

Abstract
Bone morphogenetic proteins (BMPs) are regulated by extracellular antagonists of the DAN (differential screening-selected gene aberrative in neuroblastoma) family. Similar to the BMP ligands, certain DAN family members have been shown to interact with heparin and heparan sulfate (HS). Structural studies of DAN family members Gremlin-1 and Gremlin-2 (Grem2) have revealed a dimeric growth factor-like fold where a series of lysine residues cluster along one face of the protein. In the present study, we used mutagenesis, heparin-binding measurements, and cell surface-binding analysis to identify lysine residues that are important for heparin/HS binding in Grem2. We determined that residues involved in heparin/HS binding, while not necessary for BMP antagonism, merge with the heparin/HS-binding epitope of BMP2. Furthermore, the Grem2–BMP2 complex has higher affinity for heparin than the individual proteins and this affinity is not abrogated when the heparin/HS-binding epitope of Grem2 is attenuated. Overall, the present study shows that the Grem2 heparin/HS and BMP-binding epitopes are unique and independent, where, interestingly, the Grem2–BMP2 complex exhibits a significant increase in binding affinity toward heparin moieties that appear to be partially independent of the Grem2 heparin/HS-binding epitope.
Introduction
Bone morphogenetic proteins (BMPs), a subfamily of the greater transforming growth factor-β (TGF-β) superfamily, were originally discovered based on their profound roles in bone generation/regeneration [1]. BMPs have been shown to play critical roles in numerous developmental and physiological processes, where misregulation in their signaling can lead to severe disease (e.g. diabetic kidney nephropathy, symphalangism, cardiovascular diseases, and pulmonary arterial hypertension) and numerous cancer phenotypes [2–11].
BMP ligands signal by localizing pairs of serine/threonine kinase receptors, termed Type I and Type II. Formation of the extracellular ligand Type I/Type II receptor complex activates the intracellular kinase domain of the Type I receptor that initiates a signaling cascade through phosphorylation and activation of SMAD transcription factors (SMAD-1/5/8). Subsequently, context-dependent signaling occurs through the interaction of activated SMAD proteins with various co-activators and co-repressors to promote highly specific genetic outcomes [1,12].
Extracellularly, BMPs are regulated by a variety of secreted antagonists, including Noggin, Follistatin, Chordin, and members of the DAN family, which have been shown to directly bind to and neutralize the BMP ligands. While diverse in structure, extracellular antagonists have been shown to utilize conserved mechanisms to bind and block specific epitopes on BMPs that are used to engage their target Type I and Type II receptors [2–12]. In addition, most BMP antagonists contain variable epitopes of positively charged amino acids that are important for their ability to bind heparin and heparan sulfate (HS) glycosaminoglycans [13–16]. Binding to heparin/HS can localize antagonists to cellular surfaces and is thought to provide a canopy for certain cells to limit BMP signaling and form potentially important ligand/antagonist gradients during development [17]. In addition, many antagonists have been shown or suggested to utilize this interaction with heparin/HS to regulate and enhance endocytosis of the antagonist–ligand complexes, probably leading to rapid BMP signal degradation [13,18,19].
The DAN family represents a large and diverse collection of BMP antagonists that includes the members Gremlin-1 (Grem1), Gremlin-2 (Grem2 or PRDC), Cerberus, Coco (Grem3), NBL1 (Dan), USAG-1 (Wise), and SOST [20]. Several members of the DAN family have been shown to directly bind to HS. However, since DAN family members lack a conserved heparin-binding motif (BBXB and BxBXXB, where B is basic amino acid), it has been difficult to predict the epitopes necessary to confer heparin/HS binding within individual members. NMR studies were used to identify amino acids in SOST that are involved in heparin/HS binding [21]. Furthermore, a recent study has identified residues in Grem1 involved in this interaction [22]. However, DAN family members exhibit significant sequence diversity in the number and placement of positively charged residues. As such, characterizing of these motifs within the DAN family appears to be specific for individual members [13,20].
In this study, we identify a significant portion of the heparin/HS-binding motif within Grem2 and show its exclusivity from the BMP-binding epitope. Furthermore, we show how this motif affects binding to the cellular surface and show that Grem2–BMP2 complexes maintain a higher binding affinity to heparin/HS than either protein alone, suggesting a potential enhancement of BMP inhibitor activities.
Materials and methods
Protein expression and purification
Grem2 wild-type (WT) and Grem2 variants were cloned, expressed, refolded, and purified following previously published protocols [23]. Briefly, mutations in Grem2 (pET21a) were introduced using QuikChange site-directed mutagenesis. Proteins were expressed in BL21 (DE3) Escherichia coli and refolded following purification from inclusion bodies as previously described [24]. Refolded proteins were then purified by heparin-affinity chromatography and C18 reverse-phase chromatography.
Size-exclusion chromatography
Size-exclusion chromatography (SEC) of recombinant Grem2 WT and lysine mutant proteins was carried out on a Superdex 75 column (GE Healthcare) equilibrated with 20 mM HEPES (pH 7.5), 0.8 M NaCl.
Heparin-affinity chromatography
Grem2 proteins (∼150 μg) were suspended or diluted in 20 mM HEPES (pH 7.5) and loaded onto a 1 ml heparin-Sepharose column (GE Healthcare) equilibrated in the same buffer. After washing, proteins were eluted with a 0–1 M NaCl linear gradient (100 column volumes) and absorbance was monitored at 280 nm. Each experiment was conducted in duplicate. The Grem2–BMP2 complex was made by mixing equal molar ratios of the two proteins and adding 0.5% CHAPS in the sample and running buffers. Controls for complex analysis were also performed using CHAPS in the methodology.
Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were performed at room temperature using a Biacore T200 as previously described [24,25]. Recombinant mature BMP2 was purified as previously described and diluted in sodium acetate buffer (pH 4.5) prior to immobilization on a CM5 sensor chip using the standard EDC/NHS primary amine coupling chemistry and according to the manufacturer’s protocol [25,26]. To determine the binding kinetics of Grem2 WT and mutants, serial dilutions in HBS–EP buffer [20 mM HEPES (pH 7.4), 150 mM NaCl, 3.4 mM EDTA, and 0.005% v/v P20 surfactant] were injected at a constant flow rate of 30 μl/min for an association of 3 min and allowed to dissociate for 10 min. Chip surfaces were regenerated by injecting short pulses of 2 M GnHCl. Binding curves were corrected for background and bulk refractive index contribution by subtraction of the reference flow cell. Kinetic and binding parameters, kon, koff, and KD, were calculated by globally fitting the data to a 1:1 interaction model using the BIAEvaluation software package.
Cell culture
All cell lines were cultured in 5% CO2 at 37°C. The CHO-K1 cell line and its derivative deficient in HS biosynthesis, CHO-HSD (pgsD-677), were obtained from American Type Culture Collection (Manassas, VA) and grown in Ham’s F-12 medium (F-12) containing 10% (v/v) fetal bovine serum (FBS) and penicillin (100 μg/ml) plus streptomycin (100 units/ml). The BRITER osteoblast cell line was cultured in αMEM containing 10% FBS and 100 μg/ml Hygromycin B, and supplemented with streptomycin (100 μg/ml) and penicillin (100 units/ml) [27,28].
