

Abstract
Nanoparticle-mediated delivery of chemotherapeutics has demonstrated potential in improving anti-cancer efficacy by increasing serum half-life and providing tissue specificity and controlled drug release to improve biodistribution of hydrophobic chemotherapeutics. However, sub-optimal drug loading, particularly for solid core nanoparticles (NPs), remains a challenge that limits their clinical application. In this study we formulated a NP coated with a pH-sensitive polymer of O6-methylguanine-DNA methyltransferase (MGMT) inhibitor analog, dialdehyde modified O6-benzylguanosine (DABGS) to achieve high drug loading and with polyethylene glycol (PEG) to ameliorate water solubility while maintaining NP stability. The base nanovector consists of an iron oxide core (9 nm) coated with hydrazide functionalized PEG (IOPH). DABGS and PEG-dihydrazide were polymerized on the iron oxide nanoparticle surface (IOPH-pBGS) through acid-labile hydrazone bonds utilizing a rapid, freeze-thaw catalysis approach. DABGS polymerization was confirmed by FTIR and quantitated by UV-Vis spectroscopy. IOPH-pBGS demonstrated excellent drug loading of 33.4 ± 5.1% by weight while maintaining small size (36.5 ± 1.8 nm). Drug release was monitored at biologically relevant pHs and demonstrated pH dependent release with maximum release at pH 5.5 (intracellular conditions), and minimal release at physiological pH (7.4). IOPH-pBGS significantly suppressed activity of MGMT and potentiated temozolomide (TMZ) toxicity in vitro, demonstrating the potential as a new treatment option for glioblastomas (GBMs).
Keywords: Iron oxide, drug delivery, glioblastoma, controlled release, nanomedicine
Graphical abstract
A pH-sensitive alternating copolymer of MGMT inhibitor, dialdehyde modified O6-benzylguanosine, and polyethylene glycol was polymerized on the surface of iron oxide nanoparticles to achieve high drug loading and improve water solubility of the hydrophobic chemotherapeutic. The highly stable nanoparticle conjugate released MGMT inhibitor in a pH-dependent manner suppressing MGMT activity and potentiated temozolomide toxicity.

Introduction
GBMs are highly aggressive, infiltrative brain tumors affecting 14,000 individuals a year in the United States and present many treatment challenges.1 The tumor is afforded variable protection within the blood-brain barrier (BBB) and can develop chemoresistance due to efflux and upregulation of DNA repair factors.2 Even with aggressive treatment comprising surgery, chemotherapy, and radiation, mean survival is 12–15 months,3, 4 with a 5-year survival of < 5%.5 The DNA methylating agent temozolomide (TMZ) has become the standard-of-care, but the majority of patients suffer from the resistance to TMZ, induced by MGMT, a DNA repair protein that eliminates the cytotoxic O6-methylguanine DNA lesions produced by TMZ. GBM resistance to TMZ may, in theory, be mitigated by inclusion of MGMT inhibitors such as BG in current drug regemins.6 However, incorporation of conventional BG formulations in TMZ treatments reduces the maximum tolerated dose of TMZ by 50% while leading to significant meyolosupression, and the treatment has shown no significant increase in patient survival or quality of life.7
Recent advances in nanotechnology have provided tools to address the limitations of current chemotherapeutic treatments of GBM. NP drug formulations can increase blood circulation time, protect therapeutic payloads from degradation, mitigate resistance to drug efflux, provide controlled drug release, and minimize off-target drug accumulation.8, 9 Previously, we reformulated BG in combination with a redox-responsive theranostic superparamagnetic iron oxide NP (SPION) platform to improve intracellular delivery of BG to GBM cells while minimizing drug accumulation in healthy tissue.9 This improved BG formulation demonstrated a significant reduction in MGMT activity and potentiated TMZ cytotoxicity in vitro while increasing survival 3-fold over untreated controls in an orthotopic primary human GBM xenograft mouse model. However, the relatively low drug loading of the nanoparticle BG formulation required site specific administration via intratumoral convection enhanced delivery (CED) to achieve efficacious drug concentrations. While CED is emerging as a promising drug administration technique, it is highly invasive and requires surgical intervention. Improved drug loading may allow for a less invasive intravenous injection that would simplify administration and mitigate the potential side effects associated with CED.
In an effort to improve control over temporal and spatial release of therapeutics from SPION based theranostic agents, many researchers have turned to either external or physiologically induced stimuli-responsive drug release mechanisms.10 These approaches can further reduce off target accumulation of drug by minimizing release in the blood or extracellular matrix. Many strategies have been explored to create NP formulations that respond to environmental stimuli such as temperature, pH, ionic strength, redox potential, and electrical, or magnetic fields.11 Reversible hydrazone linkages formed by reaction of hydrazides with ketones or aldehyde functionalities have been extensively studied as a pH responsive mechanism for controlled intracellular release of doxorubicin.8, 12, 13 These formulations have shown favorable release profiles, minimizing release at physiological pH, yet exhibiting controlled degradation of drug linkages at intracellular pH, demonstrating the potential of this chemistry in cancer treatment.
A major constraint of covalent loading of drug on solid core NPs is the limited surface area and reactive sites for conjugation of chemotherapeutic drugs. The number of chemical reactive sites on an NP surface is typically the limiting factor for covalent drug loading as conventionally, only one chemotherapeutic molecule can be bound to each NP reactive site. Additionally, increased loading of drug with poor water solubility may destabilize the NP formulation, leading to a greater hydrodynamic size and shorter serum half-life, hampering performance in vivo.14, 15 By chemically modifying existing chemotherapeutic drugs, the moieties can be rendered more water soluble and amicable to polymerization on the surface of solid core NPs, thereby increasing drug loading while retaining NP stability.
