

Abstract
Dopaminergic medications ameliorate many of the motor impairments of Parkinson’s disease (PD). However, parkinsonism is often only partially reversed by these drugs, and they can have significant side effects. Therefore, a need remains for novel treatments of parkinsonism. Studies in rodents and preliminary clinical evidence have shown that T-type calcium channel (TTCC) antagonists have antiparkinsonian effects. However, most of the available studies utilized non-selective agents. We now evaluated whether systemic injections of the specific TTCC blocker ML218 have antiparkinsonian effects in MPTP-treated parkinsonian Rhesus monkeys. The animals were treated chronically with MPTP until they reached stable parkinsonism. In pharmacokinetic studies we found that ML218 reaches a peak CSF concentration 1–2 hrs after s.c. administration. In electrocardiographic studies, we found no effects of ML218 on cardiac rhythmicity. As expected, systemic injections of the dopamine precursor L-DOPA dose-dependently increased the movements in our parkinsonian animals. We then tested the behavioral effects of systemic injections of ML218 (1, 10 or 30 mg/kg) or its vehicle, but did not detect specific antiparkinsonian effects. ML218 (3 or 10 mg/kg) was also not synergistic with L-DOPA. Using recordings of electrocorticogram signals (in one animal), we found that ML218 increased sleep. We conclude that ML218 does not have antiparkinsonian effects in MPTP-treated parkinsonian monkeys, due at least in part, to the agent’s sedative effects.
Keywords: Non-human primates, parkinsonism, T-type calcium channel, ML218, MPTP, pharmacokinetic, behavior
Graphical Abstract
INTRODUCTION
Current pharmacologic therapies for parkinsonism almost exclusively rely on restoring dopaminergic transmission in Parkinson’s disease (PD) patients, either by providing them with the dopamine precursor L-DOPA, or by administration of dopamine receptor agonists. However, dopaminergic therapy does not fully reverse parkinsonism in many patients, and can lead to unwanted side effects. Thus there remains a need to explore other therapeutic options for patients with PD.
The loss of the dopamine innervation to the striatum and other basal ganglia in PD triggers the appearance of altered firing patterns throughout the basal ganglia-thalamocortical circuits.1 Among such abnormalities, in PD patients and in animal models of parkinsonism, neurons in the basal ganglia and in the motor thalamus show an increased tendency to fire in bursts (e.g., ref. 2, 3–10). In parkinsonian animals, the increased bursting parallels the development of parkinsonism.11, 12 Effective antiparkinsonian treatments, such as deep-brain stimulation (DBS) or dopamine replacement therapy reduce bursting,13–15 highlighting the behavioral importance of this firing abnormality.
In the basal ganglia and thalamus, a particular type of bursting activity, the so called ‘rebound’ bursts, occur after profound membrane hyperpolarization and depend on T-type calcium channels (Cav3, e.g., ref. 16). T-type calcium channels (TTCC) inactivate by depolarization and de-inactivate after a period of hyperpolarization, generating bursting activity.17–19. This type of bursting could also contribute to the pathophysiologically relevant increased oscillations in the basal ganglia in PD (e.g., ref. 20). Blockade of rebound bursting by TTCC antagonists could therefore have antiparkinsonian properties. In fact, local administration of non-selective TTCC inhibitors (Ni2+, mibefradil, NNC 55-0396 and efonidipine) in the subthalamic nucleus (STN) reduced rebound bursting and ameliorated motor deficits in 6-OHDA-treated parkinsonian rats.21
Systemic injections of the anti-epileptic drug ethosuximide reduced parkinsonian tremor in aged rats22 and in an MPTP-treated parkinsonian monkey23. While the effect of ethosuximide was not replicated in patients with PD,24 another anti-epileptic compound, zonisamide, has been shown to ameliorate parkinsonism in PD patients.25, 26 Both ethosuximide and zonisamide have TTCC inhibitory activity, but also act at several other pharmacological targets,27–35 complicating the interpretation of the mechanisms of action behind the antiparkinsonian effects of these drugs.
Recently, we have developed new highly specific TTCC blockers with high brain permeability,36, 37 including voltage-independent and voltage-dependent drugs.38–40 One of the voltage-independent agents, ML218, has demonstrated anti-cataleptic properties in the haloperidol-induced catalepsy model in rats,38 a commonly used model to test antiparkinsonian effects of drugs. Here we investigated the potential antiparkinsonian properties of ML218 in MPTP-treated stable parkinsonian Rhesus monkeys. We also studied sedative and cardiac effects of ML218 that could interfere with potential antiparkinsonian actions.
RESULTS AND DISCUSSION
Pharmacokinetic studies
We examined the pharmacokinetic profile of ML218 after systemic administration to Rhesus monkeys. Following a 10 mg/kg subcutaneous (s.c.) injection of ML218 (in 10% EtOH/40% PEG/50% saline), we observed a typical absorption-phased-limited disposition of ML218 in vivo as evidenced by a rapid time to achieve a maximal plasma concentration (Tmax) and a systemic concentration of the parent compound that plateaued through 24 hours (fig. 1). An in vitro assessment of the permeability properties of ML218 in MDCK-MDR1 (MDCK-Pgp) cells indicated that the compound was freely diffusing across a cellular monolayer (Papp AB, 2.6e− 6 cm/sec) and was not a substrate for efflux mediated by Pgp (Efflux Ratio, 0.9). Coupled with a relatively high fraction of unbound compound in Rhesus monkey plasma (Fu, 0.09), we predicted that ML218 would distribute into the brain in animals receiving the drug by s.c. administration (as previously reported in rats38). Indeed, the time-concentration profile of ML218 depicted in figure 1 indicates that the compound distributes to the CNS, as it achieves CSF levels that reach a Tmax (1–2 hr) similar to that observed in plasma. Importantly, the CSF concentrations observed experimentally mirror our predicted CSF time-concentration profile (figure 1, dotted line) that was generated using the calculated unbound concentration of ML218 in plasma (data not shown). These data are consistent with the principle that, for compounds that are freely diffusible and not substrates for efflux proteins, the unbound plasma concentration will reflect the unbound brain and CSF concentrations at equilibrium.41, 42
Figure 1. Pharmacokinetics of ML218 after systemic administration.

Plasma and CSF time-concentration profiles of ML218 in Rhesus monkeys following a single s.c. dose (10 mg/kg) of ML218). Data are means + SEM
For subsequent studies, ML218 was administered using a 10% Tween saline mixture to avoid unwanted side effects common with formulations high in ethanol content.