Luciferase reporter assay
A BMP-responsive luciferase reporter osteoblast cell line (BRITER), kindly provided by Dr Amitabha Bandyopadhyay (Indian Institute of Technology, Kanpur, India), was used to measure BMP activity as previously reported [27,28]. Cells were grown overnight in a 96-well plate and treated with DMEM/Hi Glucose for 5–6 h and then treated with either BMP alone or mixed with WT or mutant Grem2 for 3 h. Subsequently, the cells were lysed and monitored for luminescence. IC50 values were calculated using Prism Graphpad. All experiments were performed in triplicate and statistical significance was determined using Student’s t-test.
Cell surface labeling with Alexa Fluor 488-conjugated proteins
Grem2 WT and Grem2 select mutants (0.5 mg) were buffer exchanged into 0.1 M sodium bicarbonate (pH 8.5) and labeled with Alexa Fluor 488 cadaverine at a molar ratio of 1:10 (protein:dye) according to the manufacturer’s protocol (Thermo Fisher). Subsequently, Grem2 proteins were dialyzed into 10 mM HCl and stored at −80°C until experimentation.
To determine binding of labeled Grem2 WT and mutant proteins to the cellular surface, CHO-K1 cells (standard and HS deficient) were grown in 10 cm plates for 18 h (∼75% confluence), washed with PBS, and then trypsinized. Detached cells were centrifuged, washed, and resuspended in PBS and subsequently treated with labeled Grem2 WT or mutant proteins in the presence or absence of 0.3 M sodium chlorate for 30–60 min. Following incubation, cells were washed and assessed by flow cytometry.
Flow cytometry (fluorescence-activated cell sorting)
Cells were diluted to 106 cells/ml and passed through a 35 μm nylon mesh (BD Biosciences) to obtain single-cell suspensions. Fluorescence analysis was performed on a FACSCalibur flow cytometer with a 15 mW, 488 nm air-cooled argon ion laser and analyzed by the CELLQUEST software (version 3.3; Becton–Dickinson, U.S.A.). Interaction of Alexa 488-conjugated proteins with CHO-K1 and pgsD-677 cell surfaces was determined as the percentage of cells emitting a fluorescent signal. The boundary between cells that stained positive and negative for Alexa 488 was determined according to the fluorescence distribution of positively stained versus unstained samples. A minimum of 20,000 events per sample was acquired using the CellQuest software and analyzed by FlowJo (version 6.4.1; Tree Star, San Carlos, CA).
Results
Analysis of potential heparin-binding motifs on the structure of Grem2
Structural studies have shown that Grem2 exists as a stable noncovalent dimer strengthened by an extensive hydrogen-bonding network between opposing monomers [23]. Each monomer has a growth factor-like fold with a central cystine knot. Starting from the N-terminus, a short helix is followed by a concession of long, antiparallel β-strands, termed finger 1 and finger 2, and a wrist region that joins the two fingers (Figure 1C). Grem2 features 26 surface-oriented arginine (12) and lysine (14) residues per monomer, conferring a strong basic character for the protein with an apparent pI of 9.3 (Figure 1B). Similar to other DAN family members, Grem2 has been shown to interact with heparin/HS [23]. However, these proteins lack a common consensus heparin/HS-binding motif, making it difficult to predict which residues are important for heparin interactions (Figure 1A).
Figure 1. DAN family sequence alignment and electrostatics of Grem2.
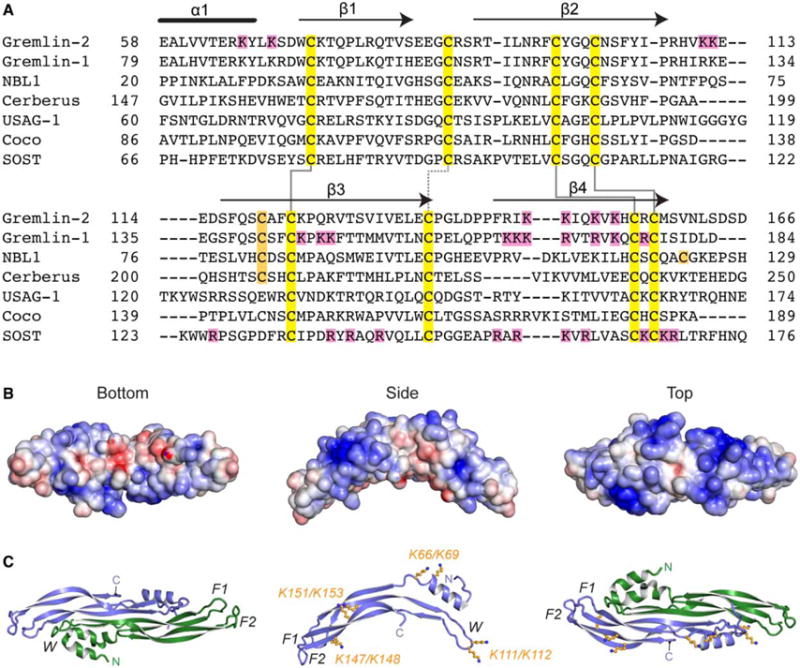
(A) Multiple sequence alignment of members from the DAN family. Alignment is based on those amino acids and ranges present in the structure of Grem2 (PDB: 4JPH). Black lines above the Grem2 sequence indicate the secondary structure. Yellow, family conserved cysteines; orange, extra cysteines; magenta, lysines in Grem2 selected for mutagenesis and those amino acids identified in Grem1 and SOST shown to be important in heparin binding. Solid black lines show disulfide bonds forming the cystine knot and the dotted black line shows the disulfide bond linking the fingers in the DAN family fold. (B and C) Structure of Grem2 shown from the bottom (concave), side, and top (convex) perspectives. (B) Electrostatic surface area predicted by APBS with blue as positive and red as negative and colored from −5 to +5 kbt/ec. (C) Ribbon representation of Grem2 dimer with chains colored green and blue. Right and left panels represent similar views as in (B). Middle panel depicts a single chain with the orange sticks depicting the lysines selected for mutagenesis studies. F1, finger 1; F2, finger 2; W, wrist region.
For most proteins, heparin/HS-binding motifs either occur as linear stretches of basic residues or as three-dimensional, well-positioned clusters of positively charged residues on the protein surface [29,30]. While the primary sequence of Grem2 lacks a consensus heparin/HS-binding motif, examination of the electrostatic surface potential of Grem2 shows that the convex surface of the protein dimer is highly positively charged, while the concave surface of the protein is more acidic (Figure 1B). This positively charged surface is composed of multiple patches of lysine residues that are separated in the primary sequence but spatially localized in the three-dimensional structure. Splitting apart these lysines, three main clusters can be described: (1) K66 and K69 located within the N-terminus; (2) K111 and K112 located within the wrist region; and (3) K147, K148, K151, and K153 located on finger 2 (Figure 1B,C). While there is significant sequence divergence across the DAN family, a few of these lysines are conserved between Grem2 and the known heparin/HS-binding motifs in Grem1 and SOST, suggesting their potential importance in Grem2 (Figure 1A).