Here we report the development of a new SPION formulation for delivery of a ribose modified BG, O6-benzyl guanosine (BGS), comprised of a superparamagnetic iron oxide core coated with a polymer shell of polyethylene glycol (PEG) functionalized with hydrazides to facilitate polymerization of homobifunctional dialdehyde BGS (DABGS) and PEG di-hydrazide (PDH) monomers via hydrazone linkages (IOPH-pBGS). DABGS was produced through oxidation of the ribose moiety to yield two aldehyde functional groups for reaction with hydrazide functionalized SPIONs and with PDH to facilitate polymerization on the SPION surface. IOPH-pBGS size, zeta potential, drug loading and release kinetics, in vitro cellular uptake, ablation of MGMT activity, and potentiation of TMZ cell kill were evaluated. Significantly, this new polymerized BGS SPION formulation may provide a less invasive, yet more effective treatment strategy than current clinical GBM treatment methods and lead to improved clinical outcomes.
Results and discussion
Formulation and characterization of SPIONs
An overview of the synthesis scheme for PEG coated, hydrazide functionalized nanoparticles (IOPH) attached with polymerized DABGS (IOPH-pBGS) is illustrated in Figure 1a. IOPH consists of a 9 nm iron oxide core synthesized from oleic acid coated SPIONs produced through thermal decomposition of iron-oleate.16 The hydrophobic oleic acid coating was exchanged with a combination of long chain methoxy-PEG and short chain PEG-dihydrazide (PDH) to render the SPIONs water soluble via an established ligand exchange process.17 It should be noted that the carrier's core material, iron oxide, is biocompatible and biodegradable, and enables the monitoring of drug trafficking and delivery by magnetic resonance imaging (MRI) when administrated both in vitro and in vivo. The lower MW hydrazide functionalized PEG was used to allow polymerization of BGS within the 2000 MW PEG layer, minimizing crosslinking and improving IOPH-pBGS water solubility. To facilitate polymerization of BGS on the SPION surface (Figure 1b), di-aldehyde groups were introduced by cleaving the carbon-carbon bonds between vicinal diols of the ribose moiety with sodium (meta) periodate (Supplementary Information, Figure S1). DABGS and PDH were slowly introduced into a solution of IOPH in MES buffer pH 5.5 at 175 μL/min. Hydrazone catalysis by way of two freeze thaw cycles was utilized to increase drug loading while reducing reaction time.18
Figure 1.
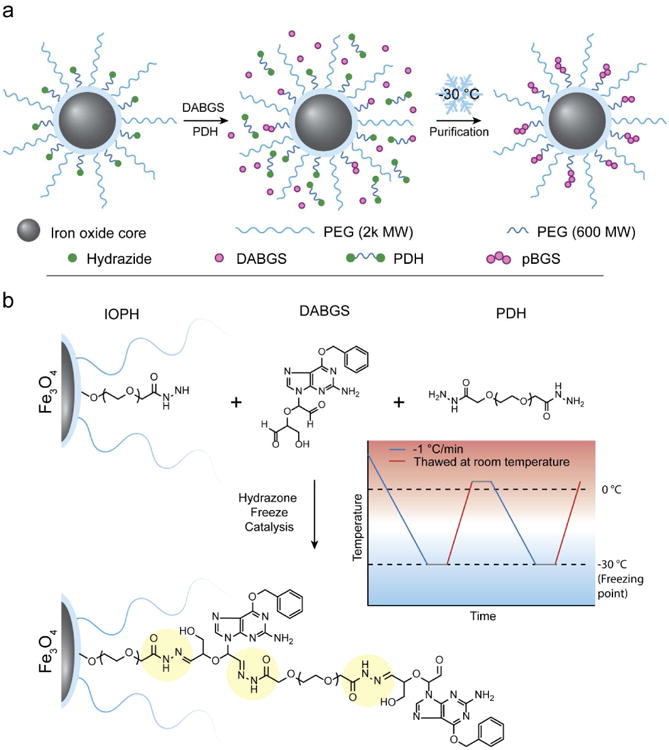
Synthesis of IOPH-pBGS. a) Illustrative overview of IOPH-pBGS synthesis. Titration of DABGS and PDH was followed by a 2 step freeze catalysis process to rapidly form IOPH-pBGS. b) The freeze catalysis process of DABGS polymerization on IOPH surface and detailed chemical structure of pBGS. Acid-labile hydrazone linkages are highlighted in yellow.
TEM images showed that IOPH-pBGS maintained spherical morphology with core sizes of roughly 9 nm (Supplementary Figure S2) both after ligand exchange to produce PEG coated water soluble SPIONs and surface polymerization (Figure 2a). Importantly, no aggregation was observed in IOPH and IOPH-pBGS indicating that no inter-particle crosslinking was produced during PEG coating or BGS polymerization.
Figure 2.

Primary physicochemical characterization of IOPH and IOPH-pBGS. TEM analysis of a) IOPH (i) IOPH-pBGS (ii). The inset in a) shows the lattice fringe of the SPION (Scale bar = 5 nm). b) Intensity based hydrodynamic size distribution of IOPH and IOPH-pBGS in PBS, pH 7.4. c) Zeta potential distribution of IOPH and IOPH-pBGS in HEPES buffer, pH 7.4. d) FTIR spectra of IOPH and IOPH-pBGS indicating the presence of aromatic structures associated with BGS.