Assessment of electrocardiographic effects
We next examined whether ML218 affects electrical cardiac activities which could limit the usefulness of this drug. These experiments were carried out in three of the animals, using s.c. injections of ML218 (10 mg/kg and 30 mg/kg) or vehicle, under ketamine sedation. We found that, at the doses studied, ML218 had minor negative chronotropic effects (lengthening the R-R interval in all animals at the 30 mg/kg dose), but no consistent effects on the other parameters studied (fig. 2). There were no systematic effects on the shape/amplitude of any component of the ECG in these experiments.
Figure 2. Electrocardiographic effects of ML218.

Shown are measurements of RR, PR and RT intervals (means + SEM) in 3 animals (vehicle, 10 mg/kg, 30 mg/kg). Asterisks indicate significant differences between the data obtained with vehicle injections and those obtained with ML218.
Behavioral studies
We examined if ML218 has antiparkinsonian effects when administered to MPTP-treated parkinsonian monkeys. Six monkeys received weekly administrations of MPTP, until stable parkinsonism developed. At the beginning of these studies, at least 6 weeks have passed after the last MPTP administration and all monkeys were considered stable parkinsonian according to our behavioral evaluations (see Methods). To test the behavioral effects of systemic drug administrations, the monkey was transferred to an observation cage, in which the spontaneous movements of the monkey were monitored via infrared beam break counting, or by an observer who counted the number and type of movements by using a computer-assisted method (see Methods). In each session, the animal was allowed to habituate to the cage for at least 15 minutes.
Antiparkinsonian effects of L-DOPA
First, we assessed the anti-parkinsonian effects of L-DOPA in this group of monkeys. After habituation to the cage, a baseline of motor behavior was established for that session (15 min before drug injection, see fig. 3A). We tested the effects of L-DOPA at 5 mg/kg, 10 mg/kg, 15 mg/kg and 20 mg/kg (in combination with benserazide) to identify doses providing significant (but not maximal) antiparkinsonian effects for the subsequent experiments testing the combination of L-DOPA and ML218.
Figure 3. Time course of behavioral experiments.

The arrows indicate the times when L-DOPA (i.m.) or ML218 (s.c.) were systemically administered. A, B and C indicate sessions in which L-DOPA, ML218 or a combination of both drugs were administered.
The results are shown in figure 4 (in figures 4 and 5, changes in movement are shown as seen by the observer (figure 4A and 5A) and as monitored via beam break counting (figure 4B and 5B)). As expected, L-DOPA had strong, dose-dependent antiparkinsonian effects in all animals, significantly different from the vehicle for all L-DOPA doses tested in the number of movements (Mann-Whitney U=159.5, 64, 11, and 29, for 5, 10, 15 and 20 mg/kg respectively, all p<0.001) and in the number of beam breaks (activity counts, Mann-Whitney U=215, p=0.007 for 10 mg/kg, U=114, p<0.001 for 15 mg/kg; U=38, p<0.001 for 20 mg/kg).
Figure 4. Behavioral responses of parkinsonian animals to the administration of L-DOPA, ML218, or a combination of ML218 and L-DOPA.

Each column represents the average (+ SEM) of data from 6 monkeys. For each experiment we calculated the median of all observation periods (5 consecutive periods of 15 min each). The baseline activity on the day of testing was subtracted from the post-injection values. Shown are number of movements (A) and activity counts (B). Statistical difference from respective vehicle: ** p<0.01; *** p<0.001 (Mann-Whitney test, followed by Bonferroni corrections).
Figure 5. Time course of behavioral changes after s.c. of L-DOPA, alone or in combination with ML218.

The data shown are mean values (± SEM) of the number of movements (A) and activity counts (B) obtained for each observation period starting 15 min after L-DOPA injection (values are corrected to the pre-injection baseline and normalized to the number of minutes per observation period). ML218 (or the vehicle Tween 80) were administered 60 min before L-DOPA administration. The two experimental conditions were compared at each time point (Mann-Whitney test), no significant differences were found.
Behavioral effects of ML218
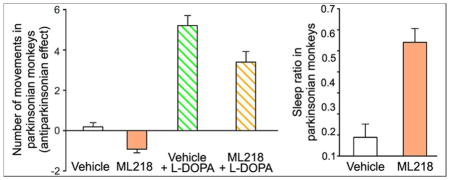
Once established that the parkinsonism in these animals was reduced with systemic L-DOPA injections, we evaluated the effects of ML218. Based on our pharmacokinetic studies (figure 1), we examined the effect of ML218 1 hour after the systemic administration of the compound (fig. 3B), using concentrations of 1, 10, and 30 mg/kg (based on published rodent studies38). Figure 4 shows the effects of ML218 on motor behavior (center bars in figures 4A and 4B). The compound had no antiparkinsonian effects at any of the doses tested. In fact, in some cases, the drug reduced the number of movements (Mann-Whitney U=24, p=0.006, for 10 mg/kg) and the activity counts (Mann-Whitney U=20, p=0.002 for 10 mg/kg), compared to baseline.
We hypothesized that, even if ML218 on its own did not show antiparkinsonian effects in our monkeys, the combination of this compound with L-DOPA could enhance the effects of a submaximal dose of L-DOPA. The animals were first observed for 15 minutes as described above, and then exposed to vehicle (Tween80) or ML218 (s.c., 3 mg/kg). Sixty minutes later, they received L-DOPA (i.m., 10 mg/kg, plus 2.5 mg/kg benserazide, fig. 3C). The animals were examined for a 15 minute period immediately before the L-DOPA exposure, and for 5 additional 15-minute periods thereafter. The relatively low dose of ML218 was chosen because higher doses induced mild sedation in some animals (an effect that we further investigated with ECoG recordings, see below), and the dose of L-DOPA was chosen because it had moderate antiparkinsonian effects when given alone (fig. 4). L-DOPA had the expected antiparkinsonian effect, more noticeable in the number of movements (figs. 4A and 5A). However, addition of ML218 3 mg/kg did not alter the L-DOPA-mediated responses. Neither the number of movements (Mann-Whitney U=323, p=0.06) nor the activity counts (Mann-Whitney U=380, p=0.301) were significantly different when ML218 was added to the L-DOPA treatment (fig. 4). The time course of the response to L-DOPA was also unchanged (fig. 5)
Effects of ML218 on the state of arousal and ECoG signals
Given that we observed (presumed) sedative effects of ML218 administration, we examined the effects of ML218 on arousal, by recording ECoG signals after systemic injections of the compound in one MPTP-treated stably parkinsonian monkey. Examples of ECoG traces obtained during wake and sleep episodes are shown in fig. 6A.
