

Abstract
Accurate identification of low-frequency somatic point mutations in tumor samples has important clinical utilities. Although high-throughput sequencing technology enables capturing such variants while sequencing primary tumor samples, our ability for accurate detection is compromised when the variant frequency is close to the sequencer error rate. Most current experimental and bioinformatic strategies target mutations with ≥5% allele frequency, which limits our ability to understand the cancer etiology and tumor evolution. We present an experimental and computational modeling framework, RareVar, to reliably identify low-frequency single-nucleotide variants from high-throughput sequencing data under standard experimental protocols. RareVar protocol includes a benchmark design by pooling DNAs from already sequenced individuals at various concentrations to target variants at desired frequencies, 0.5%–3% in our case. By applying a generalized, linear model-based, position-specific error model, followed by machine-learning-based variant calibration, our approach outperforms existing methods. Our method can be applied on most capture and sequencing platforms without modifying the experimental protocol.
Keywords: : low frequency SNVs, machine learning, next-generation sequencing, sequencing error modeling, somatic mutation
1. Introduction
The advance of high-throughput sequencing technologies has revolutionized our capability to detect somatic mutations in primary tumor samples. However, current experimental assays and analysis methods are not sensitive enough to accurately detect somatic mutations with allele frequencies below 3%. Such low-frequency mutations commonly exist in tumor samples due to both tumor heterogeneity (Nik-Zainal et al., 2012; Walter et al., 2012) and contamination with normal tissues (Carter et al., 2012). Accurate identification of low-frequency somatic mutations carries significant clinical implications due to their function in tumor growth, drug resistance, and metastasis (Inda et al., 2010; Ding et al., 2012). In addition, circulating tumor DNAs (ctDNAs) in early detection of cancer, prognostic assessment, and detection of relapse or acquired resistance also require accurate detection of low-frequency variants (Forshew et al., 2012; Crowley et al., 2013; Bettegowda et al., 2014) since ctDNAs only compose as much as 1%–10% for patients with high-grade cancers (Diehl et al., 2005).
The sequencing artifacts can be introduced during the many stages of the experiment (Metzker, 2010), including DNA capture and amplification, library construction, sequencing, and data analysis (O'Rawe et al., 2013). Thus, identification of single-nucleotide variants (SNVs) with allele frequencies close to experimental artifact rates (usually 0.1%–1% for most of the platforms) is extremely challenging. Many bioinformatic methods have been developed to tackle this challenge. Among existing methods, VarScan2 (Koboldt et al., 2012), Strelka (Saunders et al., 2012), and mutect (Cibulskis et al., 2013) are mainly designed to target variants with lowest allele frequencies at 5% for a whole exome or several hundreds of targeted genes sequencing with average depth around hundreds. Several other studies focus on a small number of hotspot cancer genes with ultradeep sequencing (greater than 10,000 × in depth) (Harismendy et al., 2011; Wilm et al., 2012) for pushing down the detection limit. However, such methods usually take an ad hoc filtering approach and are designed to target variant identification within a small genomic region, usually less than 20,000 nucleotides. In addition, the above existing methods failed to consider differential error profiles at different genomic sequence contexts across the targeted regions and thus are suboptimal in sensitively detecting variants with allele frequencies close to the intrinsic sequencing error rate.
To effectively boost sensitivity at or lower than 1% allele frequencies without resorting to approximations or heuristics, deepSNV (Gerstung, 2012) considered the strand-specific error rate and LoFreq (Wilm et al., 2012) modeled the position-specific error rate from base or sequencing qualities. However, deepSNV used customized, high-accuracy deep-sequencing protocols not available to many laboratories. In addition, these two methods only considered a limited number of sequence contexts and their performances on sequencing platforms with potential sequence context biases were unknown.
In this study, we present a novel experimental and computational modeling framework, RareVar, which aims to push the detection limit to 0.5%–1% under standard sequencing experiment protocols. The experimental part includes a strategy to construct a benchmark sample mimicking tumor samples enriched with low-frequency variants (0.5%–3%). The computational part utilized the constructed benchmark sample to derive a genomic, position-specific error rate for sensitive low-frequency variant detection and to further refine the candidates with machine-learning models. We evaluated the performance of RareVar together with representative existing tools on an independent constructed testing benchmark. RareVar is shown to be more sensitive and also accurate than the other tools, especially for variants with less than 3% allele frequencies.