Mutagenesis and analysis of in vitro Grem2 heparin binding
To test the importance of specific residues in heparin/HS binding, we replaced the positively charged lysine residues with alanine and produced four different double mutants: K66A/K69A, K111A/K112A, K147A/K148A, and K151A/K153A. For analysis, each mutant was produced and purified similar to the WT protein, as previously published [23]. Similar to WT Grem2, each mutant was shown to exist as a stable, noncovalent dimer using size-exclusion chromatography, indicating that the lysine mutants were appropriately folded (Figure 2A). Furthermore, when analyzed by SDS–PAGE, each mutant exhibited a similar size shift when compared with WT Grem2 under nonreducing and reducing conditions. This feature is typical for disulfide-bonded proteins and is further evidence of proper folding (Figure 2B) [23,24].
Figure 2. SEC and SDS–PAGE analysis of Grem2 lysine mutants.
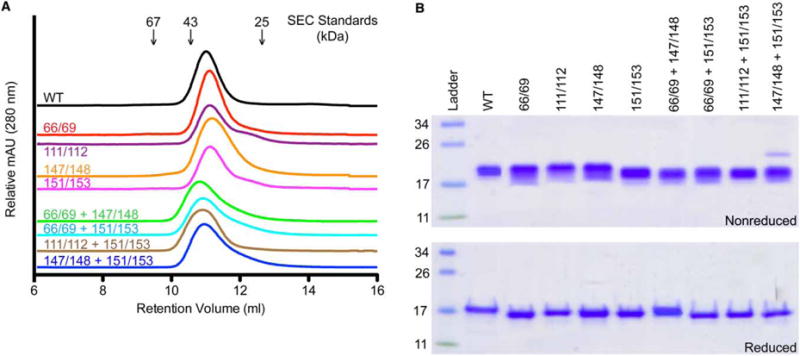
(A) SEC analysis of Grem2 WT and mutants using a Superdex 75 column. Numbers above traces indicate positions of various sizing standards. Numbers above colored lines indicate which lysines were mutated to alanine within each Grem2 mutant. (B) SDS–PAGE analysis showing the purified WT and lysine-to-alanine mutants of Grem2. Approximately 3 μg were loaded per lane. Numbers to the left of the gels indicate the molecular mass of the standards in the ladder in kDa. Numbers above gels show which lysines in Grem2 were mutated to alanine for each construct. Top gel shows nonreduced samples and the bottom gel shows BME-reduced samples.
Heparin-Sepharose chromatography is commonly used to demonstrate heparin/HS-binding affinity and the effects of mutation on this binding event [22,31]. Previously, we showed that Grem2 binds heparin with high affinity, requiring >650 mM NaCl to disrupt this interaction [23]. To examine the effect of our generated Grem2 lysine variants on heparin-binding activity, we compared the elution profile of each mutant with WT Grem2.
As can be seen, K66A/K69A and K111A/K112A had no significant effect on the heparin-binding profile of Grem2, suggesting that the wrist region and N-terminal lysines have a limited role in heparin/HS-binding affinity (Table 1 and Figure 3A). On the other hand, K147A/K148A and K151A/K153A showed a significant decrease in heparin-binding affinity, requiring only 590 and 580 mM NaCl for elution, respectively (Table 1 and Figure 3A). For comparison, Follistatin (Fst315), which also binds heparin, elutes at 719 mM NaCl, whereas NBL1, the DAN family member that lacks heparin/HS-binding affinity, does not bind to the heparin-Sepharose resin (Table 1 and Figure 3B). Taken together, these data suggest that multiple lysine clusters are important for heparin/HS binding.
Table 1.
Analysis of Grem2 mutants and proteins binding to heparin-affinity Sepharose
| Protein | NaCl (mM) | ΔNaCl (mM)a |
|---|---|---|
| Grem2 WT | 676 | 0 |
| K66A/K69A | 685 | 9 |
| K111A/K112A | 700 | 24 |
| K147A/K148A | 590 | −86 |
| K151A/K153A | 580 | −96 |
| K66A/K69A/K147A/K148A | 625 | −51 |
| K66A/K69A/K151A/K153A | 615 | −61 |
| K111A/K112A/K151A/K153A | 483 | −193 |
| K147A/K148A/K151A/K153A | 471 | −205 |
| BMP2 + Grem2 WT | 880 | 204 |
| BMP2 + K147A/K148A/K151A/K153A | 808 | 132 |
| FS315 | 719 | 43 |
| NBL1 | 0 | −676 |
ΔNaCl indicates the change in the NaCl elution concentration of WT from the variant value.
Figure 3. Analysis of Grem2 lysine mutants via heparin-affinity chromatography.
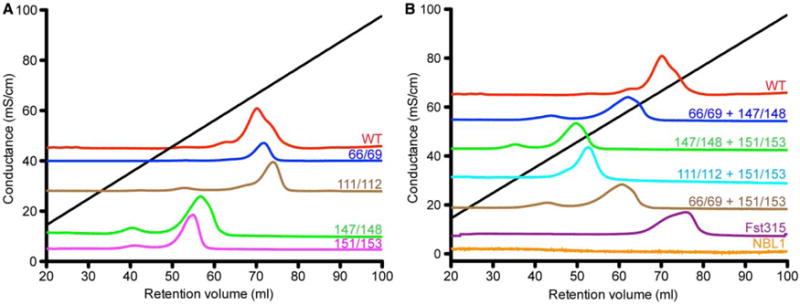
Chromatograms showing absorbance at 280 nm with conductance (mS/cm) shown on the y-axis and volume (ml) shown on the x-axis. Black line shows the linear gradient with increasing conductance. (A) Grem2 double lysine-to-alanine mutants. (B) Grem2 quadruple lysine-to-alanine mutants, NBL1, and Follistatin315.
Next, we wished to determine if these effects were combinatorial and if heparin/HS binding could be more extensively reduced. To test this, we generated the K66A/K69A/K174A/K148A, K111A/K112A/K147A/K148A, K66A/K69A/K151A/K153A, and K147A/K148A/K151A/K153A quadruple lysine mutants in Grem2. Analyzing these quadruple mutants by heparin-affinity chromatography showed that each mutant exhibited a substantially lower binding affinity when compared with the WT protein. However, only K111A/K112A/K151A/K153A and K147A/K148A/K151A/K153A showed significantly reduced heparin-binding affinity when compared with either the K147A/K148A or K151A/K153A mutations alone, suggesting that K66 and K69 are not critical in heparin/HS binding (Figure 3A,B and Table 1). Furthermore, the K147A/K148A/K151A/K153A mutant shows the most diminished heparin-binding affinity when compared with WT, only requiring 471 mM NaCl for elution. Taken together, these data suggest the importance of lysines within finger 2 in the Grem2–heparin/HS interaction (Figure 3A,B and Table 1).
