dsg1 molecules by a pathogenic anti-Dsg1 mAb is also necessary for Dsg1 clustering.
Table 2.
Correspondence of antibody injection patterns and Dsg1 clustering.
| Antibody injection patterns | Dsg1 clustering |
|---|---|
| PF1-8-15 IgG | − |
| PF1-2-6 IgG | − |
| PF1-2-22 IgG | − |
| PF1-8-15 IgG + PF1-2-6 IgG | + |
| PF1-8-15 IgG + PF1-2-22 IgG | + |
| PF1-8-15 scFv + PF1-2-6 IgG | + |
| PF1-8-15 IgG + PF1-2-6 scFv | − |
| PF1-8-15 scFv + PF1-2-6 scFv | − |
| PF1-2-6 IgG + PF1-2-22 IgG | − |
| PF1-8-15 IgG + PF1-2-6 IgG + PF1-2-22 IgG | + |
Abbreviations: Dsg1, desmoglein 1; PF, pemphigus foliaceus; scFv, single chain variable fragment.
Fig. 3.
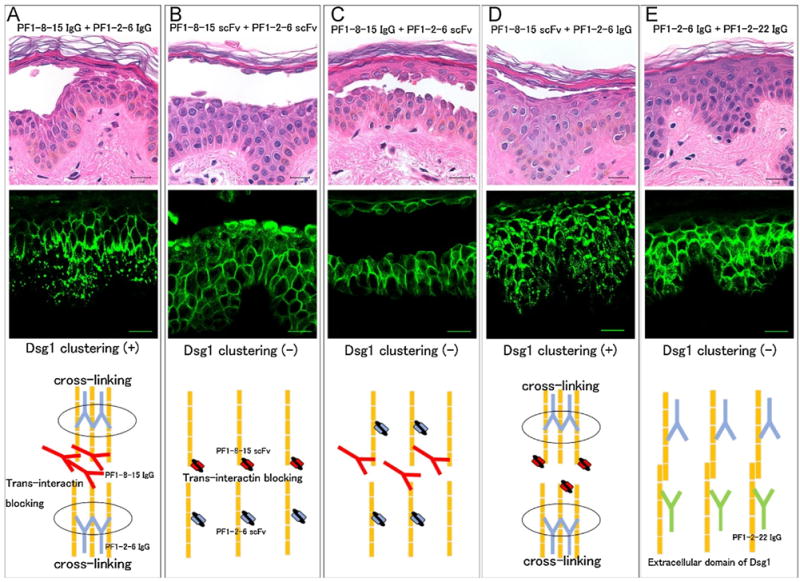
Desmoglein 1 (Dsg1) clustering needed not only a pathogenic anti-Dsg1 antibody, but also cross-linking of Dsg1 molecules by a non-pathogenic anti-Dsg1 IgG antibody. Immunofluorescence of Dsg1 staining of human skin injection assay is captured by confocal microscope. Schema show comparing the effect between IgG and single-chain variable fragment (scFv) form of anti-Dsg1 monoclonal antibodies (mAbs). A: The mixture injection of PF1-8-15 IgG and PF1-2-6 IgG (PF1-8-15 IgG + PF1-2-6 IgG) caused Dsg1 clustering and blistering. PF1-8-15 IgG disturbed trans-interaction of Dsg1 molecules. PF1-2-6 IgG caused cross-linking of Dsg1 molecules. B: The mixture injection of PF1-8-15 scFv and PF1-2-6 scFv (PF1-8-15 scFv + PF1-2-6 scFv) did not cause Dsg1 clustering, but caused blistering. ScFv type antibodies could not cause cross-linking of Dsg1 molecules because they had only one antigen binding site. C: The mixture injection of PF1-8-15 IgG and PF1-2-6 scFv (PF1-8-15 IgG + PF1-2-6 scFv) did not cause Dsg1 clustering, but caused blisteing. PF1-8-15 IgG disturbed trans-interaction of Dsg1 molecules, but did not cause cross-linking of Dsg1 molecules. D: The mixture injection of PF1-8-15 scFv and PF1-2-6 IgG (PF1-8-15 scFv + PF1-2-6 IgG) caused Dsg1 clustering and blistering. PF1-2-6 IgG caused cross-linking of Dsg1 molecules. E: The mixture injection of two different non-pathogenic anti-Dsg1 IgG mAbs (PF1-2-6 IgG + PF1-2-22 IgG) did not cause Dsg1 clustering and blistering. Bar = 20 μm.