Hydrodynamic size and zeta potential are primary factors that determine the pharmacokinetic behavior of SPIONs in vivo.19, 20 IOPH and IOPH-pBGS had an average hydrodynamic size of 26 ± 2 nm and 36.5 ± 1.8 nm in HEPES buffer (pH 7.4), respectively, which falls within the appropriate range for evasion of the mononuclear phagocyte system (Figure 2b).20 Furthermore, IOPH-pBGS remained stable in biologically relevant serum containing media for over 2 weeks (Supplementary Figure S3). Zeta potential plays a key role in non-specific SPION interactions with cell surfaces and extracellular matrices. A near neutral zeta potential is preferred for biological application of SPIONs as highly positively charged SPIONs preferentially interact with cell surfaces, whereas highly negatively charged SPIONs preferentially interact with extracellular matrices.21 The mean zeta potentials of IOPH and IOPH-pBGS were determined in HEPES buffer pH 7.4 and found to be at −12.1 ± 14.2 mV and −11.8 ± 4.0 mV, respectively (Figure 2c).
SPION surface modification was qualitatively analyzed by FTIR (Figure 2d). IR spectra of IOPH and IOPH-pBGS show characteristic bands of different vibrational modes of PEG's C-O-C bonds at 1456, 1350, 1250, 1103, and 950 cm−1 22. In addition, The IR spectra for IOPH-pBGS have additional bands at 1579, 1558, 1543, 797 cm−1 indicative of aromatic structures confirming the presence of BGS. Band designation for additional functional groups of IOPH and IOPH-pBGS can be found in Supplementary Information, Table S1.
Direct quantitation of drug loading can be difficult for SPIONs because they absorb over a broad range of UV-Vis wavelengths, while their magnetic properties cause NMR peak broadening and inconsistencies in peak ratios due to anisotropic effects.23 We utilized the mono-hydrazide PAH in excess to drive the degradation of pBGS from the SPION surface for quantitation without interference from the iron oxide core. Quantitative drug loading was determined via UV-Vis spectroscopy using a standard addition method, and percent drug loading by weight was determined to be 33.4 ± 5.1%, which is high in comparison to typical solid core nanoformulations which often only achieve 10% drug loading by weight24 Table 1 summarizes the key physicochemical properties of IOPH-pBGS.
Table 1.
Primary physiochemical properties of IOPH-pBGS.
| Core Size (nm) | Hydrodynamic Size (nm) | PDI | Zeta Potential (mV) | % Drug loading by weight |
|---|---|---|---|---|
| 9 ± 0.7 | 36.5 ± 1.8 | 0.196 | −11.8 ± 4.02 | 33.4 ± 5.1 |
pH dependent drug release
Hydrazone linkages were chosen for polymerization of DABGS due to their acid sensitive degradation profile that facilitates release of drug intracellularly while minimizing release in the blood.25, 26 The in vitro DABGS release from IOPH-pBGS was evaluated under simulated physiological conditions (PBS, pH 7.4) and in conditions mimicking acidity of endosomes/lysosomes (Acetic acid buffer at pH 4.5 and MES buffers at pH 5.5 and 6.5) (Figure 3a). It was found that DABGS release from IOPH-pBGS was greatly affected by pH of the release media. DABGS release was greatest at pH 5.5 approaching maximum release at ∼6 h. DABGS release was suppressed with only ∼30% of total drug released after 48 h for SPIONs evaluated in PBS at pH 6.5 and 7.4. Interestingly, only ∼8% of total drug release was detected at pH 4.5, indicating an optimal pH of ∼5.5 for rapid drug release. Previous studies have shown that the optimal pH for hydrazone bond formation and hydrolysis is dependent on several factors including acid/base effects of groups that neighbor the aldehyde or hydrazide group, steric effects, and macromolecular structure.27-29 The end result of these findings is that hydrazone bond formation and hydrolysis rates typically proceed through a pH dependent bell-shaped curve that yields lower rates at either end of the curve.28 BG analogs form stable tertiary structures with PEG and we speculate this effect may be enhanced at low pH where DABGS is less soluble, leading to the significant reduction of hydrolysis rates at pH 4.5. Linearized plots of percent total drug release confirm a burst release of DABGS at pH 5.5 but not at other evaluated pH's (Figure 3b). The differences in drug release at neutral and pH 5.5 can be attributed to the effect of pH on the equilibrium of hydrazone bond formation.30, 31 The high drug loading and pH-dependent release of DABGS from IOPH-pBGS under endosomal/lysosomal conditions demonstrate the potential of IOPH-pBGS as an improved MGMT inhibitor delivery platform.
Figure 3.
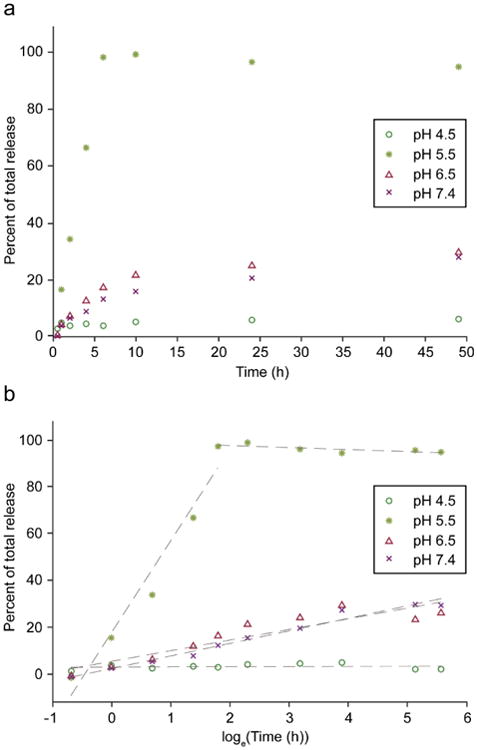
pH dependent release of DABGS from IOPH-pBGS. a) Drug release curves were determined over a range of pH's (4.5-7.4), encompassing physiological conditions (pH 7.4) and endosomal/lysosomal conditions (∼pH 5.5). b) Linearized plots of drug total release vs loge of time indicates a burst release at pH 5.5.