2. Methods
2.1. Overall framework
The RareVar protocol includes five major components: benchmark sample design, target region amplification and sequencing, position-specific error modeling (PSEM), variant identification, and machine-learning-based variant calibration (Fig. 1). A training benchmark sample was designed to contain a set of mutations at known allele frequencies in the desired capture regions. This benchmark was sequenced in parallel with the tumor/cell samples of interest using the exact same capturing and sequencing protocol. Thus, it serves as a calibration set to evaluate the accuracy of the sequencing and analysis pipeline. The invariant loci in the benchmark sample provide data for PSEM on genomic features that distinguish low-frequency SNVs from sequencing errors, while the known SNVs allow further calibration of the variant calls based on features from the particular experimental procedures.
FIG. 1.

RareVar framework overview. During the training phase, genome contexts of invariant loci are used to train a position-specific error model (PSEM). Then, the genome contexts of all loci are fed to PSEM and the resultant predictions comprise the candidate SNV loci. Sequencing qualities of those candidates are used to further calibrate their fidelity. Actual data involved in model training are highlighted in dashed lines and boxes. During the testing phase, a matched normal–tumor pair goes through the trained PSEM and recalibration model to generate high-confidence somatic SNVs. SNV, single-nucleotide variant.
2.2. Capture assay and sequencing platform
The exonic regions of 409 known cancer genes, totaling ∼1.7 million bases in 16,000 amplicons, were captured using the Ion AmpliSeq™ Comprehensive Cancer Panel. Ion Proton™ system was used to generate sequencing data. TMAP from Torrent Suite™ Software, version 4.4.2, was applied to align the sequencing reads. Only uniquely mappable reads were used in further analysis. The aligned reads with mapping quality less than 40 were filtered out.
2.3. Benchmark design
A total of 22 DNA samples from the 1000 Genomes Project for which there are genotype data (1000 Genomes Project Consortium et al., 2012) were selected and pooled as described. Two sets of 18 samples were used, one for the training benchmark set and the other for the testing benchmark set (Supplementary Table S1).
The goal of the training benchmark design was to maximize the number of low-frequency (0.5%–3%) SNVs in the exonic target regions. First, among each set of 18 DNA samples, we identified the one that has the largest number of overlapping SNVs with other samples in the target regions. The SNVs from this sample were used to represent the normal cell population. Second, the other 17 samples were mixed at varying concentrations (1%–10%); samples with larger number of unique SNVs in the target regions were assigned lower concentrations. Similarly, the testing benchmark sample was designed by mixing another set of 18 DNA samples in which 4 samples were not in the training benchmark and 3 samples with higher number of unique variants were assigned lowest concentrations serving as low-frequency testing set totally independent of the training set. For DNA samples that appeared in both training and testing, smaller concentrations were preferably assigned to samples with higher number of unique variants and also tried to assign different values from the training. The DNA mixing strategy for both training and test benchmark tumor samples is shown in Supplementary Table S1. To demonstrate the performance on normal–tumor pair design, the sample representing normal cell populations was also sequenced.
2.4. Position-specific error model
A Poisson distribution generalized linear model (PD-GLM) (Hao et al., 2016) was applied to model the relationship between sequence contexts and error rates. PD-GLM integrated nine genome context features related to sequencing errors (Bragg et al., 2013; Ross et al., 2013), including alternative base substitution types, the nucleotides immediate upstream and downstream of the variant loci, and the percentage of guanine-cytosine (GC) nucleotides in the nearby region. In addition, homopolymer features were also considered since sequencing data tend to be erroneous if the locus is near the boundary of one or more long homopolymer(s).
2.5. Variant identification
For each targeted locus, a Bayes factor was calculated by comparing the likelihood of two competing models, ME—the number of alternative reads follows sequencing error distribution—estimated by PD-GLM, and MV, the number follows the lowest intended identifiable frequency distribution. In this study, intended frequency is f = 0.5%. In Equation (1), 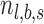 is the number of reads in location
is the number of reads in location 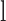 with nonreference base
with nonreference base 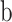 on strand
on strand 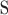 (forward or reverse) and
(forward or reverse) and 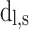 is the depth at location
is the depth at location 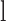 on strand
on strand 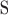 . In addition,
. In addition, 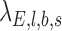 and
and 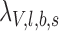 represent the expected number of alternative reads assuming the candidate locus is not an SNV (
represent the expected number of alternative reads assuming the candidate locus is not an SNV (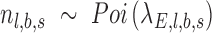 ) and is an SNV with the lowest intended identifiable allele frequency (
) and is an SNV with the lowest intended identifiable allele frequency (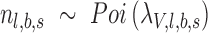 ), respectively. An observed substitution in a location is considered an SNV candidate if Bayesian factors
), respectively. An observed substitution in a location is considered an SNV candidate if Bayesian factors 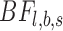 for both strands are greater than
for both strands are greater than 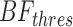 .