Analysis of Grem2 lysine mutants for functional BMP inhibition
To assess the functional activity of the purified Grem2 lysine mutants, we compared their ability to inhibit BMP2 signaling activity using a cell-based BMP-responsive luciferase assay [27]. Overall, titrating the various Grem2 mutants against 2 nM BMP2 shows that each protein construct is fully capable of inhibiting BMP signaling based on their observed IC50 values (Figure 4A and Table 2). However, while each of these mutants exhibits high affinity for BMP2, their IC50 values are slightly higher (2–3-fold) when compared with the WT Grem2 protein, with the K147A/K148A mutant showing the largest reduction in functional BMP inhibition with a roughly 5-fold increase in its observed IC50 value for BMP2. In addition, we also measured the ability of the various Grem2 lysine mutants to bind to immobilized BMP2 utilizing SPR. The binding affinity of each mutant, as determined by the apparent dissociation constant (KD), exhibits minor increases in their determined equilibrium constants (KD) when compared with WT Grem2, where K66A/K69A/K151A/K153A and K147A/K148A/K151A/K153A show the largest decreases in affinity with a 3- and 4.5-fold higher KD compared with WT, respectively (Figure 4B; Supplementary Figure S1 and Table 2). Despite these minor observed differences, we believe that these data suggest that mutation of these specific lysine residues does not significantly alter the ability of Grem2 to bind to or inhibit BMP2 signaling, suggesting that the heparin/HS-binding motif and the BMP-binding motif are probably distinct in nature. Furthermore, this is consistent with the recent structure of Grem2–GDF5, where the lysine residues mutated in the present study are not located within or near the observed Grem2–ligand interface, thus appearing to play no role in direct BMP binding or inhibition [25].
Figure 4. BMP2 inhibitory activity of Grem2 lysine-to-alanine mutants.
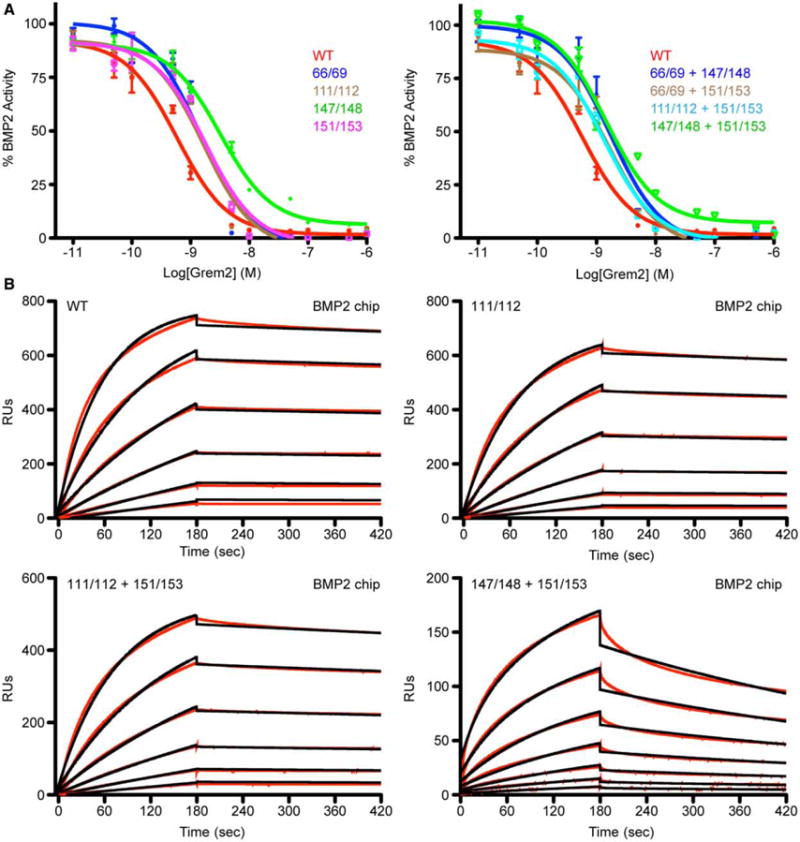
(A) Luciferase reporter assay using a BMP-responsive osteoblast cell line (BRITER). BMP2 (2 nM) was titrated against increasing amounts of Grem2 WT or lysine-to-alanine mutants. Left graph shows double lysine-to-alanine mutants and the right graph shows quadruple lysine-to-alanine mutants. (B) SPR sensorgrams using amine-coupled BMP2. Graphs show respective experimental traces (black) and corresponding fitting analysis (red) for Grem2 WT and select lysine-to-alanine mutants. RU, SPR response units.
Table 2.
SPR and luciferase analysis of Grem2 WT and mutant proteins against BMP2
| Mutant | kon (105 s−1) | koff (10−4 s−1) | KD (nM) | Fold SPRa | IC50 (nM) | Fold Lucb |
|---|---|---|---|---|---|---|
| Grem2-WT | 1.38 | 1.95 | 1.42 | 1.00 | 0.67 ± 0.06 | 1.00 |
| K66A/K69A | 1.05 | 2.53 | 2.40 | 1.69 | 1.45 ± 0.05 | 2.43 |
| K111A/K112A | 1.08 | 1.91 | 1.77 | 1.25 | 1.72 ± 0.06 | 2.78 |
| K147A/K148A | 0.87 | 1.72 | 1.98 | 1.39 | 3.17 ± 0.18 | 5.38 |
| K151A/K153A | 1.06 | 1.38 | 1.30 | 0.92 | 1.80 ± 0.06 | 2.97 |
| K66A/K69A/K147A/K148A | 3.20 | 18.1 | 5.66 | 4.00 | 1.72 ± 0.05 | 2.88 |
| K66A/K69A/K151A/K153A | 2.65 | 16.6 | 6.30 | 4.44 | 1.66 ± 0.05 | 2.78 |
| K111A/K112A/K151A/K153A | 1.07 | 2.26 | 2.11 | 1.49 | 1.31 ± 0.03 | 2.23 |
| K147A/K148A/K151A/K153A | 3.21 | 14.1 | 4.40 | 3.1 | 1.43 ± 0.08 | 2.45 |
Fold SPR determined by mutant KD divided by WT KD.
Fold Luc determined by mutant IC50 divided by WT IC50.
Analysis of the cell surface-binding properties of WT and mutant Grem2
Since the ability to bind heparin/HS localizes proteins to the cellular surface, we next wished to determine if the Grem2 lysine mutants exhibited a weaker ability to interact with the cellular surface. To test this, we labeled Grem2 WT, K111A/K112A/K151A/K153A, and K147A/K148A/K151A/K153A with Alexa 488-Cadaverine, which covalently labels proteins through carboxylic acid moieties, and tested their ability to adhere to cellular surfaces. Flow cytometric analysis [fluorescence-activated cell sorting, FACS] shows that Grem2 WT binds in a dose-dependent manner to CHO-K1 cells (Figure 5A and Table 3). However, when compared with Grem2 WT, both quadruple lysine mutants showed a significant reduction in their observed cell surface-binding characteristics (Figure 5B and Table 3). Furthermore, agreeing with our heparin-Sepharose data, K147A/K148A/K151A/K153A exhibited the most drastic decrease in cell surface binding, showing a 55% reduction in the mean fluorescence intensity (MFI) when compared with WT, while the K111A/K112A/K151A/K153A mutant only showed a corresponding 21% decrease in the MFI (Figure 5B and Table 3).