3.6. Dsg1 clustering by anti-Dsg1 mAbs is p38MAPK dependent, but not essential for blister formation in ex vivo
Several papers have demonstrated, with PV sera and PV anti-Dsg3 mAbs, that p38MAPK signaling has a key role for blistering through endocytosis and clustering of Dsg3 [12,14]. Moreover, it has been reported that phosphorylation of p38MAPK was increased in a PF patient’s skin and in PF-IgG injected mouse skin [11,23]. Therefore, we used the p38MAPK inhibitor SB202190 to determine if p38MAPK also plays a role in blistering associated with clustering of Dsg1 in the human organ culture model (Fig. 4). Treatment with SB202190 prevented Dsg1 clustering induced by the mixture of PF1-8-15IgG and PF1-2-6 IgG. However, SB202190 did not prevent blister formation by either the mixture pattern or PF1-8-15 IgG alone. This result suggests that Dsg1 clustering is p38MAPK signaling dependent but not essential for blistering in ex vivo.
Fig. 4.
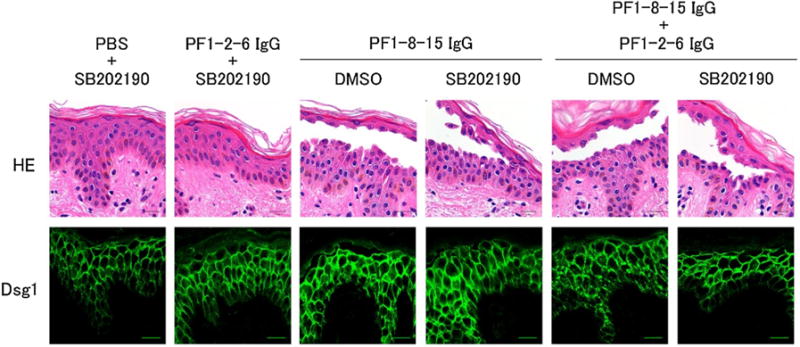
The p38 mitogen-activated protein kinase (p38MAPK) inhibitor SB202190 prevented desmoglein 1 (Dsg1) clustering, but not blistering by a mixture of pathogenic and non-pathogenic anti-Dsg1 IgG monoclonal antibodies (mAbs). Human skin specimens were pretreated with the p38MAPK inhibitor SB202190 or DMSO for 1.5 h. Pretreated skins were injected anti-Dsg1 IgG mAbs with SB202190 or DMSO and cultured for 22 h. Histology and immunofluorescence of Dsg1 staining of confocal microscopy were shown. SB202190 had no effect for Dsg1 distribution of individual anti-Dsg1 IgG mAb injected skin. On the other hand, SB202190 blocked the Dsg1 clustering induced by the mixture injection (PF1-8-15 IgG + PF1-2-6 IgG), but could not block blistering caused by PF1-8-15 IgG and the mixture injection. Bar = 20 μm.
3.7. A mixture of anti-Dsg1 IgG mAbs enhanced pathogenic activity for the loss of cell adhesion in association with p38MAPK-dependent Dsg1 clustering
To further verify results of ex vivo injection assay, we analyzed Dsg1 distribution and pathogenic strength by in vitro cultured primary keratinocytes. Primary keratinocytes were incubated in 0.5 mM calcium-containing medium for 24 h to induce Dsg1 expression and then treated with individual anti-Dsg1 IgG mAbs, individual anti-Dsg1 scFv mAbs or those antibody combinations. To evaluate additional effects of non-pathogenic anti-Dsg1 mAbs, we used same doses of PF1-8-15 IgG in both individual and mixture patterns. As expected, the mixture of PF1-8-15 IgG (50 μg/ml) and PF1-2-6 IgG (50 μg/ml) induced punctuated Dsg1 distribution, but other individual anti-Dsg1 mAbs and a mixture of PF1-8-15 IgG (50 μg/ml) and PF1-2-6 scFv (25 μg/ml) showed linear staining of Dsg1 along cell borders (Fig. 5A). Next, we used a previously described in vitro dissociation assay with some modifications to compare the pathogenic strength of the anti-Dsg1 mAbs in a quantitative manner [18]. For cell sheets to fragment in the dissociation assay, adhesion of both Dsg3 and Dsg1 must be blocked. Therefore, we incubated cells with anti-Dsg1 mAbs in the presence of AK23, an anti-Dsg3 mAb which inhibits adhesion by Dsg3, in each well. Addition of AK23 alone induced fragmentation of the cell sheet at only minimum levels (leftmost panel in Fig. 5B). Therefore, the number of fragments represents the pathogenic strength of anti-Dsg1 mAbs. The average dissociation index is shown in Fig. 5B and Supplemental Fig. 4B. The dissociation index of IgG or scFv form of non-pathogenic PF1-2-6 was almost the same as that of control PBS as expected. The dissociation index of the single PF1-8-15 IgG was almost 3 times higher than that of the single PF1-2-6 IgG. The mixture of PF1-8-15 IgG (50 μg/ml) and PF1-2-6 IgG (50 μg/ml) dissociated keratinocytes sheets more than the single PF1-8-15 IgG (50 μg/ml), while the dissociation index of the mixture of PF1-8-15 IgG (50 μg/ml) and PF1-2-6 scFv (25 μg/ml) was almost the same as the single PF1-8-15 IgG (50 μg/ml). This result suggests the addition of IgG form non-pathogenic anti-Dsg1 Ab to pathogenic anti-Dsg1 Ab can accelerate the dissociation, which was correlated with the clustering of Dsg1.