Figure 6. Results of ECoG recordings.

A, example of 10 second epoch of M1 ECoG recording representing wakefulness and sleep. The scale bars in the top trace also apply for the bottom trace. Positive changes in voltage are upward. B, proportion of sleep epoch among all epochs (‘Sleep ratio’) obtained in the 2 hour period following the injection of Tween (white) and ML218 (grey). C, number of spontaneous movements in 5 minute epochs (out of 15 minute time blocks) obtained during 2 hours following the injections. D, spectral power of the ECoG signals, integrated across the 5 frequency bands (delta, theta, alpha beta, gamma). Values are means + SEM. The differences between ML218 and vehicle injection sessions were assessed with a Mann-Whitney test. *, p<0.05; **, p<0.01.
After systemic administration of ML218 (3 mg/kg), the animal showed a larger number of sleep periods compared to the awake periods. The sleep ratio, i.e., the ratio of time within 120 minutes after ML218, was significantly higher than after vehicle injections (0.09 ± 0.06 and 0.54 ± 0.06; for vehicle and ML218 injections, respectively, p=0.008, Mann-Whitney test; fig. 6B). As in the observation cage experiments, the number of spontaneous movements was significantly reduced after ML218 treatment compared to measurements after vehicle injections (51.07 ± 6.42 and 25.39 ± 4.52 mean number of movements per 5 minute of observation in experiments using vehicle and ML218 treatments, respectively; p=0.016 Mann-Whitney test, fig. 6C). In the ECoG signals, the oscillatory power in the delta frequency range (a frequency range associated with sleep states) was significantly higher after ML218 treatment when compared to vehicle injections (1.6 × 10−4 ± 1.7 × 10−5 and 3.4 × 10−4 V ± 4.6 × 10−5 for vehicle and ML218 sessions, respectively, p=0.008, Mann-Whitney test).
Dopamine loss, as assessed by TH immunoperoxidase staining
Post-mortem histological analysis showed that TH immunolabeling in the post-commissural putamen and caudate was reduced by 87 ± 3% and 89 ± 3% (mean ± SEM), respectively, in the 7 MPTP-treated animals, compared to the mean of measurements from 3 non-treated monkeys.
In summary, our studies examined whether the selective TTCC blocker ML218 has antiparkinsonian properties in MPTP-treated Rhesus macaques. We did not detect improvements in the parkinsonian signs of these monkeys after ML218 injections, but found that the agent had sedative effects. The systemic injections of ML218, at the doses used in our study, had minimal effects on the ECG, and did not produce other adverse effects.
Our pharmacokinetic studies, coupled to assessment of permeability and efflux of ML218, indicated that the compound distributes in the brain after systemic administration. These methods allowed us to accurately predict ML218 concentrations in the CSF along time, with a minimal need of CSF collections.
ML218 is a highly specific TTCC blocker. Our recent studies of the neuronal effects of intrathalamic microinjections of ML218 in MPTP-treated animal suggested that this compound can regulate thalamocortical transmission.43 In these studies ML218 reversed some of the parkinsonian-induced abnormalities in ventral thalamic neuronal activity,43 specifically lowering the proportion of rebound burst and low-threshold spike (LTS) bursts, along with a reduction of pathological oscillations in the 3–30 Hz frequency range. Adding to the observation that ML218 could have antiparkinsonian properties are studies in the haloperidol–induced catalepsy model in rats in which the drug had strong anti-cataleptic properties,38 and that ML218 locally applied in the STN reduces parkinsonian motor symptoms in rodents.21 These findings provided a strong foundation for the hypothesis of potential anti-parkinsonian effects of systemic administration ML218 in MPTP-treated parkinsonian monkeys tested in the present study.
However, similar pro-kinetic properties of ML218 were not observed in our studies in MPTP-treated parkinsonian animals. At the doses tested, ML218 also did not enhance the behavioral effects of a subthreshold dose of L-DOPA. The lack of antiparkinsonian effects is likely not due to a low sensitivity of our behavioral observation methods, because the antiparkinsonian properties of low-dose L-DOPA injections were readily identified. Of course, it cannot be ruled out that ML218 produced subtler effects that were not tested in our studies, such as improvement in fine motor behavior or changes in motor learning.
It is possible that the drug concentration in the brain were not high enough to sufficiently block TTCCs. While our pharmacokinetic studies indicated that ML218 reached a maximal concentration in the CSF 1–2 hours after a 10 mg/kg administration, and remained at a stable CNS concentration for many hours after the administration, the average maximal concentration achieved in the CSF after the 10 mg/kg test dose of ML218 (30 nM) was lower than the concentrations used in in vitro electrophysiology experiments to block TTCCs.38 However, the dose of 10 mg/kg induced clear changes in the ECoG signals, suggesting that the concentration achieved in the CSF was sufficient to induce functional changes (because of the sedating qualities of the agent, it was impractical to use higher concentrations of this drug, see below).
We have previously shown that local injections of ML218 in the motor thalamus of parkinsonian monkeys reduces some of the rebound bursting associated with parkinsonism.43 Assuming that systemic injections of ML218 also resulted in a reduction of rebound firing in the motor thalamus, the lack of antiparkinsonian effects argues against a strong role of rebound bursting in the pathophysiology of parkinsonism. However, the outcome of the present study could also be explained by the unexpectedly strong sedative effects of the compound. We observed that some of the animals appeared more lethargic after the systemic injections of ML218 than after injection of vehicle. This impression was confirmed by recordings of ECoG signals that demonstrated an increased frequency of sleep periods and sleep-related ECoG rhythms. It is possible that these effects are mediated via the ML218-induced TTCC blockade. TTCCs have, in fact, been implicated in the regulation of wakefulness.44–46
Although our results suggest that the antiparkinsonian effects observed in early studies using nonspecific TTCCBs22, 23, 25, 26 could have been due, at least in part, to effects of the drugs at molecular targets other than TTCCs, they do not rule out that other types of selective TTCC blockers could be more effective at reducing parkinsonism. Alternatively, ML218 (or other TTCC blockers with sedative effects) could be tested in combination with a stimulant, such as modafinil or caffeine, to promote alertness in PD patients.47, 48
It could be argued that voltage-dependent TTCC blockers could be advantageous over ML218 to achieve more noticeable antiparkinsonian effects, because, by stabilizing the channel in an inactive state,39, 40 voltage-dependent drugs could help reduce parkinsonism-related pathological oscillations. However, at least one voltage-dependent TTCC, TTA-A2, promotes slow-wave sleep in mice,39, 40 which would limit is antiparkinsonian properties, as we observed for ML218.
