

Abstract
Anti-hCD20 is a therapeutic monoclonal antibody (mAb) that is clinically used to treat B-cell lymphoma. Some lymphomas are resistant to anti-hCD20; others relapse after treatment with anti-hCD20. Using a syngeneic immunocompetent mouse model, we observed that targeting lymphoma with interferon-α (IFNα) abolished resistance of B-cell lymphoma to anti-CD20 while limiting interferon (IFN)-associated systemic toxicity in the host. Control of tumors by a fusion of anti-CD20 and IFNα (anti-CD20-IFNα) depended on existing tumor-infiltrating CD8+ T cells. Although lymphomas were resistant to IFN-directed killing, IFN-exposed tumor cells became the dominant antigen-presenting cells (APCs) for the reactivation of tumor-infiltrating CD8+ T cells that then controlled those lymphomas. Anti-CD20-IFNα also abolished checkpoint blockade resistance in advanced B-cell lymphoma. Our findings indicate that anti-CD20-IFNα eradicates B-cell lymphoma by employing tumor cells as APCs to reactivate tumor-infiltrating CD8+ T cells and synergizing with anti-PD-L1 treatment.
Keywords: B-cell lymphoma, interferon, cancer immunotherapy, checkpoint blockade, tumor-infiltrating CD8+ T cells
Introduction
Lymphoma is one of the most common cancers (1). Non-Hodgkin lymphomas (NHLs) account for 80 to 90% of all lymphomas. Approximately 85% of all NHLs are of B-cell origin (B-NHLs) (2). The antibody (Ab) to hCD20, rituximab (Rituxan), which is effective against many B-NHLs (3), functions by induction of tumor apoptosis (4), Ab-dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP), Ab stimulation of complement-dependent cytotoxicity (CDC), complement-enhanced ADCC (CR3-ADCC), a vaccine effect, and a T cell-dependent immune response (5,6). Although rituximab is standard therapy for many B-NHLs, patients have an overall response rate of only 40 to 50% in relapsed or refractory low-grade B-NHLs and a median time to progression (MTTP) of approximately 9 months (7). New therapeutic options are needed to overcome anti-CD20 resistance and effectively treat B-cell lymphoma.
Interferon-α (IFNα), a protein of the type I interferon family, is used to treat NHLs and other cancers.(8) IFNα functions in cancer immunotherapy via direct anti-proliferation or pro-apoptosis of tumor cells and by up-regulating CD20 expression on tumor cells, enhancing the effect of ADCC or ADCP, and bridging the innate and adaptive immune responses (9). Use of IFNα as a maintenance therapy for follicular lymphoma (FL) improves progression-free survival (PFS). However, systemic administration of IFNα can cause severe toxicities, such as neutropenia, fatigue, liver injury, pulmonary embolism, and psychiatric disorders (10,11). The short half-life of IFNα in the blood and the wide distribution of the type I interferon receptor (IFNAR) on all nucleated cells make it difficult for IFNα to reach target tissues (12,13).
Human IFNα2b fused to a monoclonal antibody (mAb) to hCD20 (veltuzumab) had a potent antitumor effect. Treatment with the fusion protein produced a survival rate of 100%, whereas survival was 20% with either agent alone, or 40% with rituximab therapy in a human lymphoma xenograft model (14). Targeted delivery of IFNα to the tumor microenvironment by hCD20 mAb induces tumor cell apoptosis in vitro and could lead to tumor control in vivo (15). Treatment with anti-hCD20-hIFNα can reverse the rituximab resistance of B-NHL in vitro by inhibiting cell proliferation and inducing cell death (16). However, not all lymphomas respond to anti-CD20 or IFN with apoptosis. Here, we used a syngeneic immunocompetent mouse model and observed that targeting B-cell lymphoma with anti-CD20-IFNα abolished anti-CD20 resistance in B-cell lymphoma while limiting IFN-associated toxicity. Tumor regression occurred via a mechanism dependent on pre-existing tumor-infiltrating CD8+ T cells. Understanding the immune mechanism of IFN-mediated tumor control will lead to more effective treatment combinations.
Materials and Methods
Mice
Wild-type (WT) BALB/c and BALB/c nude mice were purchased from Vital River Laboratories (Beijing, China). CL-4 mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, United States). CD11c-DTR (depletion of CD11c-expressing cells via the diphtheria toxin receptor) mice were bred and housed at the Institute of Biophysics, CAS. μMT−/− mice were kindly provided by Dr. Zhihai Qin of the Institute of Biophysics, CAS. All of the mice were maintained under specific pathogen-free conditions and were used between 6 and 12 weeks of age in accordance with the experimental animal guidelines set by the Institutional Animal Care and Use Committee of the Institute of Biophysics, CAS (SYXK2014-44).
Cell lines and reagents
A20 is a murine B-cell lymphoma cell line with a BALB/c background and was purchased from ATCC in 2013. A20-HA was selected as a single clone with 2 μg/ml puromycin (InvivoGen, San Diego) after being transduced by lentivirus expressing hemagglutination antigen (HA) in 2014. BL3750 is a B-cell lymphoma cell line with a C57BL/6 background and was provided by Dr. Holbrook Kohrt (Stanford University Medical Center, Stanford) in 2015. L929 is a murine fibroblast cell line with a C3H/An background, which was provided by Dr. Zhihai Qin (Institute of Biophysics, CAS) in 2013. All cell lines were maintained according to the method used by the ATCC and were tested and found to be free of mycoplasma contamination. The cell lines were authenticated by flow cytometry and morphology. Anti–PD-L1 blocking mAb (10F.9G2) was purchased from Bioxcell. The FcγRII/III blocking Ab (clone 2.4G2), CD8-depleting Ab (clone 2.43) and CD4-depleting Ab (clone GK1.5) were produced in house. Diphtheria toxin (DT) was purchased from CALBIOCHEM (Darmstadt) and prepared according to the manufacturers’ instructions. Clophosome, which was used to deplete macrophage cells, was purchased from FormuMax (Sunnyvale).