Figure 5. FACS analysis of Grem2 WT and lysine-to-alanine mutants.
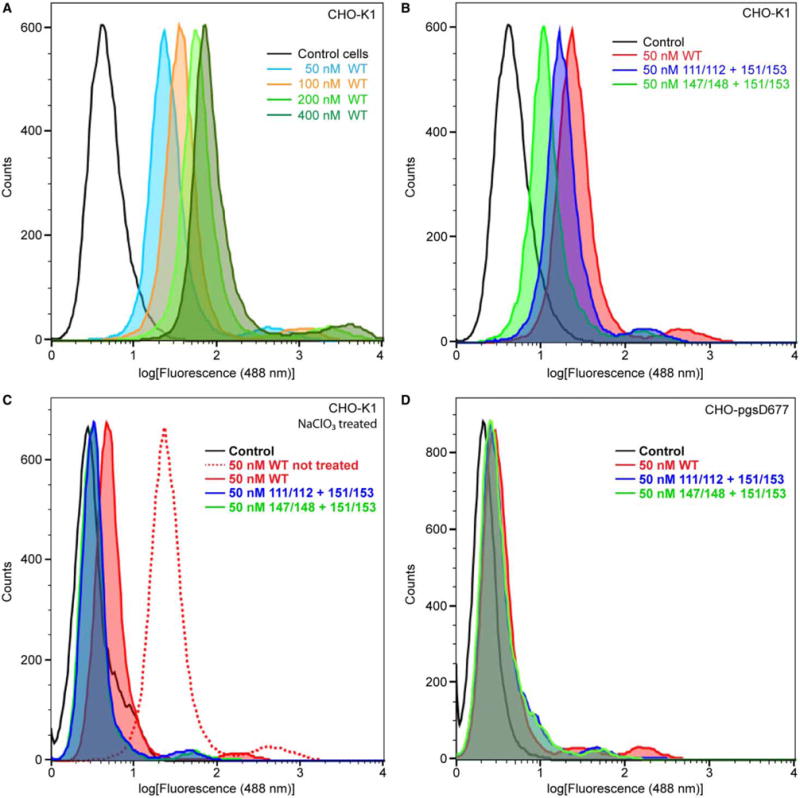
FACS sorting graphs based on fluorescence at 488 nm. Graphs show the log of the MFI of cells treated with Grem2 WT or select lysine-to-alanine mutants. (A) CHO-K1 treated with varying concentrations of Grem2 WT. (B) CHO-K1 cells treated with 50 nM Grem2 WT or select lysine-to-alanine mutants. (C) CHO-K1 cells treated with NaClO3 prior to protein treatment. (D) CHO-pgsD-677 cells, which are deficient in their ability to produce heparin/HS precursors, treated with various Grem2 proteins.
Table 3.
FACS analysis of cell surface binding of Grem2 WT and mutants
| Concentration (nM) | MFIa | |
|---|---|---|
| Figure 5A | ||
| Control cells | – | 4.8 |
| Grem2 WT | 50 | 29.2 |
| 100 | 43.3 | |
| 200 | 74.0 | |
| 400 | 111.0 | |
| Figure 5B | ||
| Control cells | – | 4.8 |
| Grem WT | 50 | 29.2 |
| K111A/K112A/K151A/K153A | 50 | 20.2 |
| K147A/K148A/K151A/K153A | 50 | 13.2 |
| Figure 5C | ||
| Control cells | – | 3.3 |
| Grem2 WT | 50 | 5.9 |
| K111A/K112A/K151A/K153A | 50 | 3.8 |
| K147A/K148A/K151A/K153A | 50 | 3.7 |
| Figure 5D | ||
| Control cells | – | 2.2 |
| Grem2 WT | 50 | 4.2 |
| K111A/K112A/K151A/K153A | 50 | 3.8 |
| K147A/K148A/K151A/K153A | 50 | 3.6 |
Abbreviations: MFI, mean fluorescence intensity.
MFI was determined using a 488 nM laser.
Next, to verify that the differences observed in the FACS data were a direct result of impaired heparin/HS-binding affinity, we determined the ability of WT and mutant Grem2 to bind to CHO-K1 cells following treatment with NaClO3. NaClO3 has previously been shown to inhibit the ability of cells to produce the functional sulfated precursors necessary in heparin/HS biosynthesis [32]. Following NaClO3 treatment, nearly all ability of the WT or mutant Grem2 constructs to bind to the CHO-K1 cell surface was removed (Figure 5C and Table 3). In addition, these results were recapitulated using the CHO-pgsD-677 cell line, which lacks the ability to produce and synthesize HS (Figure 5D and Table 3). Taken together, these data suggest that heparin/HS binding mediates the ability of Grem2 to bind to cellular surfaces, where K147, K148, K151, and K153, localized to finger 2, make up a large portion of this functional binding epitope (Figure 1C and Table 3).
Heparin-binding affinity of the Grem2–BMP2 complex
Next, we wished to determine if complexes of Grem2 with BMP, specifically BMP2, had any effect on the ability of the protein/complex to bind to heparin/HS. Similar to Grem2, BMP2 maintains the ability to bind heparin/HS using amino acids located within the N-terminus of the ligand, losing this ability upon removal of these basic residues [33]. Using our recently published Grem2–GDF5 structure, we generated a working model of the Grem2–BMP2 complex [25]. In this model, similar to our published structure, two molecules of Grem2 can be found binding to the distal ends of the ligand dimer (Figure 6A). Interestingly, the BMP2 heparin-binding motif, located on the basal surface of the ligand, lies within proximity of several lysine residues located on finger 2 of Grem2, including K147, K148, K151, and K153. As such, this generates a large and continuous electropositive surface between Grem2, BMP2, and the opposing Grem2 molecule, providing a larger area for a potential heparin/HS interaction than either Grem2 or BMP2 alone (Figure 6A). With these details in mind, we wished to determine the effect of complex formation on the ability of these two proteins to bind to heparin via a Sepharose affinity column and determine if the complex exhibits an improved ability to interact with heparin as would be predicted using our structure model.
Figure 6. Analysis of heparin-binding affinity of the Grem2–BMP2 complex.