IOPH-pBGS cellular internalization and inhibition of MGMT
Effective application of SPION-delivered chemotherapeutics requires cellular internalization of the SPION prior to drug release. Cellular uptake of fluorescently labeled IOPH-pBGS (IOPH-Cy5-pBGS) was evaluated at 4 and 37°C utilizing flow cytometry (Supplementary Figure S4a). The differences in mean fluorescent signal at 4 and 37°C demonstrated a nearly 90% reduction in cell uptake at both 5 and 20 μg/mL of IOPH-Cy5-pBGS at low temperature (Supplementary Figure S4b). The energy-dependent uptake of IOPH-Cy5-pBGS indicates endocytosis of the SPION, likely facilitated by the negative charge of IOPH-Cy5-pBGS.32, 33 NPs that enter cells through endocytosis mainly localize in the acidic environment of endosomes or lysosomes which is advantageous for degradation of acid labile drug conjugates.34
Repair of cytotoxic DNA damage by repair protein MGMT has been shown to be a major component in the development of resistance of GBMs to chemotherapeutic alkylating agents such as TMZ.6 It has also been shown that MGMT levels in healthy brain are relatively low and that the high level of the repair protein in tumors is associated with tumorigenesis.35 Therefore, the ablation of MGMT activity in combination with TMZ is a promising treatment strategy for GBMs. Although BGS is simply a ribose modified analog of BG, there are no reports of its use as an MGMT inhibitor in literature. Furthermore, upon degradation of pBGS, DABGS is the analog that presents itself intracellularly and must serve as a potent MGMT inhibitor. To examine if BGS and DABGS function as MGMT inhibitors, a quantitative MGMT inhibition assay was performed using the MGMT+ human GBM cell line SF767. Figure 4 shows the effects of BG, BGS, and DABGS on MGMT activity of SF767 cells. Activity was determined at 2 and 24 h in untreated cells and cells treated with 10 or 20 μM of the inhibitor. Untreated cells had an activity of 25 fmol/106 cells or ∼15,000 MGMT molecules/cell. All treatment conditions were normalized as the percent activity of untreated cells. Treatment of SF767 cells with 10 μM BGS led to a reduction in MGMT activity of ∼3.9 and ∼83.3-fold (25.6% and 1.2% activity of untreated) at 2 and 24 h, respectively. Treatment at 20 μM BGS reduced activity ∼7.2 and ∼63.7-fold (13.8% and 1.6% activity of untreated) at 2 and 24 h, respectively. DABGS treated cells showed a reduction of ∼11.8 and ∼34.4-fold (8.5% and 2.9% activity of untreated) at 2 and 24 h, respectively. Treatment at 20 μM DABGS reduced activity ∼34.5 and ∼119-fold (2.9% and 0.8% activity of untreated) at 2 and 24 h, respectively. As expected, BG was a potent inhibitor of MGMT with reduction in activity at 10 μM of ∼17.8 and ∼18.5-fold (5.6% and 5.4% activity of untreated) at 2 and 24 h, respectively. Treatment at 20 μM BG reduced activity ∼18.1 and ∼85.5-fold (5.5% and 1.2% activity of untreated) at 2 and 24 h, respectively. The difference in MGMT suppression at 2 h between BG, BGS, and DABGS, particularly at 20 μM is likely explained by the increased hydrophilicity of BGS and DABGS. BG is capable of efficiently partitioning into lipid bilayers which facilitates rapid uptake in vitro but leads to poor pharmacokinetic profiles in vivo. Importantly, BGS and DABGS, in comparison to BG, demonstrated similar reduction in MGMT levels at 24 h indicating their utility as MGMT inhibitors.
Figure 4.

Inhibition of MGMT activity by BGS, DABGS, and IOPH-pBGS. Suppression of MGMT activity in SF767 cells treated with BG, BGS or DABGS at 10 and 20 μM Cells were harvested 2 h and 24 h after inhibitor addition. MGMT activity was determined from protein extractions by quantitating transfer of radioactivity from O6-[3H]methylguanine to MGMT. Data represent the results of a single preparation of IOPH-pBGS and are representative of results obtained from independent preparation of IOPH-pBGS.
Inhibition of MGMT by IOPH-pBGS was evaluated at the same treatment conditions described above (Figure 4). MGMT activity of IOPH-pBGS was compared to IOPH at doses of 10 and 20 μM BGS. The IOPH dose was normalized to the Fe equivalent of IOPH-pBGS. For treatment at 10 μM BGS, IOPH-pBGS showed a reduction of ∼1.5-fold (∼68.7% activity of untreated) and ∼9.9-fold (∼10.1% activity of untreated) at 2 and 24 h, respectively. For treatment at 20 μM BGS, IOPH-pBGS showed a reduction of ∼3.9-fold (∼25.6% activity of untreated) and ∼40.8-fold (∼2.45% activity of untreated) at 2 and 24 h, respectively. At the equivalent iron doses, IOPH had a minimal effect on MGMT activity. These results confirm that BGS and DABGS serve as inhibitors of MGMT and that DABGS maintains its inhibitor activity when formulated as IOPH-pBGS demonstrating the potential clinical relevance of the IOPH-pBGS formulation.