ML218 blocks the three subtypes of TTCC (Cav3.1, Cav3.2 and Cav3.3).38 Given that sleep effects might be modulated by thalamocortical networks,40 while antiparkinsonian effects could be more closely related to STN oscillations,21 it would be of interest to explore TTCC blockers with enhanced effect at STN over corticothalamic neurons. This may be achieved with subtype-selective TTCC blockers. STN neurons, at least in rodents, express high levels of mRNA for Cav3.3 and Cav3.2, while thalamic relay neurons have high expressions of Cav3.1.43, 49
We conclude that ML218 does not have usable antiparkinsonian effects in MPTP-treated parkinsonian monkeys at the doses tested here. Our results suggest that further studies of antiparkinsonian potential of TTCC blockers should take into consideration possible sedative effects of these drugs.
METHODS
Animals
All experiments were conducted according to the “Guide for the Care and Use of Laboratory Animals” 50 and the United States Public Health Service Policy on the Humane Care and Use of Laboratory Animals (amended 2002). The experiments were approved by the Animal Care and Use Committee and the Biosafety Committee of Vanderbilt and Emory University. The studies were done in 7 Rhesus macaques (macaca mulatta, 6 females, 1 male, mean age at the start of experiments 12.2 years [range 6.7 – 17.5 years]). The monkeys were obtained from the Yerkes National Primate Research Center colony and had free access to food and water and received vegetables and fruits daily.
Pharmacokinetics and biochemical studies
Reagents
Solvents or reagents were of the highest purity commercially available and purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Rhesus monkey plasma and whole brain tissue were obtained from Bioreclamation (Westbury, NY). Minimum essential medium (MEM), phosphate-buffered saline (PBS) and Hank’s Balanced Salt Solution (HBSS) were obtained from VWR International (Radnor, PA). Corning plastic tubes, serological pipettes, T-25 and T-75 flasks, BD Falcon™ cell culture inserts (transparent PET membrane, 0.4 μm pore size and 0.33 cm2 surface areas), 24-well insert companion plates, and Thermo Scientific Hyclone™ fetal bovine serum (FBS) were purchased from ThermoFisher Scientific (Waltham, MA). Versene (0.48 mM, with EDTA, in PBS) was obtained from Life Technologies (ThermoFisher Scientific). ML218 utilized in the present studies was prepared and characterized (identity/purity) in the Medicinal Chemistry Laboratory of the Vanderbilt Center for Neuroscience Drug Discovery (Nashville, TN).
Pharmacokinetics of ML218
Three female monkeys (from the same cohort of animals used for the behavioral experiments) received an injection of ML218 (10 mg/kg s.c., in 10% EtOH/40% PEG/50% Saline, 10 mg/mL dosing solution). To collect blood and cerebrospinal fluid (CSF) samples, the monkeys were anesthetized with Ketamine (10 mg/kg IM) or Telazol (3–5 mg/kg, i.m.) Blood collections via the femoral vein were performed 30 minutes, 1 hour, 2 hours, 8 hours and 24 hours post administration. CSF samples were obtained via lumbar punctures, 1 hour and 2 hours after administration. The samples were collected into chilled, EDTA-fortified tubes, centrifuged for 10 minutes (3000 RCF, 4 °C), and the resulting plasma stored at −80 °C until LC/MS/MS analysis. Pharmacokinetic parameters were obtained from non-compartmental analysis (NCA, WinNonLin, v5.3, Pharsight Corp./Certara, Princeton, NJ) of individual animal concentration-time profiles following the in vivo administration of ML218 to rats and monkeys.
Liquid-Chromatography-Mass Spectrometry Bioanalysis
Plasma and CSF samples originating from in vivo studies were analyzed with electrospray ionization by an AB Sciex Q-TRAP 5500 (Foster City, CA) that was coupled to a Shimadzu LC-20AD pumps (Columbia, MD) and a Leap Technologies CTC PAL auto-sampler (Carrboro, NC). Samples containing ML218 were subjected to a gradient elution using a C18 column (3 × 50 mm, 3 μm; Fortis Technologies Ltd, Cheshire, UK) that was thermostated at 40 °C. For the HPLC, the mobile phase A was 0.1% formic acid in water (pH unadjusted), and the mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). A 30% B gradient was held for 0.2 minutes and was linearly increased to 90% B over 0.8 minutes, with an isocratic hold for 0.6 minutes, prior to transitioning to 30% B over 0.1 minutes. The column was re-equilibrated (1 minute) prior to the next sample injection. The total run time was 2.5 minutes and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500 °C and mass spectral analyses were performed using multiple reaction monitoring (MRM) of transitions specific for the test articles and metabolites, and utilizing a Turbo-IonsprayR source in positive ionization mode (5.0 kV spray voltage). All data were analyzed using AB Sciex Analyst 1.5.1 software.
Plasma Protein Binding
The extent of plasma protein binding ML218 was determined in vitro in male SD rat plasma and Rhesus monkey plasma via rapid equilibrium dialysis (RED; ThermoFisher Scientific). A 96 well plate containing plasma and an individual test article (5 μM) was vortex mixed. A portion (200 μl) of the mixture was transferred to the cis chamber of the RED insert and dialyzed against phosphate buffer (350 μl, 25 mM, pH 7.4) in the trans-chamber and incubated (37 °C) with shaking (4 hours). Following the incubation, an aliquot from each chamber was diluted (1:1; v/v) with either plasma or buffer from the cis- or trans-chamber, respectively, and transferred to a new 96 well plate. Protein precipitation of the matrices was executed by the addition of ice-cold acetonitrile containing carbamazepine (50 nM) as an internal standard for LC-MS/MS analysis. The plate was centrifuged (3000 RCF, 10 minutes) and the supernatants transferred to a new 96 well plate where they were diluted in H2O (1:1; v/v). The plate was then sealed for LC/MS/MS analysis.