Production of the anti-CD20-IFNα fusion protein
The variable region sequence of the mAb to mouse CD20 (18B12, Biogen) was synthesized by Invitrogen and cloned into the pEE12.4 expression plasmid (Lonza, Basel, Switzerland) as a single-chain variable fragment (ScFv) with a human IgG1 Fc in the C-terminal region. Murine IFNα4a was inserted into the N-terminal region of ScFv with a (SG4)4 linker to make the anti-CD20-IFNa fusion protein. The plasmid was transiently transfected into FreeStyle 293-F cells, and the fusion protein in the supernatant was purified using a protein A-Sepharose column (GE Healthcare). The non-targeting control protein IFNα-IgG1 was obtained in a similar manner except that the variable region sequence of the Ab was from anti-HBsAg H25B10 hybridoma cells. Please refer to the supplementary material for all protein gene sequences
Tumor growth and treatments
A total of 2 × 106 to 3 × 106 A20 tumor cells were subcutaneously (s.c.) transplanted into the flanks of the mice. Tumor volumes were measured along three orthogonal axes (a, b, and c) and calculated as tumor volume = abc/2. Tumors were grown for 9 to 14 days to reach a size of 100 mm3 and then treated with anti-CD20-IFNα or control hIgG intratumorally (i.t.) or intravenously (i.v.). For the CD8-depletion experiment, 200 μg of anti-CD8 Ab (clone 2.43) was injected intraperitoneally (i.p.) one day before anti-CD20-IFNα treatment. For the CD4-depletion experiment, 200 μg of CD4 mAb (clone GK1.5) was injected intraperitoneally (i.p.) one day before anti-CD20-IFNα treatment. To block lymphocyte trafficking, 25 μg of FTY720 was injected i.p. one day before anti-CD20-IFNα treatment. A total of 10 μg of FTY720 was administered every day to maintain the blockade. For the PD-L1 blockade experiment, 50 μg of anti-mouse PD-L1 Ab (clone 10F.9G2) was injected i.t. simultaneously with anti-CD20-IFNα treatment.
Binding assay
A20 cells (2 × 105) were stained with indicated proteins and single-cell suspensions of A20 cells (2 × 105) were incubated with anti-CD16/32 (anti-FcγIII/II receptor, clone 2.4G2) for 20 minutes. Anti-CD20, anti-CD20-IFNα and hIgG were serially diluted and added to the cells to achieve final dose of 0–500 ng. Cells were incubated on ice for 20 min, washed twice, and then incubated with anti-human IgG Fcγ-PE for 20 min on ice. After washing twice, asmples were run on a FACSCalibur (BD). Data were analyzed using FlowJo software (TreeStar).
Flow cytometric sorting and analysis
Single-cell suspensions were incubated with anti-CD16/32 (anti-FcγIII/II receptor, clone 2.4G2) for 20 min and then stained with conjugated Abs. All fluorescent-labeling mAbs were purchased from BioLegend or eBioscience. Samples were analyzed on a FACSCalibur (BD) or FACSFortessa flow cytometer (BD), and cells were sorted on a FACSAria III Cell Sorter (BD). Data were analyzed using FlowJo software (TreeStar).
Apoptosis assay
A total of 5 × 104 A20 cells and BL3750 cells were incubated with mitomycin C (1 μg/ml) or 2,000 pM hIgG, IFNα-IgG, anti-CD20 and anti-CD20-IFNα in 96 round-well plates at 37°C for 48 hours. Cells were stained with Annexin V-Alexa Fluor® 647 and propidium iodide (PI) to distinguish populations of early apoptotic (Annexin V+PI−), late apoptotic (Annexin V+PI+) and necrotic (Annexin V−PI+) cells. The percentage of apoptotic cells was calculated for each sample as the sum of early apoptotic and late apoptotic cells.
Measurement of IFNγ-secreting tumor-infiltrating CD8+ T cells by ELISPOT assay
A20-bearing mice were i.t. treated with 3 μg of anti-CD20-IFNα and control Abs on day 16. Two days later, tumor tissues were digested, and CD8+ T cells were purified by FACS sorting. Approximately 2 × 104 CD8+ T cells and 5 × 104 WT BALB/c mice splenocytes were mixed together with 0.1 mg/ml mitomycin C-treated A20 cells for 44 hours. IFNγ production was determined with an IFNγ ELISPOT assay kit according to the manufacturer’s manual (BD). The visualized cytokine spots were quantified using the ImmunoSpot Analyzer (CTL).
Generation of bone marrow chimeras
WT BALB/c mice were irradiated with a single dose of 1,000 rad. Irradiated mice were adoptively transferred i.v. with 5 × 106 CD11c-DTR Tg of donor bone marrow cells the next day. Mice were maintained on sulfamethoxazole and trimethoprim (Bactrim) antibiotics diluted in drinking water for 4 weeks after reconstitution. Mice were injected with tumor cells approximately 12 weeks after reconstitution.
APC presentation assay
For the ex vivo APC cross-presentation assay, A20-HA tumor-bearing BALB/c nude mice were treated i.t. with 3 μg of anti-CD20-IFNα or anti-CD20 Ab on day 12. Two days later, the mice were sacrificed, and the tumor cells were digested with 1 mg/ml collagenase IV (Sigma-Aldrich, St. Louis) and 20 U/ml DNase I (Sigma-Aldrich, St. Louis) at 37°C for 40 minutes. Tumor cells (CD45+B220+), DCs (CD45+B220−CD11c+F4/80−), and macrophages (CD45+B220−CD11c−F4/80+) were sorted using a FACSAria III Cell Sorter (BD). A total of 2 × 104 purified CL4 T cells were mixed with an approximate number of APCs in ratios of 4:1, 8:1 and 16:1. Two days later, the supernatants were collected, and IFNγ was measured by ELISA. For the in vitro tumor cell presentation assay, 2.5 × 105 A20-HA cells and 5 × 104 purified CL4 T cells were incubated with PMA/ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin, obtained from Sigma-Aldrich, St. Louis) or 50 pM anti-CD20 and anti-CD20-IFNα in 96 round-well plates at 37°C. Eighteen hours later, brefeldin A was added to the supernatants at a final concentration of 5 μg/ml. Six hours later, the cells were first stained with APC-conjugated anti-mouse CD8α Ab (53-6.7) as a surface CD8 marker before fixation/permeabilization and intracellularly stained for IFNγ (XMG1.2). All of the reagents and Abs were purchased from eBioscience, and surface and intracellular staining was performed according to the manufacturer’s protocols.