To investigate to what extent p38MAPK signaling or p38MAPK-dependent Dsg1 clustering contributes to loss of cell adhesion, we treated the cells with the p38MAPK inhibitor SB202190. Immunofluorescence staining showed SB202190 prevented Dsg1 clustering caused by the mixture of anti-Dsg1 IgG mAbs, but did not affect Dsg1 distribution in cells treated with the single anti-Dsg1 IgG mAb (Fig. 6A). Next we performed the in vitro dissociation assay in the presence or in the absence of p38MAPK inhibitor. Treatment of SB202190 failed to prevent loss of adhesion in cells incubated with the single PF1-8-15 IgG, indicating that PF1-8-15 IgG does not require p38MAPK signaling to inhibit cell adhesion. On the other hand, SB202190 partially prevented fragmentation of cell sheets induced by the mixture of PF1-8-15 IgG and PF1-2-6 IgG (Fig. 6B). Furthermore, dissociation index of the mixture pattern with SB202190 was almost the same as that of the single PF1-8-15 IgG with SB202190, suggesting that Dsg1 clustering induced additive effect. These findings also indicated that p38MAPK-dependent Dsg1 clustering enhanced the loss of cell adhesion, but it was not required for blister formation.
4. Discussion
In this study, by comparing individual anti-Dsg1 IgG mAbs and a mixture of anti-Dsg1 mAbs, mAbs, we found that a polyclonal mixture enhances pathogenicity in PF. Importantly, non-pathogenic mAbs, that do not induce blisters by themselves can enhance pathogenic activity associated with clustering of Dsg1. However, inhibition of p38MAPK-dependent Dsg1 clustering is not sufficient to prevent blister formation, suggesting that p38MAPK- independent mechanism, presumably trans-interaction blocking of Dsg1 molecules or other signaling pathways, may be a primary mechanism of blister formation, and p38MAPK-dependent Dsg1 clustering has an additive effect in inducing pathology in PF.
The above studies were done in human skin organ culture, but we confirmed in an in vitro dissociation assay, modified for detection of pathogenicity of anti-Dsg1 Abs. Our findings are consistent with some reports about PV autoantibody showing that polyclonal conditions affect pathogenicity [24,25]. In trying to determine how anti-Dsg1 mAbs interfered with cell adhesion, we examined the major adhesion junction in keratinocytes, the desmosome. We observed that the length of desmosomes was predominantly shorter in the skin injected with the mixture of anti-Dsg1 IgG mAbs than that with the single anti-Dsg1 IgG mAbs in the basal and spinous layers of epidermis at the Nikolsky positive non-blister area. This is consistent with the previous finding that smaller desmosomes were seen at the basal and spinous layer of Nikolsky positive PF patients’ skin [22]. In addition to Dsg1 clustering, clustering of PG and Dsc1 was also observed in the lower epidermis of the skin injected with the mixture of anti-Dsg1 IgG mAbs. Those findings suggested that polyclonal condition of PF IgG promotes desmosome disassembly and in turn augments the loss of cell adhesion.
Dsg1 clustering was more clearly observed in the lower epidermis where blisters in PF usually do not occur. The desmosome structure change in electron microscopy and alteration of sublocalization of desmosomal molecules were also more prominent in the lower epidermis. The reason of the discrepancy is not clear. One possible reason is that as IgG mAbs was injected at dermis in organ-cultured human skin, the lower layers may shows the earliest change after IgG antibodies bind Dsg1. The possibility is supported by the finding that Dsg1 clustering was more evident in whole layers of epidermis at incubation times more than 24 h (Supplemental Fig. 2). It is possible that the desmosomal length in granular layers may be shortened at these longer incubation times as it has been reported that hypoplastic desmosomes were observed in lesional skin in PF patients by electron microscopy [26]. Or it may be possible that keratinocytes in lower epidermis may be more susceptible to clustering by IgG binding as less differentiated state compared to upper layers in epidermis. Furthermore, the in vitro dissociation assay may reflect the strength of cell adhesion of whole layers because the cultured keratinocytes contains both differentiated and undifferentiated cells. Higher dissociation index by the mixture of anti-Dsg1 mAbs may reflect weaker adhesion in the lower epidermis in addition to upper epidermis.