Permeability and bidirectional efflux assessment
Madin-Darby Canine Kidney wild type cells (MDCK-WT) and P-glycoprotein (Pgp; MDR1) transfected cell lines were obtained from the Netherland Cancer Institute (Amsterdam, Netherlands). Cells were cultured in MEM supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 1% L-glutamine, and 1% Penicillin-Streptomycin antibioticantimycotic. Cells were incubated (37 °C, 5% CO2) in a T-75 culture flask for three to four days to reach 80–90% confluence. Cells were passaged with 0.25% trypsin-EDTA solution after washing with PBS and Versene. Transwell inserts in a 24-well apparatus were pre-incubated with culture medium overnight and subsequently seeded at a density of 160,000 cells/ml and a total volume of 0.3 ml per insert. The medium was changed every other day. All uni- and bidirectional efflux experiments were performed with cells obtained from passage numbers 4 through 18. Just prior to a bidirectional efflux assessment, culture medium was replaced with HBSS (pH 7.4) containing glucose (15 mM) and HEPES (25 mM). The transepithelial electrical resistance (TEER) values were obtained using a Millicell ESR-2 Volt-Ohm Meter from EMD Millipore (Billerica, MA). Cells were subsequently pre-incubated with fresh HBSS buffer for 1 hour (37 °C) with shaking (50 rpm). For each experiment within the 24-well plate apparatus, 0.3 and 1.0 ml of HBSS buffer was added to the apical and basolateral wells, respectively. Working solutions of test articles (2 or 5 μM in HBSS) were prepared from DMSO stock solutions (10 mM). Following 1 hour of incubation, the HBSS solution was removed and test article was added to the donor well and HBSS buffer was added to the receiver well. The transwell plates were then placed in the 37 °C shaker (50 rpm) and allowed to incubate for 2 hours. At times 0, and 2 hours, 20 and 50 μL of samples were collected from donor and receiver sides, respectively. The samples were transferred to 96-well plates. To make sample volume and matrix equal, the donor samples (20 μl) were diluted with 30 μl of HBSS. A series of quantitative standard solutions were prepared in HBSS buffer in the concentration range of 2.5 nM to 5 μM in preparation for LC/MS/MS bioanalysis. Standard solutions (50 μL) were then transferred to the 96-well plate. Lastly, the 96-well plates were fortified with 150 μl of chilled acetonitrile containing carbamazepine (50 nM) for protein and cell membrane precipitation, followed by vortexing and centrifugation (10 minutes at 4000 RCF, 20 °C). The resulting supernatant (50 μl) was transferred to a fresh 96-well plate containing 100 μl of de-ionized H2O. The plate was sealed and bioanalysis carried out with conventional ESI-LC/MS/MS. Propranolol was used as a reference standard, reflective of passive transcellular diffusion. Loperamide was used as a reference substrate for Pgp. The permeability (Equation 1) and bidirectional efflux ratio (Equation 2) of a test article was determined by the following relationships:
| (1) |
| (2) |
where Papp represents the apparent permeability of the test article across a monolayer of cells; dQ/dt or (VdC)/dt is the rate at which the test article traverses the cell monolayer; V is the sample volume and A is the exposed cell monolayer surface area; and C0 is the initial concentration fortified in the donor compartment. The efflux ratio (ER) is the ratio of Papp,BA (basolateral [B] to apical [A]) to that of the Papp,AB (apical [A] to basolateral [B]), which stand for the drug permeability from either side of the cell monolayer. The integrity of the cell monolayers was verified by TEER values and the permeability of propranolol. The function of Pgp was confirmed by measuring the ER of loperamide. In our experiments, TEER > 80 Ω·cm2 and PappAB of propranolol in the range of (2–10) × 10−6 cm/sec were acceptable for MDCK-WT and MDCK-Pgp cells. An ER of loperamide > 3 was considered as acceptance criteria for Pgp functioning in MDCK-Pgp cells
In vivo studies in primates
MPTP treatment and assessment of parkinsonism
The animals received weekly administrations of MPTP (Natland International, Morrisville, NC; 0.2–0.8 mg/kg, i. m.). The MPTP treatment was carried out for 6–13 months and the total amount of MPTP delivered ranged from 4.2–17.5 mg/kg. Each week, the degree and stability of parkinsonism was evaluated as previously described.51–53 We used a parkinsonism rating scale (rating bradykinesia, limb akinesia, posture, action tremor, finger dexterity, home cage activity, balance and freezing, each on a 0–3 scale, to a maximum of 30 points) and observations of the spontaneous motor behavior in an observation cage. The cage was equipped with 8 infrared beam/reflector pairs, mounted in two rows of 4 beam/reflector pairs, front-to-back and side-to-side of the cage. Beam crossings generated TTL pulses. The pulses were time-stamped, and the timing information collected to computer disk, thus allowing us to quantify the number of times the beams were broken as a measure of the animal’s movement. The animals were also observed via a CCTV system and their movements logged by an observer who pressed keys on a keyboard coding movements of specific body parts. The key presses from an observer can be more sensitive to detect small movements (such as finger movements or small head rotations) that may not be detectable by counting of infrared beam breaks. To be considered moderately parkinsonian, a monkey had to score >10 on the parkinsonism rating scale and show a reduction of >50% in the number of movements performed in the observation cage compared to pre-MPTP assessments, for at least 4 consecutive weeks after the last MPTP injection.
Effects of ML218 on cardiac function
To examine whether ML218 affects electrical cardiac activities, we injected ML218 (s.c., 10 and 30 mg/kg) in three monkeys. Two hours after the ML218 injection, the animals were sedated with ketamine (10 mg/kg, i. m.) and a standard three-lead ECG was obtained. The ECG records were visually inspected, and PR-, RR-, and RT-intervals were measured for statistical evaluation.
Effects of systemic injections of L-DOPA or ML218 on motor behavior in parkinsonian monkeys
For behavioral assessments we used the cage observation methods described above. For each experimental session, the monkey was transferred to the observation cage and allowed to habituate for at least 15 minutes, followed by a baseline observation period of 15 minutes. The drug was injected after gently moving the monkey to the front of the cage using a sliding back panel of the cage. Immediately after the injection the back panel was released. The animal was observed for 75 minutes after the L-DOPA administration. For ML218 experiments, the observations started one hour after the injection (to study the effects of the maximal ML218 plasma and CSF concentrations, based on the pharmacokinetic data obtained, see Results), and lasted for 75 minutes. In experiments where the combined effects of LDOPA and ML218 were evaluated, the animals were observed for a 15 minute (drug-free) baseline period, followed by administration of ML218. A second (pre-L-DOPA) baseline was obtained 45 minutes later, followed by the L-DOPA injection (fig. 3C). Subsequent to these L-DOPA injections, the animals were observed for 75 minutes.