Statistical analyses
Data were analyzed using Prism 6.0 software (GraphPad) and presented as the mean ± SEM. The P values were assessed using two-tailed unpaired Student’s t-test or two-way analysis of variance with the following thresholds for statistical significance: *P < 0.05; **P < 0.01 and ***P < 0.001.
Results
Delivery of IFNα to the tumor inhibits B-cell lymphoma growth
Both anti-CD20 (3,5) and IFNα (9) are therapeutically beneficial in the treatment of NHLs. We asked whether combined anti-CD20 and IFNα treatment would synergistically reduce the CD20+ A20 B-cell lymphoma burden in a syngeneic immunocompetent mouse model. BALB/c mice were inoculated s.c. with A20 cells. Nine days later, 100 μg of anti-CD20 was administered i.v. to deplete most normal B cells (Supplementary Fig. S1). On days 10–13, mice were daily given 10 μg of IFNα-IgG1, anti-CD20, or control PBS via i.v. injection. Systemic administration of anti-CD20 or IFNα-IgG1 could not control the tumor growth (Fig. 1A). This result indicates that this model is an appropriate for mimicking clinical anti-CD20 treatment resistance. Considering that all nucleated cells express IFNAR (13), and that less than 0.01% of s.c.-injected IFNα would accumulate in the target tissue (12), targeting IFNα to the tumor environment could enhance tumor control. To test this hypothesis, we administered 3 μg of IFNα-IgG1, anti-CD20, or control hIgG i.t. on days 12 and 14. The intratumoral delivery of this dose of IFNα-IgG1 eliminated the tumor (Fig. 1B). Thus, administration of IFNα directly to the tumor controlled tumor growth more effectively than did systemic administration. This suggests that the target for IFNα-induced tumor control is likely inside the tumor microenvironment.
Figure 1. Targeting delivery of IFNα to the tumor microenvironment using anti-CD20 efficiently enhances the antitumor effect.

(A) WT BALB/c mice (n = 5/group) were injected subcutaneously (s.c.) with A20 cells (3 × 106). Then, 100 μg of anti-CD20 was administered intravenously (i.v.) on day 9. A total of 10 μg of IFNα-IgG1, anti-CD20, or control PBS was administered i.v. on days 10, 11, 12, and 13. (B) WT BALB/c mice (n = 5/group) were injected s.c. with A20 cells (3 × 106) and treated intratumorally (i.t.) with 3 μg of IFNα-IgG1, anti-CD20, or control hIgG on days 12 and 14. (C) Structure of the anti-CD20-IFNα fusion protein. (D) WT BALB/c mice (n = 5/group) were injected s.c. with A20 cells (3 × 106); then, 100 μg of anti-CD20 was administered i.v. on day 9. The average tumor size was 68 mm3. A total of 10 μg of IFNα-IgG1, anti-CD20, anti-CD20-IFNα, or control PBS was administered i.v. on days 10, 11, 12, and 13. Data represent mean ± SEM of two (B, D) or three (A) independent experiments. **P < 0.01, ***P < 0.001.
We proposed that targeting the delivery of IFNα to lymphoma by linking IFNα to anti-CD20 Ab could enhance antitumor activity. Anti-CD20-IFNα fusion proteins were constructed (Fig. 1C) and purified (Supplementary Fig. S2A) as described in the methods section. Binding of the fusion proteins to murine CD20 (mCD20) was assessed by flow cytometry using the A20 cell line. The anti-CD20-IFNα fusion protein and anti-CD20 both bound mCD20-expressing murine lymphoma cells with similar avidity (Supplementary Fig. S2B). We analyzed IFNα bioactivity of the anti-CD20-IFNα fusion protein using a VSV-GFP (Vesicular stomatitis virus expressing green fluorescent protein) infection assay. Anti-CD20-IFNα and IFNα-IgG equally inhibited VSV-GFP infection (Supplementary Fig. S2C). We concluded that our anti-CD20-IFNα fusion protein maintained the binding activity of anti-CD20 and the bioactivity of IFNα.
We asked whether delivery of IFNα to the tumor site through anti-CD20 Abs could overcome anti-CD20 and IFNα resistance. In tumor-bearing mice i.v. treated with the fused anti-CD20-IFNα (Fig. 1D), growth of A20 lymphoma was inhibited; neither anti-CD20 Ab nor IFNα systemic treatment alone had any therapeutic effect. After fusion protein treatment, 40% of tumors were eradicated. Thus targeted delivery of IFNα by anti-CD20 to the tumor microenvironment is more effective than anti-CD20 or IFNα-hIgG1 treatment alone.
Tumor regression mediated by anti-CD20-IFNα depends on adaptive immunity
Anti-CD20-IFNα administered i.v. can effectively control B cell lymphoma (Fig. 1D). We administered anti-CD20-IFNα i.t or i.v., to determined whether peripheral lymphoid tissue, or the tumor, was the primary location where anti-CD20-IFNα functions. The antitumor effect was better after i.t., rather than the i.v., administration of anti-CD20-IFNα (Fig. 2A), similar to the effect observed with IFNα-IgG1 in both A20 and BL3750 tumor models (Supplementary Fig. S3A–B). This indicates that the tumor tissue is the main site for the effect of anti-CD20-IFNα. To study how Ab-IFN works inside tumor tissues and rule out effects from peripheral tissues, we used i.t. injection. IFNα can induce tumor cell apoptosis and cause tumor regression (15). We tested the sensitivity of A20 cells to IFN-mediated apoptosis using an in vitro culture system. In contrast to BL3750 cells, which are B-cell lymphoma cells with a C57BL/6 background and are sensitive to IFNα-induced apoptosis, IFNα did not induce apoptosis of A20 cells (Fig. 2B and Supplementary Fig. S4). This raises the possibility that IFN-mediated tumor regression may depend not on direct IFN-mediated killing but instead on host immune-mediated tumor clearance. To test this hypothesis, we s.c. inoculated A20 cells into syngeneic BALB/c nude mice (T-cell deficiency). Three doses of intratumoral anti-CD20-IFNα had no effect on tumor growth in nude mice (Fig. 2C); this result indicates that T cells are essential for the therapeutic effect of anti-CD20-IFNα.