Potentiation of TMZ cytotoxicity in human GBM cells
Human GBM cell line SF767 has pronounced resistance to TMZ (LD10 ∼487 μM), facilitated significantly by upregulation of MGMT.36 IOPH-pBGS suppression of MGMT activity and subsequent TMZ-mediated reduction in clonogenic survival of SF767 is illustrated in Figure 5a. Cells were exposed to IOPH-pBGS equivalent to 20 μM free drug for 24 h followed by incubation with TMZ for 24 h in the presence of inhibitor analogs. Controls were treated with 20 μM free BG, BGS, DABGS and IOPH or with an equivalent volume of DMSO. A least squares linear regression analysis was performed on the linearized survival curves to determine LD10. IOPH-pBGS reduced the resistance by approximately 40-fold (LD10 = 12.1 ± 01.5 μM), a potentiation of cytotoxicity comparable to that produced by free BG (LD10 = 9.2 ± 2.0 μM). BG analogs, BGS and DABGS, reduced resistance 19-fold (LD10 = 34.4 ± 8.9 μM) and 11-fold (LD10 = 43.7 ± 4.0 μM), respectively. The increase in LD10 for BGS and DABGS as compared to BG may be explained by hydrophobicity differences that affect localization of inhibitor. BG has been shown to provide prolonged knockdown of MGMT in vitro likely due to partitioning of hydrophobic BG in to lipid bilayers that act as a drug reserve that can slowly release BG as equilibriums dictate.37 The increased hydrophilicity of BGS and DABGS may reduce this retention and release effect, and negatively affect prolonged MGMT knockdown. Importantly, IOPH-pBGS retains excellent reduction in LD10 despite releasing DABGS as its MGMT inhibitor. The internalization of hydrophilic IOPH-pBGS and the pH dependent release of drug due to the hydrazone linkages provides a mechanism for long-term MGMT knockdown and the potential for improved in vivo biodistribution, pharmacokinetics and efficacy. These results demonstrate that IOPH-pBGS mediated suppression of MGMT is accompanied by greater sensitivity to TMZ in human GBM cells.
Figure 5.

Suppression of MGMT activity by treatment with IOPH-pBGS induces apoptosis and increases TMZ cell killing in the GBM line SF767. a) Survival was determined by a clonogenic colony-forming assay on cells treated with TMZ alone (no BG analog) or exposed to 20 μM BG, BGS, DABGS or IOPH-pBGS containing 20 μM DABGS. The insert shows a finer scale to better visualize the differences between BG, its analogs, and IOPH-pBGS on cell kill at low TMZ concentrations. b) Flow cytometry analysis of apoptosis in SF767 cells treated with 20 μM BG, IOPH-pBGS containing 20 μM DABGS or IOPH at an Fe equivalency to IOPH-pBGS, followed 24 h later by treatment with 0 or 100 μM TMZ. All experiments were performed in triplicate.
The combined treatment of GBM cells with MGMT inhibitor/TMZ should produce pro-apoptotic effects due to the inability of cells to efficiently eliminate the cytotoxic O6-methylguanine DNA lesions produced by TMZ.38 To further evaluate the effects on GBM cells in vitro from the combined treatment of IOPH-pBGS/TMZ, SF767 cells were treated with 20 μM BG, IOPH-pBGS equivalent to 20 μM free drug, or with IOPH as a non-inhibitor containing control. 24 h after treatment with MGMT inhibitor, cells were incubated with 0 or 100 μM TMZ for 48 h before staining with Annexin V to quantitate apoptosis by flow cytometry (Figure 5b). As expected, the IOPH control (no MGMT inhibitor) showed minimal apoptosis with or without TMZ addition (5.0% and 5.6% late apoptotic cells, respectively). It should be noted that 100 μM TMZ falls within the resistance shoulder of the toxicity curve and was not expected to cause significant apoptosis without addition of MGMT inhibitor (Figure 5a). Similarly, free BG and IOPH-pBGS treatment without TMZ demonstrated minimal apoptosis (8.3% and 9.1%, late apoptotic cells, respectively). However, the addition of 100 μM TMZ to cells pretreated with free BG or IOPH-pBGS led to significant increases in pro-apoptotic behavior 48 h after TMZ addition (40.7% and 37.9% late apoptotic cells, respectively). These results confirm the combined treatment of IOPH-pBGS with TMZ, which inhibits cell division through inclusion of methyl lesions on DNA, is pro-apoptotic in nature.
Conclusions
Iron oxide SPIONs carrying a BGS payload for treatment of GBMs were successfully prepared. BGS and DABGS were evaluated in vitro as MGMT inhibitors and were shown to be as potent as BG after 24 h exposure. DABGS was successfully polymerized on the base SPION surface to produce IOPH-pBGS which exhibited proper size and surface charge for in vivo application. FTIR analysis of IOPH-pBGS confirmed DABGS loading by presence of bands associated with the aromatic structures of BGS. IOPH-pBGS achieved high drug loading and released drug through a pH-dependent mechanism that favors release of drug intracellularly. Importantly, in vitro MGMT activity assays confirmed the successful suppression of MGMT activity and potentiation of TMZ cell kill via apoptosis by endocytosed IOPH-pBGS on par with free BG. The combination of high drug loading, pH controlled release and ability to efficiently suppress MGMT demonstrates the potential of IOPH-pBGS to serve as an improved MGMT inhibitor formulation that can provide minimally invasive i.v. administration and improved patient prognosis.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. 3-(triethoxysilyl)propyl succinic anhydride (SATES) was purchased from Gelest (Arlington, VA, USA). 2000 MW mono-amine functionalized polyethylene glycol (mPEG2K-NH2) was purchased from Laysan Bio (Arab, AL, USA). 600 MW hydrazide-PEG-hydrazide (PEG600DH) was purchased from Creative PEGWorks (Chapel Hill, NC, USA). O6-Benzylguanosine (BGS) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). NHS-Cy5 was purchased from Lumiprobe Corporation (Hallandale Beach, FL, USA). S-200 size exclusion resin was purchased from GE Healthcare (Picscataway, NJ, USA).