ML218 was administered s.c. at doses of 1–30 mg/kg (1–10 mg/ml in 10% Tween80/90% saline). L-DOPA was combined in a 4:1 ratio with benserazide (Sigma-Aldrich), dissolved in saline, and injected i.m. Each drug and dose (or combination of drugs) was tested ≥ 2 times in at least 5 of the 6 animals, except for L-DOPA doses 15 and 20 mg/kg and ML218 30 mg/kg, which were tested in 4 of the 6 animals. Throughout these studies, individual animals were treated only a few times with L-DOPA, so that dyskinesias did not develop.
During each behavioral session we obtained two principal measures of the animal’s movements, the number of infrared beam breaks (“Activity Counts”) and the number of movements performed by the animal as counted by key presses by an observer (“Number of Movements”). Both measurements were normalized to the duration of the observation period, and reported as per-minute values. To take into account the day-to-day variability in behavior, the baseline data from each session was subtracted from the data obtained after the drug administration. We then used the median of all time points measured during the observation session (5 periods of 15 minutes each) for statistical comparisons. The results were compared with non-parametric Mann-Whitney tests (SPSS, IBM). Bonferroni correction for multiple comparisons was applied.
Electrocorticogram studies
Surgery, systemic injections and recording methods
Studies of simultaneously sampled video and Electrocorticogram (ECoG) signals were done to study the effects of ML218 on arousal. These experiments were carried out in one MPTP-treated, parkinsonian monkey. The monkey (male, 11 kg) was first habituated to be handled by the experimenter and to sit in a primate chair and then underwent an aseptic surgery under isoflurane anesthesia (1–3%) for placement of epidural bone screw electrodes which were embedded, along with a stainless steel head holder, into an acrylic ‘cap’. Four epidural screw electrodes (diameter: 0.25 mm, length 0.4 mm) were inserted bilaterally over the primary motor cortex (M1, two electrodes on each side) through small holes drilled in the skull. Wires from the ECoG electrodes were soldered to a 9-pin connector that was also embedded in the acrylic. The monkey received post-surgical analgesics and prophylactic antibiotics. Several months after the surgery, we carried out s.c. injections of ML218 or the corresponding vehicle (10% Tween80/90% saline). A total of 10 recording sessions were performed, 5 with ML218 and 5 with vehicle injections. The injections were done at approximately the same time of day and separated by at least 48 hours. The ECoG recordings started 15 minutes after the injection and lasted for 2 hours. During the recordings, the head of the animal was fixed. The animal’s behavior was continually monitored through a streaming video system. ECoG signals were amplified, band-pass filtered (0.1–3kHz) and sampled at 1 kHz. The video signals, synchronized to the ECoG signals were stored to computer disk with a data acquisition interface (Power 1401; CED, Cambridge, UK) and commercial software (Spike2, CED).
Scoring of state of arousal and motor behavior
The animal’s state of arousal during the ECoG sampling sessions was manually scored, using the off-line “sleep score” Spike2 script (CED) on consecutive 10 second epochs of synchronized face video and ECoG data 52. Epochs with artifacts were not considered. For each epoch, the animal was classified as “awake” if it was attentive to its surroundings with eyes open, and showed low-amplitude ECoG. “Sleep” episodes were defined as periods during which the monkey was quiet with its eyes closed while the ECoG signals exhibited high amplitude slow activity52 (fig. 6A). A “sleep ratio” was then calculated for each 2-hour recording session by dividing the number of epochs scored as representing sleep by the total number of epochs. The video footage was used to evaluate the spontaneous movement of the monkey during the ECoG sessions. An observer unaware of the experimental condition counted the number of movements (trunk, left leg, left arm, right leg, and right arm) during 5 minute epochs, out of each 15 minute time block. We calculated the mean of all 5 minute epochs during the 2 hours of observation for statistical comparison between ML218 and vehicle injection.
Spectral analysis
Spectral analyses were done off-line with custom-written MATLAB scripts (MATLAB version 7.6, The Mathworks, Natick, MA, USA). The ECoG signals (down-sampled to 500 Hz) were band-pass-filtered with a second order Butterworth filter (0.1–100Hz) and line noise artifacts were removed using a 60 Hz second order Butterworth notch filter. Power spectral density was computed using the Welch technique, with Hamming windowing, and a fast Fourier transform segment length of 256 samples with no overlap, resulting in a final spectral resolution of 1.9 Hz. The spectral power was calculated for 5 frequency bands: 0–3.9 Hz (delta band), 3.9–7.8 Hz (theta band), 7.8–11.7 Hz (alpha band), 11.7–29.3 Hz (beta band), and 29.3–99.6 Hz (gamma band). The spectral analyses were done on ‘sleep’ and ‘awake’ epochs combined (see above) and were done for the ECoG signals from the left M1 (data from the right side were similar [not shown]). Data from ML218 injection sessions were compared to data from vehicle injection sessions with a Mann-Whitney rank sum test, using a significance criterion of p < 0.05.
Termination of the experiments and post-mortem assessment of dopamine loss
Once the experiments were completed, the animals were euthanized with pentobarbital (100 mg/kg), and perfused transcardially with Ringer solution followed by a fixative solution (4% paraformaldehyde/0.1% glutaraldehyde in PBS 0.01 M, pH 7.4). The brains were then removed and cut in the coronal plane in 60 μm sections with a vibrating microtome. Sections containing the striatum were immunolabeled for tyrosine hydroxylase (TH) to confirm the dopamine loss induced by MPTP treatment. Brain sections from normal animals (available from our tissue bank) were processed in parallel and served as controls. The histological processing and analysis of the TH immunostaining was done as reported in detail before.51, 54
Acknowledgments
This project was supported through grants from NIH/NINDS (R01-NS054976 [TW] and P50-NS071669 [Udall Center grant, TW]), a grant from the Michael J. Fox foundation, and a grant from the NIH/ORIP to the Yerkes Center (P51 OD011132).