Figure 2. The anti-tumor effects of anti-CD20-IFNα depend on T cells.

(A) WT BALB/c mice (n = 6/group) were injected subcutaneously (s.c.) with A20 cells (3 × 106) and treated intratumorally (i.t.) with 3 μg of anti-CD20-IFNα or control hIgG on days 11 and 14. (B) BL3750 (n = 5/group) or A20 cells (n = 8/group) were incubated with 2,000 pM of the respective treatment for 48 hours. The percentage of apoptotic cells was quantified for each sample as the sum of early apoptotic and late apoptotic cells. (C) BALB/c nude mice (n = 5/group) were injected subcutaneously (s.c.) with A20 cells (2 × 106) and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 11, 13, and 15. Data represent mean ± SEM of two (B, C) or three (A) independent experiments. ***P < 0.001.
Tumor-infiltrating CD8+ T cells play a key role in eliminating tumors
To determine which subsets of T cells are involved in tumor regression mediated by anti-CD20-IFNα, WT BALB/c mice bearing established A20 tumors were i.t. treated with fusion protein with CD8+ or CD4+ T cells ablated by i.p. administration of anti-CD8 or anti-CD4 mAbs. CD8+ T cell depletion abolished the therapeutic effect of anti-CD20-IFNα administered either i.v. (Supplementary Fig. S5) or i.t. (Fig. 3A). Depletion of CD4+ T cells did not affect treatment efficacy (Supplementary Fig. S6). Thus, CD8+ T cells but not CD4+ T cells are essential for mediated tumor regression mediated by anti-CD20-IFNα.
Figure 3. The therapeutic effect of anti-CD20-IFNα is dependent on tumor-infiltrating CD8+ T cells.

(A) WT BALB/c mice (n = 5/group) were injected subcutaneously (s.c.) with A20 cells (3 × 106) and treated intratumorally (i.t.) with 3 μg of anti-CD20-IFNα or control hIgG on days 14 and 18. CD8+ T cell depleting Ab (clone 2.43; 200 μg/mouse) was administered intraperitoneally (i.p.) twice a week starting on day 13. (B) WT BALB/c mice (n = 5/group) were injected s.c. with 3 × 106 A20 cells on the right flank and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 10 and 12. One day before administration of the fusion proteins, tumor-bearing mice received 25 μg of FTY720 i.p., and the dose was reduced to 10 μg per mouse every two days starting on day 9. A total of 20 μg of CD8-depleting Ab was administered i.t. on days 10, 12, and 14. (C) WT BALB/c mice (n = 5/group) were injected s.c. with A20 cells (3 × 106) on the right flank. Two days after treatment with 3 μg of IFNα-IgG1, anti-CD20, anti-CD20-IFNα, or control hIgG1 on day 16, tumor-infiltrating CD8+ T cells were sorted, and an IFNγ ELISPOT assay was performed with mitomycin C-treated A20 cells and naïve splenocytes from WT mice. (D) WT BALB/c mice (n = 15/group) were injected s.c. with A20 cells (3 × 106) on the right flank and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 14, 16, and 18. Tumor-infiltrating CD8+ T cells were sorted on day 24. BALB/c nude mice (n = 5/group) were injected s.c. with A20 cells (2 × 106), and the cells were transferred with previously sorted CD3+CD8+ T cells (5 × 105) on day 1. Data represent mean ± SEM of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Since i.t. injection of the IFN-fusion protein resulted in a more potent and rapid antitumor effect than did systemic delivery of IFNα, IFNα might induce antitumor immunity inside tumor tissues through reactivating exhausted T cells. We investigated whether the pre-existing TILs in the A20 model were sufficient for the responsiveness of this model to anti-CD20-IFNα treatment. FTY720, a sphingosine 1-phosphate (S1P) receptor agonist, can potently inhibit the egression of naïve and effector lymphocytes from the LN into the circulation and peripheral tissues (17) and thus limit migration of primed T cells into tumor tissues. One day after FTY720 treatment initiation, there was a nearly 90% reduction in peripheral T cells. The number of circulating T cells was further decreased after long-term administration of FTY720 (Supplementary Fig. S7A–B). Antitumor effects of the fusion protein remained potent in the presence of FTY720 (Fig. 3B). Furthermore, we depleted tumor-infiltrating CD8+ T cells by i.t. administration of low doses (20 μg) of anti-CD8 mAb. In the absence of tumor-infiltrating CD8+ T cells after FTY720 treatment, the therapeutic effect of anti-CD20-IFNα was abrogated (Fig. 3B). To explore whether anti-CD20-IFNα treatment could enhance the tumor-specific T cell response in the tumor microenvironment, we compared CD8+ T cell IFNγ production after anti-CD20-IFNα or control Ab treatment. Mice harboring established A20 tumors were treated i.t. with anti-CD20-IFNα or control Ab, and CD8+ T cells were sorted from the tumor and restimulated with mitomycin-C-treated A20 tumor cells. IFNγ+ cells were measured by an enzyme-linked immunospot (ELISPOT) assay 2 days after co-culture. More IFNγ spot-forming cells were observed in the IFNα-treated group than in the anti-CD20 treatment group (Fig. 3C). In addition, to explore the activity of CD8+ T cells after anti-CD20-IFNα treatment, mice harboring established A20 tumors were treated i.t. with anti-CD20-IFNα or control hIgG on days 14, 16 and 18. CD8+ T cells were sorted from the tumor tissue on day 24 and transferred to tumor-bearing nude mice. The CD8+ T cells from the anti-CD20-IFNα-treated immunocompetent mice effectively inhibited tumor growth in immune-deficient mice (Fig. 3D). Thus, tumor-infiltrating CD8+ T cells play a key role in the eradication of B-cell lymphoma.