IOPH synthesis and coating
Oleic acid coated iron oxide particles (IOOAs) were synthesized following a previously reported procedure 16. An established ligand exchange process was utilized with a few modifications as noted below for production of IOPH.17 50 mg of IOOA was suspended in 43 mL of anhydrous toluene followed by addition of 50 μL of triethylamine in a 3 neck round-bottom flask fitted with a Graham condenser and a vacuum adapter. The flask was sealed with rubber septa and purged with nitrogen before heating the solution to 100°C. SATES (100 μL) was then added to the flask. mPEG2K-NH2 (187.5 mg) was dissolved in 7 mL of anhydrous toluene and added to the flask 15 minutes after the addition of SATES. An additional 50 μL of SATES was injected 1 h after the mPEG2K-NH2 injection, and the solution was reacted for an additional 6 h and 45 minutes. The solution was transferred to a single neck round-bottom flask and SPIONs were precipitated with hexanes. The SPION precipitate was dispersed in tetrahydrofuran (THF), sonicated for 10 minutes, and precipitated with hexanes. The resultant SPION pellet was suspended in 10 mL anhydrous THF and sonicated for 10 minutes. 102.5 mg of mPEG2K-NH2 and 160 mg of PEG600-DH were dissolved in 12 mL anhydrous THF and added to the SPION solution. The flask was then sealed with a septum and purged with nitrogen. 12.5 mg of N,N1-dicyclohexylcarbodiimide (DCC) was dissolved in 2 mL anhydrous THF and added to the flask, and the reaction solution was placed in a sonication bath at 25°C and allowed to react for 16 h. Fully PEGylated SPIONs were precipitated with hexane, redispersed in 20 mL ethanol, sonicated for 10 minutes, and precipitated again with hexanes. The pellet was fully dried and dispersed in PBS with sonication for 10 minutes. The particles were purified through size exclusion gel chromatography.
Synthesis of DABGS
BGS was dissolved in DMSO (26.8 mM) before being mixed at an equal volume ratio with an aqueous NaIO4 solution (3.2 M). The mixture was protected from light and reacted for 48 h on a rocker.
IOPH-pBGS synthesis
IOPH (1 mg, 8.67 mg/mL in 100 mM MES, pH 5.5) was diluted in aqueous MES buffer (100 mM, 594 μL). DABGS (5 mg/mL, 133.3 μL) was titrated in at a constant rate of 175 μL/min. Titration of PDH (6.45 μg/μL, 66.7 μL) started after half of the DABGS was added. After the completion of titration, the reaction mixture was subjected to two freeze-thaw cycles utilizing an isopropanol cooling bath to cool (−1 °C/min) before purification through size exclusion chromatography with S-200 resin into water or PBS. Purified particles were collected and analyzed for size, zeta potential, and drug loading using DLS, UV, and FTIR.
IOPH-Cy5-pBGS for cellular uptake experiments were prepared in a way similar to the above-mentioned method, with Cy5 added prior to pBGS addition. IOPH was reacted with NHS-Cy5 at an 8:1 w/w ratio in 100 mM sodium bicarbonate buffer, pH 8.5 for 1 h before purification in MES buffer by size exclusion chromatography. IOPH-Cy5 was then subjected to freeze catalysis in the presence of DABGS and PDH as detailed in the previous paragraph.
SPION size and zeta potential characterization
The hydrodynamic size and zeta potential of IOPH and IOPH-pBGS were analyzed at 100 μg/mL in 20 mM HEPES buffer (pH 7.4) using a DTS Zetasizer Nano (Malvern Instruments, Worcestershire, UK).
TEM characterization of SPION core size and morphology
TEM images were acquired with an FEI TECNAI F20 TEM (Hillsboro, OR) operating at 200 kV. SPION core diameters were analyzed with ImageJ software and the size distribution, mean diameter and standard deviation was calculated from 100 SPION measurements.
Evaluation of BGS loading
Absolute quantization of drug loading was performed through UV absorbance utilizing the standard addition method. IOPH-BGS (150 μg) was freeze dried and re-suspended in a solution of propanoic acid hydrazide (10 mg/mL) in 100 mM MES buffer (pH 5.5, 375 μL). The solution was sonicated for 24 h before filtration through an Amicon 10k MWCO spin filter (EMD Millipore, Billerica, MA, USA). The supinate (100 μL) was added to equal volumes of BGS standards and UV absorbance was measured at 282 nm and total concentration calculated from the standard curve.
Drug release
IOPH-BGS (0.5 mg) was diluted with buffer at a 1:4 ratio and dialyzed using a 10k MWCO slide-a-lyzer mini dialysis device (ThermoFisher Scientific, Waltham, MA, USA) against 45 mL of four buffer solutions at 100 mM concentrations: Acetic Acid (pH 4.5), MES (pH 5.5 and 6.5), and HEPES (pH 7.4). Each mixture was stored at room temperature. Aliquots (2 mL) were removed at specified time points and measured for UV absorbance. The volume against which dialysis occurred was held constant by adding back the removed volume of media for each time point.
Cell culture
SF767, a human GBM cell line obtained from the tissue bank of the Brain Tumor Research Center (University of California–San Francisco, San Francisco, CA), was maintained in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic at 37°C and 5% CO2.