Consistent with our results, Oktarina et al. reported that purified IgG from PF serum but not Fab fragments induced clustering in human organ culture skin [27]. In addition, similar results have been reported in PV, in which a mixture of anti-Dsg3 IgG mAbs induced Dsg3 clustering in cultured keratinocytes [14]. We propose the mechanism and role of Dsg1 clustering as follows. First, blockage of trans-interaction of Dsg1 molecules is necessary for clustering. Second, the blocked Dsg1 molecules are cross-linked by divalent anti-Dsg1 IgG. Finally, the clustering enhances the pathogenic effect of antibodies.
Signaling pathways including p38MAPK have been studied in PV pathogenesis. Studies using a p38MAPK inhibitor in PV have suggested that p38MAPK signaling was linked to both Dsg3 endocytosis and the loss of keratinocyte adhesion in response to PV IgG [12]. On the other hand, a recent study using PV mAbs indicated that p38MAPK activation may not be essential for the loss of cell adhesion in PV but may function downstream of antibody binding to augment blistering via Dsg3 endocytosis [28]. Saito et al. showed p38MAPK signaling dependent and independent mechanisms in PV blister formation [14].
Roles of p38MAPK in the pathogenesis for PF, as opposed to PV, have not been well studied. In previous studies, phosphorylation of p38MAPK was increased in a PF patient skin and PF-IgG injected mouse skin [11,23]. Our study, for the first time, showed a relationship between p38MAPK signaling and Dsg1 clustering. Clustering of Dsg1 in response to the mixture of pathogenic and non-pathogenic anti-Dsg1 IgG mAbs was prevented by treating with SB202190. However, SB202190 was not sufficient to block blister formation by the mixture of anti-Dsg1 IgG mAbs in the ex vivo skin culture. In addition, SB202190 only partially reduced fragmentation of cell sheets in the in vitro dissociation assay. These findings indicated that p38MAPK-dependent Dsg1 clustering is not essential for blister formation in PF. Furthermore, SB202190 could not prevent blister formation in ex vivo skin culture or loss of cell adhesion in vitro dissociation assay by the single pathogenic PF1-8-15 IgG mAb. These findings indicate that the pathogenic mAb may cause loss of cell adhesion by a p38MAPK-independent mechanism, possibly by steric hindrance as mentioned in previous PV studies [7,14] or other signaling pathways. Our data is contradictory to a previous study using a passive transfer mouse model with purified PF IgG, in which blister formation was blocked by SB202190 [11] or a study using atomic force experiment that purified IgG from two PF patients did not disrupt trans-adhesion of Dsg1 [29]. The difference may be due to the source of IgG; a half- and-half mixture of anti-Dsg1 IgG mAbs in our study and purified PF IgG from sera in the previous studies, which should contain pathogenic and non-pathogenic anti-Dsg1 IgG in various mixture ratios among different PF patients. It may be possible that the mixture ratio between pathogenic and non-pathogenic anti-Dsg1 IgG influences the pathogenicity. An alternative explanation is that the difference of study design may affect the result.
In summary, our results suggest that, as in PV, in PF the polyclonal nature of anti-Dsg1 antibody is a factor that influences pathogenic activity in blister formation through p38MAPK-dependent clustering of Dsg1. Not only pathogenic Abs but also non-pathogenic Abs coordinately contribute to blister formation in PF. However, at least one pathogenic antibody that directly interferes cell adhesion may be necessary for the clustering and blister formation.
Supplementary Material
Acknowledgments
We thank Ms. Mayumi Ishii and Ms. Yoko Araki for technical assistance. This study was supported by Grants-in-Aid for Scientific Research and Strategic Research Basis Formation Supporting Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan, JSPS KAKENHI Grant Number (15K09749), grants from Toho University School of Medicine, Japan and a grant (NIH R01 AR052672) to JRS. Use of patient skins was approved by Toho university ethics committee (Number 25079).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jdermsci.2016.12.010.
Footnotes
This work was done at Tokyo, Japan.
Conflict of interest
The authors have no conflict of interest to declare.