Conventional APCs are not essential for tumor control
The IFNAR on DCs is essential for anti-EGFR-IFNβ-mediated tumor regression, The DC is a major APC for cross-presentation.(18) To test whether DCs are the dominant APCs activating tumor-infiltrating CD8+ T cells, we used CD11c-DTR-reconstituted BALB/c mice bearing A20 tumors. Anti-CD20-IFNα eradicated the tumor even in the absence of DCs (Fig. 4A). Anti-CD20-IFNα therapy generated protection against rechallenge (Fig. 4B), suggesting development of a protective memory response. The antitumor vaccine effect of anti-human CD20 mAb is engaged by macrophages, which selectively transfer antigens to DCs to promote long-term immunity against cancer.(19) Therefore, we used Clophosome, a drug that can deplete macrophages, to determine whether macrophages play a key role in controlling tumor growth. However, the antitumor effect of anti-CD20-IFNα was observed in the absence of macrophages (Fig. 4C), along with the establishment of a protective memory response (Fig. 4D). To exclude the compensatory effects between DCs and macrophages, we depleted both of these cells simultaneously. No differences were observed for tumor growth in the mice deficient in both DCs and macrophages (Fig. 4E). Given that B cells may function as APCs, we used μMT−/− mice (20), which are deficient in mature B cells, to study whether B cells are required for tumor regression. Host normal B cells also did not function as APCs (Fig. 4F). Thus, traditional APCs are not essential for controlling tumor growth in A20 B-cell lymphoma.
Figure 4. Conventional APCs are not required for CD8+ T cell-dependent tumor control.

(A) A20-bearing CD11c-DTR-reconstituted BALB/c mice (n = 5/group) were injected subcutaneously (s.c.) with A20 cells (1 × 107) and treated intratumorally (i.t.) with 3 μg of anti-CD20-IFNα or control hIgG on days 18 and 21. DT was administered every two days starting on day 17. (B) Approximately 45 days after the tumor rejection from (A), the mice were rechallenged with A20 cells (3 × 107). Naïve WT BALB/c mice were used as the control. (C) WT BALB/c mice (n = 8/group) were injected s.c. with A20 cells (3 × 106) and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on day 10. The macrophage-depleting reagent (Clophosome, 200 μg/mouse) or control liposome was administered intraperitoneally (i.p.) on days 9 and 12. (D) Approximately 45 days after the tumor rejection in (C), the mice were rechallenged with A20 cells (1.5 × 107). Naïve WT BALB/c mice were used as the control. (E) A20-bearing CD11c-DTR-reconstituted BALB/c mice (n = 5/group) were injected s.c. with 1 × 107 A20 cells and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 10 and 13. DT was administered i.p. on days 9, 11, and 13. Clophosome (200 μg/mouse) or control liposome was administered i.p. on days 9 and 13. (F) μMT−/− BALB/c mice (n = 6/group) were injected s.c. with A20 cells (3 × 106) on the right flank and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 10 and 12. Data represent mean ± SEM of two independent experiments. ***P < 0.001.
Tumor cells as APCs activate the tumor-infiltrating CTL response
The antitumor effect started early on day 3 post-treatment (Figs. 2A and 3C), in an intratumor CTL-dependent manner, suggesting that A20 B-cell lymphoma cells could act as APCs. We confirmed that CD86 and MHC class I expression levels on tumor cells increased in the presence of IFNα (Fig. 5A–B). Next, we evaluated whether these activated tumor cells could be APCs and stimulate antigen-specific CD8+ T cells. CL4 T cells, whose TCR is specific for the hemagglutinin antigen (HA) CD8 epitope, were incubated with A20 or A20-HA (HA-expressing) cells in the presence of PMA/ionomycin, anti-CD20, and anti-CD20-IFNα. Anti-CD20-IFNα alone could not activate CL4 T cells in the absence of its antigens. However, anti-CD20-IFNα induced CL4 T cells to produce IFNγ only in the presence of A20-HA cells (Fig. 5C). These data indicate that anti-CD20-IFNα can stimulate self-presentation by tumor cells, which then activate antigen-specific tumor-infiltrating CTLs.
Figure 5. The direct antigen-presenting ability of tumor cells is dramatically enhanced after anti-CD20-IFNα treatment.

(A–B) A20 cells (5 × 104) per well were incubated with 20 pM indicated antibodies in 96 round-well plates (n = 6/group). 48 hours later, tumor cells were collected and the expression of CD86 (A) or MHC class I (B) was analyzed by flow cytometry. (C) Splenocytes and inguinal LN cells were collected from CL4 Tg mice and Thy1.1+TCRVβ8+CD8+ T cells were sorted. Sorted T cells were co-cultured with A20-HA cells in the presence of PMA/ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin) or 50 pM anti-CD20 and anti-CD20-IFNα in 96 round-well plates for 24 hours (n = 8/group). Six hours before flow cytometry detection, brefeldin A (5 μg/ml) was added. Flow cytometry patterns and gate frequencies in percentages (left panel), and statistical results (right panel). (D) BALB/c nude mice (n = 5/group) were injected subcutaneously (s.c.) with 2 × 106 A20-HA cells and treated intratumorally (i.t.) with 3 μg of anti-CD20 or anti-CD20-IFNα on day 12. Two days later, macrophages (CD45+B220−CD11c−F4/80+), DCs (CD45+B220−CD11c+F4/80−), and tumor cells (CD45+B220+) were sorted from tumor tissues. A total of purified CL4 T cells (2 × 104) were mixed with an approximate number of APCs in ratios of 4:1, 8:1 and 16:1. Two days later, the supernatants were collected, and IFNγ was measured by ELISA. Data represent mean ± SEM of two independent experiments. ***P < 0.001.