MGMT activity Assay
The MGMT activities of protein extracts of human GBM SF767 cells were measured in a standard biochemical assay that quantifies the transfer of radioactivity from a DNA substrate containing [methyl3H]O6-methylguanine (specific activity, 80 Ci/mmol) to protein, as detailed previously.37 SF767 cells (2 × 106) were plated in 6 well dishes and incubated with 10 or 20 μM free BG, BGS or DABGS for 2 or 24 h with IOPH-pBGS equivalent to 10 and 20 μM BG in fully supplemented medium.
Cellular uptake assay
SF767 cells were seeded into 6-well plates with a density of 250,000 cells per well and incubated overnight. Cells were then incubated with IOPH-Cy5-pBGS for 1 hr (5 and 20 μg/mL) at either 37°C or 4°C, followed by washing thrice with cold PBS. Cells were then trypsinized, fixed in 4% formaldehyde, washed, and suspended in PBS. IOPH-Cy5-pBGS uptake indicated by Cy5 fluorescence was determined by flow cytometry (FACSCanto II, BD Biosciences) and the data were analyzed using FlowJo software (Treestar, Inc., San Carlos, CA, USA).
Clonogenic survival assay
Determination of proliferative survival of SF767 by clonogenic assay was performed as described previously.9, 36 Briefly, 6-well plates inoculated with 2 mL of supplemented medium containing 500 cells were incubated overnight at 37°C to allow reattachment to well plates. Cells were then incubated for 24 h with IOPH-pBGS equivalent to 20 μM BG prior to exposure to TMZ. Incubation was continued for 24 h before changing cells to fresh, drug-free medium to allow formation of colonies ≥ 50 cells. Controls included cells treated with IOPH (Fe equivalent to IOPH-pBGS) or with 20 μM free BG or BG analogs; untreated controls received an equivalent volume of DMSO solvent. Survival (mean ± SD) is the ratio of colony-forming ability of treated cells to that of untreated cells. Cytotoxicity was quantitated by a least square linear regression analysis of plots of log surviving fraction vs TMZ dose to obtain the dose required to reduce survival to 10%, LD10. Clonogenic survival was determined in triplicate experiments.
Cellular apoptosis assay
SF767 cells were seeded in 6-well plates at a density of 75,000 cells per well and incubated overnight. 20 μM MGMT inhibitor (Free BG or IOPH-pBGS) or Fe equivalent of IOPH was added to cells and incubated for 24 h. TMZ (0 or 100 μM) was then added and the cell were incubated for an additional 48 h. Cells were then trypsanized, aspirated, and washed once with PBS. Cells were then counted and suspended in 0.1 mL annexin V binding buffer containing 50 μg/mL propidium iodide and 5 μL FITC-Annexin V reagent. The cells were incubated for an additional 15 min in the dark at room temperature. 0.4 mL annexin V binding buffer was added prior to analysis by flow cytometry. Data acquisition was performed on a FACSCanto II and analyzed by FlowJo software (Treestar, Inc., San Carlos, CA).
Supplementary Material
Acknowledgments
This work is supported by NIH grants R01CA161953. We acknowledge lab assistance from D. Kahn, C. Yen, M. Gebhart and the use of resources at the Molecular Engineering and Science Institute's Molecular analysis facility.
Abbreviations
- NP
solid core nanoparticle
- SPION
superparamagnetic iron oxide nanoparticle
- BG
O6-benzylguanine
- BGS
O6-benzylguanosine
- DABGS
dialdehyde functionalized O6-benzylguanosine
- PEG
polyethylene glycol
- IOPH
iron oxide nanoparticle coated in hydrazide functionalized polyethylene glycol
- IOPH-pBGS
IOPH conjugated to polymerized BGS
- MGMT
O6-methylguanine-DNA methyltransferase
- TMZ
temozolomide
- GBM
Glioblastoma
- PDH
PEG600-dihydrazide
Footnotes
Supplementary Information Available. Schematic representation of sodium periodate cleaving of BGS. Size analysis of NP TEM images, long-term stability of IOPH-pBGS, and FTIR analysis. This material is free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Cancer Report 2014. WHO Press; 2014. [Google Scholar]
- 2.Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, Wick W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. 2008;14:2900–2908. doi: 10.1158/1078-0432.CCR-07-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130:2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozoloide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Gallego O. Nonsurgical treatment of recurrent glioblastoma. Curr Oncol. 2015;22:E273–E281. doi: 10.3747/co.22.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silber JR, Bobola MS, Blank A, Chamberlain MC. O6-Methylguanine-DNA methyltransferase in glioma therapy: Promise and problems. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. doi: 10.1016/j.bbcan.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009;27:1262–7. doi: 10.1200/JCO.2008.18.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kievit FM, Wang FY, Fang C, Mok H, Wang K, Silber JR, Ellenbogen RG, Zhang MQ. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. Journal of Controlled Release. 2011;152:76–83. doi: 10.1016/j.jconrel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephen ZR, Kievit FM, Veiseh O, Chiarelli PA, Fang C, Wang K, Hatzinger SJ, Ellenbogen RG, Silber JR, et al. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS nano. 2014;8:10383–95. doi: 10.1021/nn503735w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Ke HT, Dai ZF, Liu Z. Nanoscale theranostics for physical stimulus-responsive cancer therapies. Biomaterials. 2015;73:214–230. doi: 10.1016/j.biomaterials.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Caldorera-Moore ME, Liechty WB, Peppas NA. Responsive theranostic systems: integration of diagnostic imaging agents and responsive controlled release drug delivery carriers. Acc Chem Res. 2011;44:1061–70. doi: 10.1021/ar2001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, Li Y. Smart pH-Sensitive and Temporal-Controlled Polymeric Micelles for Effective Combination Therapy of Doxorubicin and Disulfiram. ACS nano. 2013;7:5858–5869. doi: 10.1021/nn4010796. [DOI] [PubMed] [Google Scholar]