References
- 1.Galvan A, Devergnas A, Wichmann T. Alterations in neuronal activity in basal ganglia-thalamocortical circuits in the parkinsonian state. Front Neuroanat. 2015;9:5. doi: 10.3389/fnana.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol. 2006;95:2120–2133. doi: 10.1152/jn.01013.2005. [DOI] [PubMed] [Google Scholar]
- 3.Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- 4.Raz A, Vaadia E, Bergman H. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J Neurosci. 2000;20:8559–8571. doi: 10.1523/JNEUROSCI.20-22-08559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zirh TA, Lenz FA, Reich SG, Dougherty PM. Patterns of bursting occurring in thalamic cells during parkinsonian tremor. Neuroscience. 1998;83:107–121. doi: 10.1016/s0306-4522(97)00295-9. [DOI] [PubMed] [Google Scholar]
- 6.Magnin M, Morel A, Jeanmonod D. Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neuroscience. 2000;96:549–564. doi: 10.1016/s0306-4522(99)00583-7. [DOI] [PubMed] [Google Scholar]
- 7.Filion M. Effects of interruption of the nigrostriatal pathway and of dopaminergic agents on the spontaneous activity of globus pallidus neurons in the awake monkey. Brain Res. 1979;178:425–441. doi: 10.1016/0006-8993(79)90704-2. [DOI] [PubMed] [Google Scholar]
- 8.Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp Brain Res. 1999;125:397–409. doi: 10.1007/s002210050696. [DOI] [PubMed] [Google Scholar]
- 9.Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. Role of external pallidal segment in primate parkinsonism: comparison of the effects of MPTPinduced parkinsonism and lesions of the external pallidal segment. J Neurosci. 2004;24:6417–6426. doi: 10.1523/JNEUROSCI.0836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guehl D, Pessiglione M, Francois C, Yelnik J, Hirsch EC, Feger J, Tremblay L. Tremor-related activity of neurons in the ‘motor’ thalamus: changes in firing rate and pattern in the MPTP vervet model of parkinsonism. Eur J Neurosci. 2003;17:2388–2400. doi: 10.1046/j.1460-9568.2003.02685.x. [DOI] [PubMed] [Google Scholar]
- 11.Breit S, Bouali-Benazzouz R, Popa RC, Gasser T, Benabid AL, Benazzouz A. Effects of 6-hydroxydopamine-induced severe or partial lesion of the nigrostriatal pathway on the neuronal activity of pallido-subthalamic network in the rat. Exp Neurol. 2007;205:36–47. doi: 10.1016/j.expneurol.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, Hirsch EC. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur J Neurosci. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- 13.Xu W, Zhang J, Russo GS, Hashimoto T, Vitek JL. STN DBS differentially modulates neuronal activity in the pallidal and cerebellar receiving areas of the motor thalamus. Mov Disord. 2008;23:S298–S298. [Google Scholar]
- 14.Shi LH, Luo F, Woodward DJ, Chang JY. Basal ganglia neural responses during behaviorally effective deep brain stimulation of the subthalamic nucleus in rats performing a treadmill locomotion test. Synapse. 2006;59:445–457. doi: 10.1002/syn.20261. [DOI] [PubMed] [Google Scholar]
- 15.Filion M, Tremblay L, Bedard PJ. Effects of dopamine agonists on the spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:152–161. [PubMed] [Google Scholar]
- 16.von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- 17.Destexhe A, Neubig M, Ulrich D, Huguenard J. Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci. 1998;18:3574–3588. doi: 10.1523/JNEUROSCI.18-10-03574.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jahnsen H, Llinas R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol. 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman SM, Koch C. The control of retinogeniculate transmission in the mammalian lateral geniculate nucleus. Exp Brain Res. 1986;63:1–20. doi: 10.1007/BF00235642. [DOI] [PubMed] [Google Scholar]
- 20.Bevan MD, Wilson CJ, Bolam JP, Magill PJ. Equilibrium potential of GABA(A) current and implications for rebound burst firing in rat subthalamic neurons in vitro. J Neurophysiol. 2000;83:3169–3172. doi: 10.1152/jn.2000.83.5.3169. [DOI] [PubMed] [Google Scholar]
- 21.Tai CH, Yang YC, Pan MK, Huang CS, Kuo CC. Modulation of subthalamic T-type Ca(2+) channels remedies locomotor deficits in a rat model of Parkinson disease. J Clin Invest. 2011;121:3289–3305. doi: 10.1172/JCI46482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buzsaki G, Smith A, Berger S, Fisher LJ, Gage FH. Petit mal epilepsy and parkinsonian tremor: hypothesis of a common pacemaker. Neuroscience. 1990;36:1–14. doi: 10.1016/0306-4522(90)90345-5. [DOI] [PubMed] [Google Scholar]
- 23.Gomez-Mancilla B, Latulippe JF, Boucher R, Bedard PJ. Effect of ethosuximide on rest tremor in the MPTP monkey model. Mov Disord. 1992;7:137–141. doi: 10.1002/mds.870070207. [DOI] [PubMed] [Google Scholar]
- 24.Pourcher E, Gomez-Mancilla B, Bedard PJ. Ethosuximide and tremor in Parkinson’s disease: a pilot study. Mov Disord. 1992;7:132–136. doi: 10.1002/mds.870070206. [DOI] [PubMed] [Google Scholar]
- 25.Murata M, Hasegawa K, Kanazawa I. Zonisamide improves motor function in Parkinson disease: a randomized, double-blind study. Neurology. 2007;68:45–50. doi: 10.1212/01.wnl.0000250236.75053.16. [DOI] [PubMed] [Google Scholar]
- 26.Bermejo PE. Zonisamide in patients with essential tremor and Parkinson’s disease. Mov Disord. 2007;22:2137–2138. doi: 10.1002/mds.21717. [DOI] [PubMed] [Google Scholar]
- 27.Lory P, Chemin J. Towards the discovery of novel T-type calcium channel blockers. Expert Opin Ther Targets. 2007;11:717–722. doi: 10.1517/14728222.11.5.717. [DOI] [PubMed] [Google Scholar]
- 28.Greenhill SD, Morgan NH, Massey PV, Woodhall GL, Jones RS. Ethosuximide modifies network excitability in the rat entorhinal cortex via an increase in GABA release. Neuropharmacology. 2012;62:807–814. doi: 10.1016/j.neuropharm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Leresche N, Parri HR, Erdemli G, Guyon A, Turner JP, Williams SR, Asprodini E, Crunelli V. On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. J Neurosci. 1998;18:4842–4853. doi: 10.1523/JNEUROSCI.18-13-04842.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi T, Hirai H, Iino M, Fuse I, Mitsumura K, Washiyama K, Kasai S, Ikeda K. Inhibitory effects of the antiepileptic drug ethosuximide on G protein-activated inwardly rectifying K+ channels. Neuropharmacology. 2009;56:499–506. doi: 10.1016/j.neuropharm.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Leppik IE. Zonisamide: chemistry, mechanism of action, and pharmacokinetics. Seizure. 2004;13(Suppl 1):S5–9. doi: 10.1016/j.seizure.2004.04.016. discussion S10. [DOI] [PubMed] [Google Scholar]