To verify whether tumor cells could be the dominant APCs, we sorted out various APCs from the Ab-treated tumor tissues and co-incubated them with CL4 T cells in the presence of 20pM Abs for 48 hours. Compared to the group treated with anti-CD20, the antigen-presenting ability of APCs was improved after anti-CD20-IFNα treatment (Fig. 5D). Tumor cells, which make up more than 90% of the CD45+ population in the tumor microenvironment, displayed the most powerful antigen-presenting ability among three types of APCs on a per cell basis (Fig. 5D). Thus, tumor cells, but not conventional APCs, are the dominant APCs responsible for the anti-CD20-IFNα-mediated tumor control.
Anti-PD-L1 therapy could enhance anti-CD20-IFNα treatment
Persistent antigenic stimulation leads to CD8+ T cell exhaustion (21). Advanced tumors might develop adaptive resistance and exhaust TIL over time. Classic Hodgkin lymphoma (cHL) frequently exhibits genetic alterations that lead to overexpression of the programmed death-1 (PD-1) ligands; such tumors might be vulnerable to PD-1 blockade. Indeed, anti-PD-L1 Ab is associated with a favorable safety profile, but complete response is still rare (22,23). Type I and II IFNs can induce PD-L1 up-regulation in B cells (24). Consistent with this report, our in vitro (Fig. 6A) and in vivo (Fig. 6B) results demonstrated that PD-L1 levels were increased in tumor cells after IFNα treatment. This increase explains the difficulty in controlling advanced cancers using the fusion protein and raises the possibility that PD-L1 may be an adaptive resistance mechanism that allows lymphoma to escape immune destruction and resist IFN therapy. We proposed that the combination therapy of PD-L1 checkpoint blockade and IFNα might overcome the resistance of large tumors to a single treatment. To test the efficacy of this treatment strategy, WT BALB/c mice bearing established A20 tumors that were not responsive to single treatment were treated with the fusion protein and anti-PD-L1 Abs simultaneously. PD-L1 blockade further enhanced the antitumor efficacy of anti-CD20-IFNα for the advanced large tumors, and approximately 40% of the tumors were eradicated (Fig. 6C). Mice with complete tumor regression after combinational treatment resisted the A20 tumor rechallenge with 5 times the lethal dose; this result suggests long-term memory protection from relapse (Fig. 6D). Together, these data indicate that PD-L1 checkpoint blockade can enhance the therapeutic effect of anti-CD20-IFNα.
Figure 6. Anti-PD-L1 can enhance the therapeutic effect of anti-CD20-IFNα to control advanced B-cell lymphoma.

(A) A20 cells (5 × 104) per well were incubated with 20 pM of indicated antibodies in 96 round-well plates (n = 6/group). 48 hours later, tumor cells were collected and the expression of PD-L1 was analyzed by flow cytometry. (B) WT BALB/c mice (n = 5/group) were injected subcutaneously (s.c.) with A20 cells (3 × 106). A total of 3 μg of IFNα-IgG1, anti-CD20, anti-CD20-IFNα, or control hIgG was administered intratumorally (i.t.) on day 16. The average tumor size was 193 mm3. Two days later, tumor cells were collected and PD-L1 expression was analyzed by flow cytometry. (C) WT BALB/c mice (n = 13/group) were injected s.c. with A20 cells (3 × 106) and treated i.t. with 3 μg of anti-CD20-IFNα or control hIgG on days 16, 18 and 20. 50 μg of anti-PD-L1 (10F.9G2) or Rat IgG (rIgG) was administered i.t. on the same time. (D) Approximately 45 days after the tumor rejection from (C), the mice were rechallenged with A20 cells (1.5 × 107). Naïve WT BALB/c mice were used as the control. Data represent mean ± SEM of two independent experiments. **P < 0.01, ***P < 0.001.
Discussion
Some patients with B-cell lymphoma respond to treatment with either anti-CD20 or IFN; others do not respond or develop resistance. Toxicity with systemic delivery limits clinical use of IFN. Human IFNα2b fused to anti-hCD20 Ab (veltuzumab) exhibited more potent antitumor efficiency than either agent alone or in combination in human lymphoma xenografts (14). Linking type I IFN to tumor antigen-associated Abs increased the antitumor effect of type I IFN in a xenograft model by direct killing of IFNα-sensitive 38C13-hCD20 tumor cells (15). Treatment with anti-hCD20-hIFNα reversed rituximab resistance of B-NHL in vitro, resulting in inhibition of cell proliferation and induction of cell death (16). Because many lymphomas are resistant to anti-CD20- or IFN-mediated apoptosis, we wondered whether targeting tumor cells with IFNα could mobilize the adaptive immune system for tumor control. Using a syngeneic tumor model of IFNα-resistant A20 B-cell lymphoma, we observed that tumor regression mediated by anti-CD20-IFNα depends on adaptive immunity. Anti-CD20-IFNα enhanced the antigen-presenting function of lymphoma cells to reactivate tumor-infiltrating CD8+ T cells, which play a key role in eliminating the tumor. However, anti-CD20-IFNα also induced adaptive resistance after several doses of administration. Therefore, we used a combination of immune checkpoint blockade and anti-CD20-IFNα to overcome treatment-induced adaptive resistance. Indeed, anti-CD20-IFNα enhanced anti-PD-L1 treatment to enhance the antitumor effect for advanced B-cell lymphoma (Fig. 7).
Figure 7. Proposed model for anti-CD20-IFNα mediated tumor regression.