References
- 1.Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355(17):1800–1810. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- 2.Owen GR, Stokes DL. Exploring the nature of desmosomal cadherin associations in 3D. Dermatol Res Pract. 2010;2010:930401. doi: 10.1155/2010/930401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 2013;116:95–118. doi: 10.1016/B978-0-12-394311-8.00005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol. 2001;167(9):5439–5448. doi: 10.4049/jimmunol.167.9.5439. [DOI] [PubMed] [Google Scholar]
- 5.Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170(4):2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 6.Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115(4):888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 2008;181(3):1825–1834. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- 8.Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, Didona B, Cianchini G, Hertl M, Eming R, Amagai M, Ohyama B, Hashimoto T, Sloostra J, Sallusto F, Zambruno G, Lanzavecchia A. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. 2012;122(10):3781–3790. doi: 10.1172/JCI64413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y. Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. 1995;105(3):329–333. doi: 10.1111/1523-1747.ep12319948. [DOI] [PubMed] [Google Scholar]
- 10.Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103(34):12855–12860. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. 2008;173(6):1628–1636. doi: 10.2353/ajpath.2008.080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, Rubenstein DS. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 2010;285(12):8936–8941. doi: 10.1074/jbc.M109.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waschke J, Spindler V. Desmosomes and extradesmosomal adhesive signaling contacts in pemphigus. Med Res Rev. 2014;34(6):1127–1145. doi: 10.1002/med.21310. [DOI] [PubMed] [Google Scholar]
- 14.Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, Amagai M, Kowalczyk AP. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One. 2012;7(12):e50696. doi: 10.1371/journal.pone.0050696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128(4):939–948. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokouchi M, Saleh MA, Kuroda K, Hachiya T, Stanley JR, Amagai M, Ishii K. Pathogenic epitopes of autoantibodies in pemphigus reside in the amino-terminal adhesive region of desmogleins which are unmasked by proteolytic processing of prosequence. J Invest Dermatol. 2009;129(9):2156–2166. doi: 10.1038/jid.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne AS, Siegel DL, Stanley JR. Targeting pemphigus autoantibodies through their heavy-chain variable region genes. J Invest Dermatol. 2007;127(7):1681–1691. doi: 10.1038/sj.jid.5700790. [DOI] [PubMed] [Google Scholar]
- 18.Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol. 2005;124(5):939–946. doi: 10.1111/j.0022-202X.2005.23714.x. [DOI] [PubMed] [Google Scholar]
- 19.Rader C, Popkov M, Neves JA, Barbas CF., 3rd Integrin alpha(v)beta3 targeted therapy for Kaposi’s sarcoma with an in vitro evolved antibody. FASEB J. 2002;16(14):2000–2002. doi: 10.1096/fj.02-0281fje. [DOI] [PubMed] [Google Scholar]
- 20.Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 2006;281(11):7623–7634. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- 21.Ko CJ, McNiff JM. Punctate pemphigus: an underreported direct immunofluorescence pattern. J Cutan Pathol. 2014;41(3):293–296. doi: 10.1111/cup.12272. [DOI] [PubMed] [Google Scholar]
- 22.van der Wier G, Pas HH, Kramer D, Diercks GF, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J Invest Dermatol. 2014;134(8):2287–2290. doi: 10.1038/jid.2014.140. [DOI] [PubMed] [Google Scholar]
- 23.Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 2008;128(3):738–740. doi: 10.1038/sj.jid.5701080. [DOI] [PubMed] [Google Scholar]
- 24.Kawasaki H, Tsunoda K, Hata T, Ishii K, Yamada T, Amagai M. Synergistic pathogenic effects of combined mouse monoclonal anti-desmoglein 3 IgG antibodies on pemphigus vulgaris blister formation. J Invest Dermatol. 2006;126(12):2621–2630. doi: 10.1038/sj.jid.5700450. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem. 2007;282(24):17866–17876. doi: 10.1074/jbc.M607963200. [DOI] [PubMed] [Google Scholar]
- 26.van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of acantholysis in pemphigus foliaceus re-examined from the current perspective. Br J Dermatol. 2012;167(6):1265–1271. doi: 10.1111/j.1365-2133.2012.11173.x. [DOI] [PubMed] [Google Scholar]
- 27.Oktarina DA, van der Wier G, Diercks GF, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol. 2011;165(3):552–562. doi: 10.1111/j.1365-2133.2011.10463.x. [DOI] [PubMed] [Google Scholar]
- 28.Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. 2011;286(2):1283–1291. doi: 10.1074/jbc.M110.172874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 2005;115(11):3157–3165. doi: 10.1172/JCI23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.