- 32.Mimaki T, Suzuki Y, Tagawa T, Karasawa T, Yabuuchi H. Interaction of zonisamide with benzodiazepine and GABA receptors in rat brain. Med J Osaka Univ. 1990;39:13–17. [PubMed] [Google Scholar]
- 33.Ueda Y, Doi T, Tokumaru J, Willmore LJ. Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Mol Brain Res. 2003;116:1–6. doi: 10.1016/s0169-328x(03)00183-9. [DOI] [PubMed] [Google Scholar]
- 34.Gluck MR, Santana LA, Granson H, Yahr MD. Novel dopamine releasing response of an anti-convulsant agent with possible anti-Parkinson’s activity. J Neural Transm. 2004;111:713–724. doi: 10.1007/s00702-004-0107-1. [DOI] [PubMed] [Google Scholar]
- 35.Yamamura S, Ohoyama K, Nagase H, Okada M. Zonisamide enhances delta receptor-associated neurotransmitter release in striato-pallidal pathway. Neuropharmacology. 2009;57:322–331. doi: 10.1016/j.neuropharm.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, Fox SV, Kraus RL, Marino MJ, Graufelds VK, Vargas HM, Bunting PB, Hasbun-Manning M, Evans RM, Koblan KS, Renger JJ. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem. 2008;51:3692–3695. doi: 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- 37.Yang ZQ, Barrow JC, Shipe WD, Schlegel KA, Shu Y, Yang FV, Lindsley CW, Rittle KE, Bock MG, Hartman GD, Uebele VN, Nuss CE, Fox SV, Kraus RL, Doran SM, Connolly TM, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Marino MJ, Graufelds VK, DiLella AG, Reynolds IJ, Vargas HM, Bunting PB, Woltmann RF, Magee MM, Koblan KS, Renger JJ. Discovery of 1,4-substituted piperidines as potent and selective inhibitors of T-type calcium channels. J Med Chem. 2008;51:6471–6477. doi: 10.1021/jm800830n. [DOI] [PubMed] [Google Scholar]
- 38.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Hopkins CR, Xie XS, Conn PJ, Weaver CD, Lindsley CW. The discovery and characterization of ML218: a novel, centrally active T-type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson’s disease. ACS Chem Neurosci. 2011;2:730–742. doi: 10.1021/cn200090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uebele VN, Gotter AL, Nuss CE, Kraus RL, Doran SM, Garson SL, Reiss DR, Li Y, Barrow JC, Reger TS, Yang ZQ, Ballard JE, Tang C, Metzger JM, Wang SP, Koblan KS, Renger JJ. Antagonism of T-type calcium channels inhibits high-fat dietinduced weight gain in mice. J Clin Invest. 2009;119:1659–1667. doi: 10.1172/JCI36954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraus RL, Li Y, Gregan Y, Gotter AL, Uebele VN, Fox SV, Doran SM, Barrow JC, Yang ZQ, Reger TS, Koblan KS, Renger JJ. In vitro characterization of T-type calcium channel antagonist TTA-A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther. 2010;335:409–417. doi: 10.1124/jpet.110.171058. [DOI] [PubMed] [Google Scholar]
- 41.Bridges TM, Morrison RD, Byers FW, Luo S, Scott Daniels J. Use of a novel rapid and resource-efficient cassette dosing approach to determine the pharmacokinetics and CNS distribution of small molecule 7-transmembrane receptor allosteric modulators in rat. Pharmacol Res Perspect. 2014;2 doi: 10.1002/prp2.77. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 43.Devergnas A, Chen E, Ma Y, Hamada I, Pittard D, Kammermeier S, Mullin AP, Faundez V, Lindsley CW, Jones C, Smith Y, Wichmann T. Anatomical localization of Cav3.1 calcium channels and electrophysiological effects of T-type calcium channel blockade in the motor thalamus of MPTP-treated monkeys. J Neurophysiol. 2016;115:470–485. doi: 10.1152/jn.00858.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron. 2002;33:947–958. doi: 10.1016/s0896-6273(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 45.Crunelli V, David F, Leresche N, Lambert RC. Role for T-type Ca2+ channels in sleep waves. Pflugers Arch. 2014;466:735–745. doi: 10.1007/s00424-014-1477-3. [DOI] [PubMed] [Google Scholar]
- 46.Shin HS, Cheong EJ, Choi S, Lee J, Na HS. T-type Ca2+ channels as therapeutic targets in the nervous system. Curr Opin Pharmacol. 2008;8:33–41. doi: 10.1016/j.coph.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Rodrigues TM, Castro Caldas A, Ferreira JJ. Pharmacological interventions for daytime sleepiness and sleep disorders in Parkinson’s disease: Systematic review and meta-analysis. Parkinsonism Relat Disord. 2016;27:25–34. doi: 10.1016/j.parkreldis.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Postuma RB, Lang AE, Munhoz RP, Charland K, Pelletier A, Moscovich M, Filla L, Zanatta D, Rios Romenets S, Altman R, Chuang R, Shah B. Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology. 2012;79:651–658. doi: 10.1212/WNL.0b013e318263570d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garber JC, Barbee RW, Bielitzki JT, Clayton LA, Donovan JC, Hendriksen CFM, Kohn DF, Lipman NS, Locke PA, Melcher J, Quimby FW, Turner PV, Wood GA, Wurbel H. Guide for the Care and Use of Laboratory Animals. 8. The National Academies Press; Washington, D.C: 2010. [Google Scholar]
- 51.Galvan A, Hu X, Rommelfanger KS, Pare JF, Khan ZU, Smith Y, Wichmann T. Localization and function of dopamine receptors in the subthalamic nucleus of normal and parkinsonian monkeys. J Neurophysiol. 2014;112:467–479. doi: 10.1152/jn.00849.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devergnas A, Pittard D, Bliwise D, Wichmann T. Relationship between oscillatory activity in the cortico-basal ganglia network and parkinsonism in MPTP-treated monkeys. Neurobiol Dis. 2014;68:156–166. doi: 10.1016/j.nbd.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wichmann T, Kliem MA, DeLong MR. Antiparkinsonian and behavioral effects of inactivation of the substantia nigra pars reticulata in hemiparkinsonian primates. Exp Neurol. 2001;167:410–424. doi: 10.1006/exnr.2000.7572. [DOI] [PubMed] [Google Scholar]
- 54.Galvan A, Hu X, Smith Y, Wichmann T. Localization and function of GABA transporters in the globus pallidus of parkinsonian monkeys. Exp Neurol. 2010;223:505–515. doi: 10.1016/j.expneurol.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]