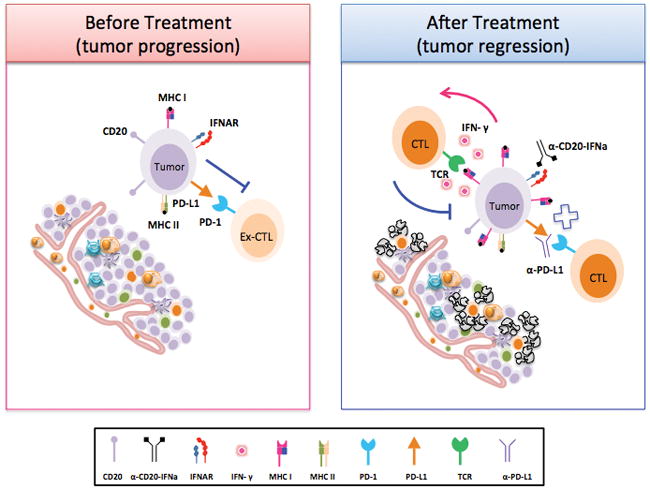
During the process of tumor establishment, infiltrating activated T cells become exhausted in the tumor microenvironment. Anti-CD20-IFNα fusion protein could deliver targeted IFNα to the tumor and dramatically enhance the antigen-presenting ability of tumor cells. In this situation, tumor cells could rapidly activate tumor-infiltrating CD8+ T cells, leading to a reduction of the tumor burden. Although both type I IFN and type II IFN could upregulate PD-L1 expression on tumor cells, anti-CD20-IFNα could also enhance anti-PD-L1 treatment to mediate the antitumor response.
Addition of rituximab or IFNα treatment to chemotherapy could increase patients’ response rates and effectively extend progression-free survival (25–27). Response to IFNα and rituximab/IFNα combination treatment is similar, and longer progression-free survival was observed in patients with extended IFNα treatment (20.9 months vs. 48.7 months) (28). Many B-cell lymphoma patients have no effective response to the current clinical treatment. Similar to many human lymphomas, A20 tumors are resistant to chemotherapy and to anti-CD20 Ab- or IFNα-induced tumor apoptosis and have poor responses to both anti-CD20 therapy and chemotherapy. These characteristics indicate that the A20 model may mimic lymphoma that is resistant to clinical treatment. Administration of IFNα i.t. reduced tumor burden; systemic administration of anti-CD20-IFNa was less effective. Thus, a proper IFNα concentration in the tumor is critical for tumor control. Therefore, we constructed an anti-CD20-IFNα fusion protein in order to achieve target delivery of IFNα into the tumor microenvironment. Some lymphomas may be more sensitive to direct killing by either anti-CD20 Ab or IFNα, whereas others depend more on adaptive immune responses.
IFN is a potent cytokine that activates antigen-presenting cells and increases cross-priming in the draining LN, which can then expand tumor-specific T cells. However, pre-existing tumor-infiltrating CD8+ T cells played a key role in eliminating B-cell lymphoma by using FTY720, an inhibitor of T-cell trafficking from the draining LN to tumor tissues. Injection of FTY720 before and during treatment did not change the therapeutic effect of anti-CD20-IFNα. This result raises the possibility that targeting the tumor microenvironment with IFNα might be sufficient to activate local APCs for TILs. DCs are the chief APCs that prime the CTL response (29,30). The antitumor vaccine effect of anti-human-CD20 Ab is engaged by macrophages via selective transfer of antigens to DCs; this result further promotes long-term immunity against cancer (19). Neither DCs nor macrophages were essential for tumor regression mediated by anti-CD20-IFNα or establishment of immune protection in our syngeneic B-cell lymphoma. In addition, anti-CD20-IFNα retained its antitumor function in the absence of host normal B cells. CD8+ T cells directly interact with antigen-expressing B lymphoma (31–33). A20 lymphoma B cells could be the APCs that affect the tumor-infiltrating CTL response. Unlike anti-CD20-treated tumor cells, the antigen-specific CD8+ T cells were activated by tumor cells treated with anti-CD20-IFNα and were the largest cell population in the tumor microenvironment. Tumor-infiltrating CD8+ T cells in the previously treatment-resistant tumor microenvironment might be activated when the APC function of lymphoma cells is significantly enhanced by treatment with the targeted Ab anti-CD20-IFNα. Our study demonstrates that tumor cells could be antigen-presenting cells but does not rule out that host cells can also contribute to antigen presentation.
During tumor progression, a single treatment loses its therapeutic efficacy because of intrinsic resistance and acquired immune tolerance (5,34,35). Many combination therapies have been proposed to treat advanced cancers, including costimulatory receptor agonists (anti-CD137) (36) or blockade of the antiphagocytic (“don’t eat me”) signal (anti-CD47) (37) in combination with CD20 mAb. Immune checkpoint therapy has led to breakthroughs in cancer therapy (38). The ligands for PD-1, PD-L1 (B7-H1), and PD-L2 (B7-DC) could also be upregulated in response to inflammation (39). Many B-cell lymphomas remain unresponsive or become unresponsive after initial favorable responses to anti–PD-L1 or IFN treatment. As a combination therapy, PD-L1 blockade could enhance the antitumor efficacy of anti-CD20-IFNα and reduce relapse rates for advanced large tumors that are resistant to either anti-CD20 Ab or the anti-CD20-IFNα fusion protein. Thus, the promising future of anti-CD20-IFNα therapy might be in combination with other immunotherapies, including anti–PD-L1, for the treatment of cancer.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by National Nature and Science Foundation of China grant (No. 81172814) to H.P (No 881202328) to Y.L, Key deployment project from Chinese Academy of Sciences (No. KFZD-SW-205) to H.P, Ministry of Science and Technology of China grant (No. 2016YFC1303400 and No. 2011DFA31250) to Y.-X.F., and National Science and Technology Major Project of China grant (No. 2012ZX10001006) to H.P. This research was in part supported by the U.S. National Institutes of Health grants CA141975 to Y.-X. F.
The authors thank Dr. Mingzhao. Zhu (Institute of Biophysics, CAS) for helpful suggestions and comments on the project. We also thank Daryl Harmon and Casey Timmerman for editing. μMT−/− mice were kindly provided by Z. Qin (the Institute of Biophysics, CAS). The authors also thank H. Su, Q. Li and S. Wei (Institute of Biophysics, CAS) for expert technical assistance.