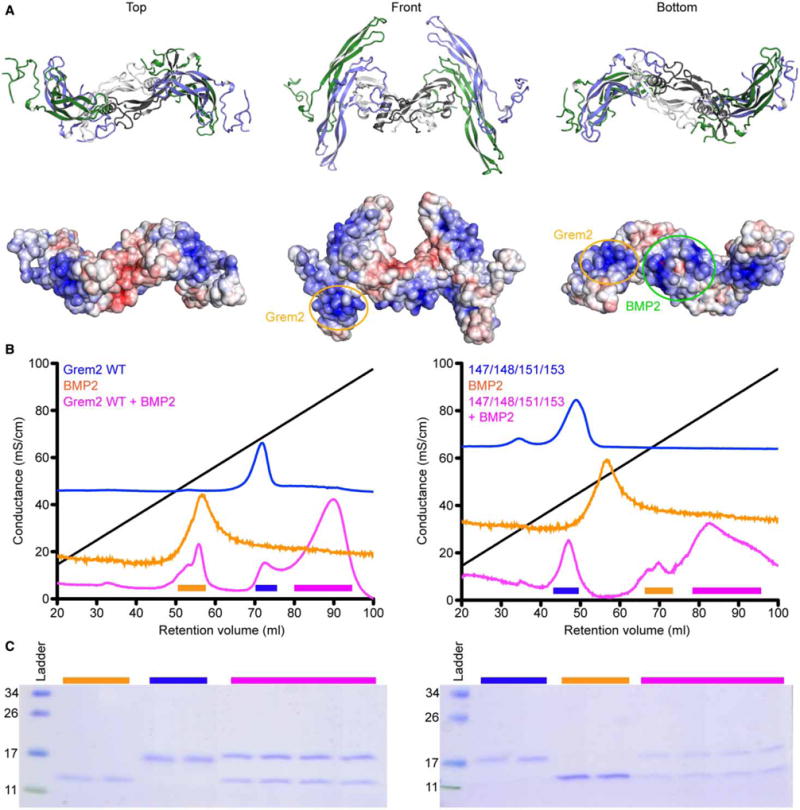
(A) Model of the Grem2–BMP2 complex generated from the recently resolved Grem2–GDF5 crystal structure (PDB: 5HK5). Complex shown in top, bottom, and front views. Top shows ribbon representation of the complex, whereas bottom shows electrostatic surface representation of the complex, colored from −5 to +5 kbt/ec, with blue showing positive surfaces and red showing negative surfaces. (B) Heparin-affinity Sepharose traces (absorbance at 280 nm) showing the elution profiles of Grem2, BMP2, and the Grem2–BMP2 complex. (C) SDS–PAGE (reduced) showing the protein composition of the peaks outlined in (B). Colored lines indicate fractions ran on the gel from the peaks indicated from (B). Numbers on the left indicate the molecular mass of the ladder in kDa. (B and C) Left graph and gel indicate analysis using Grem2 WT, whereas the right graph and gel indicate analysis using Grem2 K147A/K148A/K151A/K153A.
To test the ability of the Grem2–BMP2 complex to bind to the heparin-affinity column, we incorporated the addition of 0.5% CHAPS to our loading and running buffers. CHAPS is necessary to maintain solubility of the complex in solution as substantial and structured aggregation occurs for this complex in solution under standard conditions and as depicted in our structural analysis of Grem2–GDF5 [25]. Furthermore, SEC analysis shows that there is no difference in the elution profile of Grem2 WT and K147A/K148A/K151A/K153A in the presence of the added CHAPS detergent (Supplementary Figure S2). As such, we tested Grem2 WT and K147A/K148A/K151A/K153A, BMP2, and the corresponding Grem2–BMP2 complexes under these modified conditions.
As can be seen, the Grem2–BMP2 complex binds to heparin with higher affinity than either protein alone (580 mM NaCl for BMP2 and 700 mM NaCl for Grem2), requiring 880 mM NaCl for elution (Figure 6B,C and Table 1). Next, we tested the ability of the Grem2 K147A/K148A/K151A/K153A–BMP2 complex to bind to the heparin-affinity Sepharose. Interestingly, while the K147A/K148A/K151A/K153A mutant shows a much-reduced ability to bind to heparin in comparison with Grem2 WT or BMP2 alone, the complex shows a much-improved ability to bind to heparin, requiring nearly 808 mM NaCl to elute off of the column (Figure 6B and Table 1). While this mutant complex does elute from the heparin-Sepharose with lower NaCl when compared with the WT Grem2– BMP2 complex, the difference is much less than anticipated and suggests that the combinatorial nature of the Grem2–BMP2 interaction can overcome the reduced heparin/HS-binding affinity of the Grem2 lysine mutations.
Discussion
In the present study, we uncovered a significant portion of the Grem2 heparin-binding motif. By mutating specific surface-exposed lysines, we were able to observe the following: (1) lysines within the finger 2 region of Grem2 appear to contribute significantly to the heparin-binding affinity of the protein in vitro and on the cell surface, namely K147, K148, K151, and K153; (2) these lysines appear not to drastically affect BMP-binding affinity and antagonism, suggesting that the two epitopes are mutually exclusive; and (3) the heparin/HS-binding sites of BMP2 and Grem2 within the Grem2–BMP2 complex coalesce to form a collectively larger electropositive interface that gives the complex higher binding affinity when compared with either protein component alone.
Within the structure of Grem2, the location of lysines 147, 148, 151, and 153 can be found clustered together within finger 2, forming a strip that is ∼20–25 Å in length (Figure 1B). This length would correspond to a heparin/HS oligomer of 18–20 saccharides in length and probably represents the major epitope for this interaction. In addition, it should be noted that each lysine mutation confers removal of two lysines on two separate heparin/HS-binding epitopes as Grem2 functions as a dimer. Comparing our findings with the known heparin-binding epitopes within Grem1, many observations can be made (Figure 1A) [22]. First, both proteins appear to mainly utilize surface-exposed lysines and, to a lesser extent, arginines in finger 2 to facilitate heparin/HS binding. Outside of this region, however, Grem1 contains three additional lysines on finger 2 closer to the wrist region that are implicated in heparin binding, but to a lesser extent than those conserved with K147, K148, K151, and K153 in Grem2 (Figures 1A and 7B) [22]. Although untested, it is likely that these amino acids in Grem2 contribute to heparin/HS binding based on the high amino acid identity and similarity with Grem1, but to a lesser extent than those amino acids identified in the present study.
Figure 7. Electrostatic surface of the Grem2–BMP2 ‘daisy-chain’ and structural comparison of different DAN family heparin/HS-binding motifs.