- 13.Pourjavadi A, Hosseini SH, Alizadeh M, Bennett C. Magnetic pH-responsive nanocarrier with long spacer length and high colloidal stability for controlled delivery of doxorubicin. Colloid Surf B-Biointerfaces. 2014;116:49–54. doi: 10.1016/j.colsurfb.2013.12.048. [DOI] [PubMed] [Google Scholar]
- 14.Chu KS, Schorzman AN, Finniss MC, Bowerman CJ, Peng L, Luft JC, Madden AJ, Wang AZ, Zamboni WC, DeSimone JM. Nanoparticle drug loading as a design parameter to improve docetaxel pharmacokinetics and efficacy. Biomaterials. 2013;34:8424–9. doi: 10.1016/j.biomaterials.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 16.Park J, An KJ, Hwang YS, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat Mater. 2004;3:891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 17.Fang C, Bhattarai N, Sun C, Zhang M. Functionalized nanoparticles with long-term stability in biological media. Small (Weinheim an der Bergstrasse, Germany) 2009;5:1637–41. doi: 10.1002/smll.200801647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agten SM, Suylen DPL, Hackeng TM. Oxime Catalysis by Freezing. Bioconjugate Chem. 2016;27:42–46. doi: 10.1021/acs.bioconjchem.5b00611. [DOI] [PubMed] [Google Scholar]
- 19.Faure AC, Dufort S, Josserand V, Perriat P, Coll JL, Roux S, Tillement O. Control of the in vivo Biodistribution of Hybrid Nanoparticles with Different Poly(ethylene glycol) Coatings. Small (Weinheim an der Bergstrasse, Germany) 2009;5:2565–2575. doi: 10.1002/smll.200900563. [DOI] [PubMed] [Google Scholar]
- 20.Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3:703–717. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nature nanotechnology. 2010;5:465–72. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willis AL, Turro NJ, OBrien S. Spectroscopic characterization of the surface of iron oxide nanocrystals. Chem Mat. 2005;17:5970–5975. [Google Scholar]
- 23.Korringa J, Seevers DO, Torrey HC. Theory of Spin Pumping and Relaxation in Systems With A Low Concentration of Electron Spin Resonance Centers. Physical Review. 1962;127:1143–&. [Google Scholar]
- 24.Di Marco M, Sadun C, Port M, Guilber I, Couveur P, Dubernet C. Physicochemical characterization of ultrasmall superparamagnetic iron oxide particles (USPIO) for biomedical application as MRI contrast agents. Interanational Journal of Nanomedicine. 2007;2:609–622. [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang AA, Lee BY, Clemens DL, Dillon BJ, Zink JI, Horwitz MA. pH-Responsive Isoniazid-Loaded Nanoparticles Markedly Improve Tuberculosis Treatment in Mice. Small (Weinheim an der Bergstrasse, Germany) 2015;11:5066–78. doi: 10.1002/smll.201500937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruan S, He Q, Gao H. Matrix metalloproteinase triggered size-shrinkable gelatin-gold fabricated nanoparticles for tumor microenvironment sensitive penetration and diagnosis of glioma. Nanoscale. 2015;7:9487–96. doi: 10.1039/c5nr01408e. [DOI] [PubMed] [Google Scholar]
- 27.Kool ET, Park DH, Crisalli P. Fast Hydrazone Reactants: Electronic and Acid/Base Effects Strongly Influence Rate at Biological pH. J Am Chem Soc. 2013;135:17663–17666. doi: 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King TP, Zhao SW, Lam T. Preparation of Protein Conjugates Via Intermolecular Hydrazone Linkage. Biochemistry. 1986;25:5774–5779. doi: 10.1021/bi00367a064. [DOI] [PubMed] [Google Scholar]
- 29.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angew Chem-Int Edit. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. Nucleophilic catalysis of hydrazone formation and transimination: implications for dynamic covalent chemistry. J Am Chem Soc. 2006;128:15602–3. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- 31.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angewandte Chemie (International ed in English) 2008;47:7523–6. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomed. 2014;9:51–63. doi: 10.2147/IJN.S26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He CB, Hu YP, Yin LC, Tang C, Yin CH. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 34.Yameen B, Choi WI, Vilos C, Swami A, Shi JJ, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. Journal of Controlled Release. 2014;190:485–499. doi: 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma S, Salehi F, Scheithauer BW, Rotondo F, Syro LV, Kovacs K. Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer research. 2009;29:3759–68. [PubMed] [Google Scholar]
- 36.Blank A, Bobola MS, Gold B, Varadarajan S, DKolstoe D, Meade EH, Rabinovitch PS, Loeb LA, Silber JR. The Werner syndrome protein confers resistance to the DNA lesions N3-methyladenine and O6-methylguanine: implications for WRN function. DNA Repair. 2004;3:629–638. doi: 10.1016/j.dnarep.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Silber JR, Bobola MS, Ghatan S, Blank A, Kolstoe DD, Berger MS. O6-Methylguanine-DNA Methyltransferase Activity in Adult Gliomas: Relation to Patient and Tumor Characteristics. Cancer research. 1998;58:1068–1073. [PubMed] [Google Scholar]
- 38.Kitange GJ, Mladek AC, Schroeder MA, Pokorny JC, Carlson BL, Zhang YJ, Nair AA, Lee JH, Yan HH, Decker PA, et al. Retinoblastoma Binding Protein 4 Modulates Temozolomide Sensitivity in Glioblastoma by Regulating DNA Repair Proteins. Cell Reports. 2016;14:2587–2598. doi: 10.1016/j.celrep.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.