Footnotes
Conflict of Interest Policy: No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: J. Liao, Y.-X. Fu
Development of methodology: J. Liao, Z. Ren, Y.-X. Fu
Acquisition of data: J. Liao, X. Liu, H. Xu, D. Xue
Analysis and interpretation of data: J. Liao, H. Peng, Y.-X. Fu
Writing, review, and/or revision of the manuscript: J. Liao, H. Peng, Y.-X. Fu
Administrative, technical, or material support: Y. Luan, H. Xu, Z. Sun, K. Yang
Study supervision: H. Peng, Y.-X. Fu
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Zappasodi R, de Braud F, Di Nicola M. Lymphoma Immunotherapy: Current Status. Front Immunol. 2015;6:448. doi: 10.3389/fimmu.2015.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366:2008–16. doi: 10.1056/NEJMct1114348. [DOI] [PubMed] [Google Scholar]
- 4.Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis. 2000;26:133–43. doi: 10.1006/bcmd.2000.0287. [DOI] [PubMed] [Google Scholar]
- 5.Cartron G, Trappe RU, Solal-Celigny P, Hallek M. Interindividual variability of response to rituximab: from biological origins to individualized therapies. Clin Cancer Res. 2011;17:19–30. doi: 10.1158/1078-0432.CCR-10-1292. [DOI] [PubMed] [Google Scholar]
- 6.Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116:926–34. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 7.Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica. 2010;95:135–43. doi: 10.3324/haematol.2008.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 9.Brassard DL, Grace MJ, Bordens RW. Interferon-alpha as an immunotherapeutic protein. J Leukoc Biol. 2002;71:565–81. [PubMed] [Google Scholar]
- 10.Solal-Celigny P, Lepage E, Brousse N, Reyes F, Haioun C, Leporrier M, et al. Recombinant interferon alfa-2b combined with a regimen containing doxorubicin in patients with advanced follicular lymphoma. Groupe d’Etude des Lymphomes de l’Adulte. N Engl J Med. 1993;329:1608–14. doi: 10.1056/NEJM199311253292203. [DOI] [PubMed] [Google Scholar]
- 11.Baldo P, Rupolo M, Compagnoni A, Lazzarini R, Bearz A, Cannizzaro R, et al. Interferon-alpha for maintenance of follicular lymphoma. Cochrane Database Syst Rev. 2010:CD004629. doi: 10.1002/14651858.CD004629.pub2. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki K, Aoki K, Ohnami S, Yoshida K, Kazui T, Kato N, et al. Adenovirus-mediated gene transfer of interferon alpha improves dimethylnitrosamine-induced liver cirrhosis in rat model. Gene Ther. 2003;10:765–73. doi: 10.1038/sj.gt.3301949. [DOI] [PubMed] [Google Scholar]
- 13.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 14.Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood. 2009;114:3864–71. doi: 10.1182/blood-2009-06-228890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115:2864–71. doi: 10.1182/blood-2009-10-250555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vega GG, Franco-Cea LA, Huerta-Yepez S, Mayani H, Morrison SL, Bonavida B, et al. Overcoming rituximab drug-resistance by the genetically engineered anti-CD20-hIFN-alpha fusion protein: Direct cytotoxicity and synergy with chemotherapy. Int J Oncol. 2015;47:1735–48. doi: 10.3892/ijo.2015.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiba K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther. 2005;108:308–19. doi: 10.1016/j.pharmthera.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, et al. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell. 2014;25:37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiLillo DJ, Ravetch JV. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell. 2015;161:1035–45. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat Med. 1998;4:627–30. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- 21.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 22.Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol. 2016;34:3733–9. doi: 10.1200/JCO.2016.67.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol. 2016;34:2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen IE, Ross SD, Borden SP, Monroe MW, Kupelnick B, Connelly JE, et al. Meta-analysis to assess the efficacy of interferon-alpha in patients with follicular non-Hodgkin’s lymphoma. J Immunother. 2001;24:58–65. doi: 10.1097/00002371-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Schulz H, Bohlius JF, Trelle S, Skoetz N, Reiser M, Kober T, et al. Immunochemotherapy with rituximab and overall survival in patients with indolent or mantle cell lymphoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2007;99:706–14. doi: 10.1093/jnci/djk152. [DOI] [PubMed] [Google Scholar]
- 27.Herold M, Scholz CW, Rothmann F, Hirt C, Lakner V, Naumann R. Long-term follow-up of rituximab plus first-line mitoxantrone, chlorambucil, prednisolone and interferon-alpha as maintenance therapy in follicular lymphoma. J Cancer Res Clin Oncol. 2015;141:1689–95. doi: 10.1007/s00432-015-1963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radesi-Sarghi S, Arbion F, Dartigeas C, Delain M, Benboubker L, Herault O, et al. Interferon alpha with or without rituximab achieves a high response rate and durable responses in relapsed FL: 17 years’ experience in a single centre. Ann Hematol. 2013 doi: 10.1007/s00277-013-1934-7. [DOI] [PubMed] [Google Scholar]
- 29.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 30.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–14. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 31.Prato S, Zhan Y, Mintern JD, Villadangos JA. Rapid deletion and inactivation of CTLs upon recognition of a number of target cells over a critical threshold. J Immunol. 2013;191:3534–44. doi: 10.4049/jimmunol.1300803. [DOI] [PubMed] [Google Scholar]
- 32.Prato S, Mintern JD, Lahoud MH, Huang DC, Villadangos JA. Induction of antigen-specific effector-phase tolerance following vaccination against a previously ignored B-cell lymphoma. Immunol Cell Biol. 2011;89:595–603. doi: 10.1038/icb.2010.131. [DOI] [PubMed] [Google Scholar]
- 33.Nassef Kadry Naguib Roufaiel M, Wells JW, Steptoe RJ. Impaired T-Cell Function in B-Cell Lymphoma: A Direct Consequence of Events at the Immunological Synapse? Front Immunol. 2015;6:258. doi: 10.3389/fimmu.2015.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren Z, Guo J, Liao J, Luan Y, Liu Z, Sun Z, et al. CTLA-4 limits anti-CD20-mediated tumor regression. Clin Cancer Res. 2016;21:3597–601. doi: 10.1158/1078-0432.CCR-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359–68. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 36.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117:2423–32. doi: 10.1182/blood-2010-08-301945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 39.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.