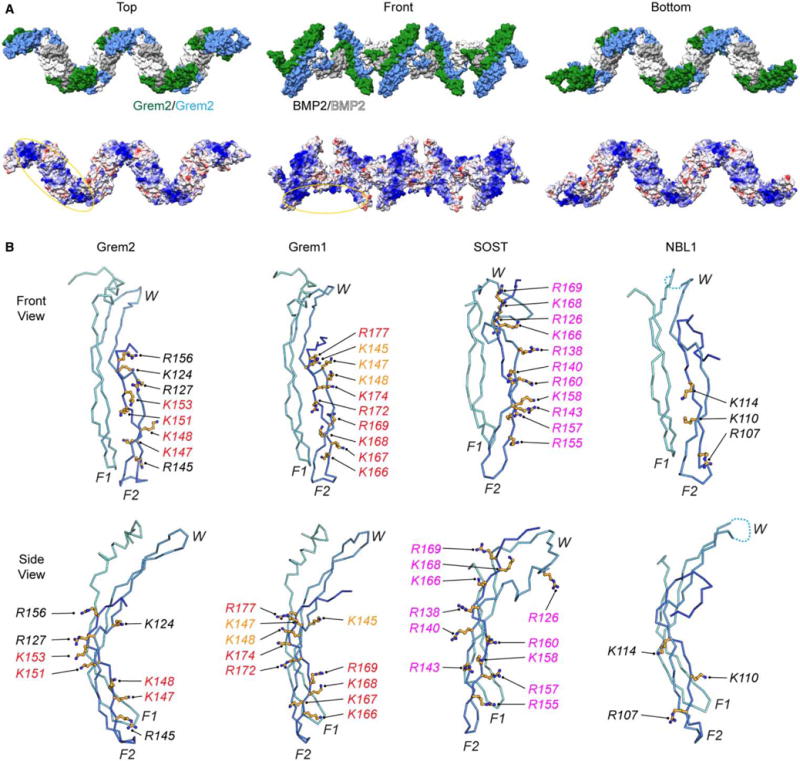
(A) Structure model of the Grem2–BMP2 polymer (‘daisy-chain’) complex made from the observed ‘daisy-chain’ complex in the Grem2–GDF5 structure (PDB: 5HK5). (Top) Surface representation of the ‘daisy-chain’ complex, with Grem2 monomers colored green and blue and BMP2 monomers colored white and gray. (Bottom) Coulombic surface coloring of the Grem2–BMP2 ‘daisy-chain’ complex as performed using UCSF Chimera. Structure is colored from −10 to +10 kbt/ec, with more negative surfaces colored in red, neutral surfaces colored in white, and positive surfaces colored in blue. Heparin/HS-binding motifs of Grem2 and BMP2 are circled. (B) Structures of Grem2 (PDB: 4JPH), Grem1 (PDB: 5AEJ), SOST (PDB: 2K8P), and NBL1 (PDB: 4X1J) shown in ribbon representation. Structures show a single monomer of each DAN family antagonist with determined or predicted heparin/HS-binding lysines or arginines shown in stick representation in orange. Orange shows amino acids in Grem1 that have been tested with moderate importance in heparin/HS binding, red shows high importance, and black shows untested amino acids that are synonymous with amino acids in other DAN family proteins shown to be important. Magenta shows amino acids in SOST that were shown to bind directly to heparin via NMR spectroscopy.
Comparison with SOST shows that several lysines and arginines are locally conserved with those identified in Grem2 and Grem1 (Figures 1A and 7B). However, SOST was shown to utilize additional amino acids that are not conserved in Grem1 and Grem2 located outside finger 2 (Figures 1A and 7B) [21,22]. As these amino acids were not individually mutated in SOST, it is hard to speculate their individual impact on heparin/HS binding. As such, it appears that finger 2, spanning from the fingertip to the core of the protein, appears to be the major functional region within the DAN family for imparting heparin/HS-binding affinity (Figures 1A and 7B). Finally, NBL1, which maintains no ability to bind to heparin, lacks most of the basic amino acids identified in Grem1, Grem2, and SOST (Figures 1A and 7B; Table 1). Taken together, this suggests that finger 2 is the most important region within DAN family members for conferring heparin/HS binding, but also displays some diversity in the exact positioning and conservation of these amino acids.
Interestingly, we have made the observation that the heparin/HS-binding affinity of the Grem2–BMP2 complex is substantially higher than either that of BMP2 or Grem2 alone. This finding probably extends to other ligand–antagonist complexes that maintain the ability to bind heparin/HS. What role this elevated affinity plays is unknown in regard to the DAN family and BMP signaling. However, similar observations have been made for the binding of Follistatin and Myostatin, where the complex exhibits an increased affinity for heparin/HS [34]. Furthermore, it has been suggested that this increase in binding affinity for the complex leads to corresponding increases in endocytosis [18,35]. Perhaps future studies can ascertain whether similar observations in regard to endocytosis are true for DAN–BMP complexes.
Looking at the DAN–BMP complex more closely, several factors can be seen to suggest increases in heparin/HS-binding affinity. Using the Grem2–GDF5 structure, we generated a model of the Grem2–BMP2 complex (Figure 6A). This model is useful as GDF5 lacks the ability to bind to heparin/HS, where BMP2 exhibits a high affinity for these oligosaccharides. The model shows that finger 2 of Grem2 is positioned directly adjacent to the heparin/HS-binding epitope of BMP2 [33]. As such, this combined interface would provide a more continuous binding interface explaining the increase in binding affinity of the Grem2–BMP2 complex. Furthermore, recent evidence has suggested that DAN–BMP complexes, particularly that of Grem2–BMP2 and Grem2–GDF5, form larger-order aggregates in a ‘daisy-chain’-like fashion, where one monomer of the Grem2 dimer binds to one BMP ligand, leaving the opposing Grem2 monomer open to bind another ligand [25]. As such, these aggregates form elongated polymers with repeating heparin/HS-binding motifs, both present on the apical and basal surfaces of the polymer (Figure 7A). With this in mind, perhaps the repeating nature of this complex and the increased number of motifs available to bind heparin/HS increase the apparent affinity of the Grem2–BMP2 complex. This aggregation may also explain why the K147A/K148A/K151A/K153A Grem2–BMP2 complex showed similar heparin/HS affinity when compared with the WT complex, where this Grem2 mutant alone showed a much-reduced heparin/HS-binding affinity compared with WT Grem2 (Table 3). It is also interesting to speculate that the larger Grem2–BMP2 complexes could bridge multiple heparin/HS molecules. Future studies will certainly aim to more readily determine the role aggregation plays in heparin/HS binding.
In conclusion, we have shown that Grem2 binds heparin/HS with high affinity utilizing lysines that are independent of the BMP-binding motif, suggesting that they are probably mutually exclusive. Furthermore, we have shown that mutation of these epitopes leads to the inability of Grem2 to bind heparin/HS both in the in vitro and cellular contexts. In addition, we have shown that NBL1 lacks the ability to form this interaction, perhaps elucidating a role similar to that seen between Follistatin and FSTL3, where FSTL3, which lacks a functional heparin/HS-binding motif, is more serum-available and also a less effective inhibitor in comparison [16,36,37]. Finally, we have shown that ligand–antagonist complexes of Grem2–BMP2 exhibit a higher in vitro binding affinity for heparin/HS, perhaps suggesting an additional form of regulation. Future studies will certainly need to be designed to test the biological significance of these interactions and their role in making anti-BMP therapeutics that are probably more serum-available.
Supplementary Material
Acknowledgments
Funding
This work was funded by an R01 grant [GM114640] awarded to T.B.T.
Abbreviations
- BMP
bone morphogenetic protein
- DAN
differential screening-selected gene abberative in neuroblastoma
- FBS
fetal bovine serum
- FACS
fluorescence-activated cell sorting
- Grem1
Gremlin-1
- Grem2
Gremlin-2
- HS
heparan sulfate
- MFI
mean fluorescent intensity
- SEC
size-exclusion chromatography
- SPR
surface plasmon resonance
- WT
wild type
Footnotes
Author Contribution
C.K. and K.N. performed experiments and interpreted the data. C.K., K.N., and T.B.T. designed the experiments and wrote the paper.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Urist MR, Strates BS. Bone morphogenetic protein. J Dent Res. 1971;50:1392–1406. doi: 10.1177/00220345710500060601. [DOI] [PubMed] [Google Scholar]
- 2.Hogan BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 3.Yoon BS, Lyons KM. Multiple functions of BMPs in chondrogenesis. J Cell Biochem. 2004;93:93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- 4.Blitz IL, Cho KWY. Finding partners: how BMPs select their targets. Dev Dyn. 2009;238:1321–1331. doi: 10.1002/dvdy.21984. [DOI] [PubMed] [Google Scholar]
- 5.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 6.Yang W, Cao L, Liu W, Jiang L, Sun M, Zhang D, et al. Novel point mutations in GDF5 associated with two distinct limb malformations in Chinese: brachydactyly type C and proximal symphalangism. J Hum Genet. 2008;53:368–374. doi: 10.1007/s10038-008-0253-7. [DOI] [PubMed] [Google Scholar]
- 7.Cogan J, Austin E, Hedges L, Womack B, West J, Loyd J, et al. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation. 2012;126:1907–1916. doi: 10.1161/CIRCULATIONAHA.112.106245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Bruggen CE, Happé CM, Dorfmüller P, Trip P, Spruijt OA, Rol N, et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation. 2016;133:1747–1760. doi: 10.1161/CIRCULATIONAHA.115.020696. [DOI] [PubMed] [Google Scholar]
- 9.Lowery JW, de Caestecker MP. BMP signaling in vascular development and disease. Cytokine Growth Factor Rev. 2010;21:287–298. doi: 10.1016/j.cytogfr.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehata S, Yokoyama Y, Takahashi K, Miyazono K. Bi-directional roles of bone morphogenetic proteins in cancer: another molecular Jekyll and Hyde? Pathol Int. 2013;63:287–296. doi: 10.1111/pin.12067. [DOI] [PubMed] [Google Scholar]
- 11.Massagué J. TGFβ in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinck AP, Mueller TD, Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb Perspect Biol. 2016;8:a022103. doi: 10.1101/cshperspect.a022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rider CC, Mulloy B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem J. 2010;429:1–12. doi: 10.1042/BJ20100305. [DOI] [PubMed] [Google Scholar]
- 14.Paine-Saunders S, Viviano BL, Economides AN, Saunders S. Heparan sulfate proteoglycans retain Noggin at the cell surface: a potential mechanism for shaping bone morphogenetic protein gradients. J Biol Chem. 2002;277:2089–2096. doi: 10.1074/jbc.M109151200. [DOI] [PubMed] [Google Scholar]
- 15.Jasuja R, Allen BL, Pappano WN, Rapraeger AC, Greenspan DS. Cell-surface heparan sulfate proteoglycans potentiate chordin antagonism of bone morphogenetic protein signaling and are necessary for cellular uptake of chordin. J Biol Chem. 2004;279:51289–51297. doi: 10.1074/jbc.M408129200. [DOI] [PubMed] [Google Scholar]
- 16.Sidis Y, Schneyer AL, Keutmann HT. Heparin and activin-binding determinants in Follistatin and FSTL3. Endocrinology. 2005;146:130–136. doi: 10.1210/en.2004-1041. [DOI] [PubMed] [Google Scholar]
- 17.Bier E, De Robertis EM. BMP gradients: a paradigm for morphogen-mediated developmental patterning. Science. 2015;348:aaa5838. doi: 10.1126/science.aaa5838. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto O, Nakamura T, Shoji H, Shimasaki S, Hayashi Y, Sugino H. A novel role of Follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. J Biol Chem. 1997;272:13835–13842. doi: 10.1074/jbc.272.21.13835. [DOI] [PubMed] [Google Scholar]
- 19.Hu Z, Wang C, Xiao Y, Sheng N, Chen Y, Xu Y, et al. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- 20.Nolan K, Thompson TB. The DAN family: modulators of TGF-β signaling and beyond. Protein Sci. 2014;23:999–1012. doi: 10.1002/pro.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284:10890–10900. doi: 10.1074/jbc.M807994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tatsinkam AJ, Mulloy B, Rider CC. Mapping the heparin-binding site of the BMP antagonist gremlin by site-directed mutagenesis based on predictive modelling. Biochem J. 2015;470:53–64. doi: 10.1042/BJ20150228. [DOI] [PubMed] [Google Scholar]
- 23.Nolan K, Kattamuri C, Luedeke DM, Deng X, Jagpal A, Zhang F, et al. Structure of protein related to DAN and cerberus: insights into the mechanism of bone morphogenetic protein antagonism. Structure. 2013;21:1417–1429. doi: 10.1016/j.str.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kattamuri C, Luedeke DM, Thompson TB. Expression and purification of recombinant protein related to DAN and cerberus (PRDC) Protein Expr Purif. 2012;82:389–395. doi: 10.1016/j.pep.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nolan K, Kattamuri C, Rankin SA, Read RJ, Zorn AM, Thompson TB. Structure of Gremlin-2 in complex with GDF5 gives insight into DAN-family-mediated BMP antagonism. Cell Rep. 2016;16:2077–2086. doi: 10.1016/j.celrep.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Einem S, Schwarz E, Rudolph R. A novel TWO-STEP renaturation procedure for efficient production of recombinant BMP-2. Protein Expr Purif. 2010;73:65–69. doi: 10.1016/j.pep.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Yadav PS, Prashar P, Bandyopadhyay A, Cooney AJ. BRITER: a BMP responsive osteoblast reporter cell line. PLoS ONE. 2012;7:e37134. doi: 10.1371/journal.pone.0037134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nolan K, Kattamuri C, Luedeke DM, Angerman EB, Rankin SA, Stevens ML, et al. Structure of neuroblastoma suppressor of tumorigenicity 1 (NBL1): insights for the functional variability across bone morphogenetic protein (BMP) antagonists. J Biol Chem. 2015;290:4759–4771. doi: 10.1074/jbc.M114.628412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arterioscler Thromb Vasc Biol. 1989;9:21–32. doi: 10.1161/01.ATV.9.1.21. [DOI] [PubMed] [Google Scholar]
- 30.Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268:19228–19231. [PubMed] [Google Scholar]
- 31.Daubner SC, Piper MM. Deletion mutants of tyrosine hydroxylase identify a region critical for heparin binding. Protein Sci. 1995;4:538–541. doi: 10.1002/pro.5560040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Safaiyan F, Kolset SO, Prydz K, Gottfridsson E, Lindahl U, Salmivirta M. Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J Biol Chem. 1999;274:36267–36273. doi: 10.1074/jbc.274.51.36267. [DOI] [PubMed] [Google Scholar]
- 33.Ruppert R, Hoffmann E, Sebald W. Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur J Biochem. 1996;237:295–302. doi: 10.1111/j.1432-1033.1996.0295n.x. [DOI] [PubMed] [Google Scholar]
- 34.Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 2009;28:2662–2676. doi: 10.1038/emboj.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R. Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci USA. 1987;84:8282–8286. doi: 10.1073/pnas.84.23.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sidis Y, Tortoriello DV, Holmes WE, Pan Y, Keutmann HT, Schneyer AL. Follistatin-related protein and follistatin differentially neutralize endogenous vs. exogenous activin. Endocrinology. 2002;143:1613–1624. doi: 10.1210/endo.143.5.8805. [DOI] [PubMed] [Google Scholar]
- 37.Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology. 2006;147:3586–3597. doi: 10.1210/en.2006-0089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.