.
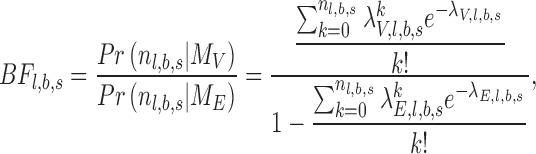 |
 |
2.6. Machine learning-based SNV calibration
SNV candidates from the variant identification step still contain a large number of false positives. Sequencing-related measurements, such as sequencing and alignment quality, have a strong influence on the accuracy of variant identity (DePristo et al., 2011). Instead of setting up a series of heuristic filters, we adopted a machine-learning-based approach to derive an optimal classification boundary between false and true positives by simultaneously modeling multiple measurements. The PSEM model focused on the sequence contexts, whereas measurements related to the experimental and analytical steps are considered here. The features included in the machine-learning model can be grouped into the following generic types: sequencing, alignment, amplicon structure, and genome context-related features from PSEM. Information gain (IG) was used to rank the classification power of all features (Supplementary Table S2).
Machine learning algorithm, random forest (Breiman, 2001), was employed to incorporate all features to best distinguish false positives from true positives. The random forest algorithm is employed with 100 trees, maximum 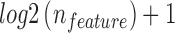 features (
features (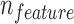 is the total number of features) to consider in each tree, and no limitation on depth of the trees.
is the total number of features) to consider in each tree, and no limitation on depth of the trees.
2.7. Performance evaluation
Exonic loci with at least five reads supporting an alternative allele are included in the evaluation. Precision, recall, and F1 score are defined in Equations (2)–(4). The allele frequency ranges were determined by observed values for precision and expected values from test benchmark for recall.
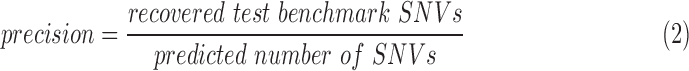 |
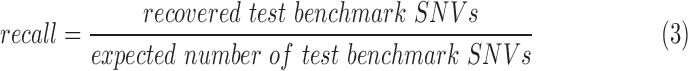 |
 |
2.8. Parameter customization for existing tools
The complete list of parameters is in Supplementary Table S3.
Torrent variant caller (TVC): change the default settings (snp-min-allele-freq, gen-min-alt-allele-freq, and downsample) to allow calling SNVs with down to 0.5% frequency. There is no option for turning off downsample, thus the maximum depth (34,223) in test benchmark data was used.
Strelka: parameter file for bwa aligner was used. Depth filter (isSkipDepthFilters) was turned off. Since low recall was observed for ≤3% SNVs in test runs, combinations of ssnvPrior and ssnvNoise were tested. The conclusion from this combinatory exploration suggested that elevated ssnvNoise decreases precision and recall, while elevated ssnvPrior increases recall with a slight drop in precision. The 1000 × bigger ssnvPrior results in ∼3% increase in recall and ∼1% drop in precision and since the extent of change is small, no further increase was attempted.
VarScan2: min-var-freq was set to 0.5% and min-reads2 was set to 5 to be consistent with RareVar. Since the percentage of DNA from test benchmark normal sample individual was 0.46, tumor-purity was set to 0.54.
deepSNV: desired significance level sig.level was set to 0.5 to be more sensitive. Multiple testing correction method, Benjamini–Hochberg procedure, was applied.
LoFreq: minimal coverage for somatic calls min-cov was set to 5. fdr was used for multiple testing correction. Parameter no-srt-qual was used to disable use of source quality in tumor.
3. Results
3.1. Benchmark data evaluation
Potential SNV allele frequency bias introduced in the pooling step was evaluated by the correlation of the detected median allele frequencies of SNVs unique to each individual with their designed frequencies. The log scale linear regression analysis showed that individuals with smaller assigned percentages tend to have slightly lower observed percentages, with R2 > 0.98 for both training and testing benchmarks (Fig. 2).
FIG. 2.

Evaluation of mixing variance in construction of the training and testing benchmarks. Numbers next to the dots represent the mixing frequencies of DNA samples; the line at 45° represents perfect pipetting (observed frequency exactly equals the expected).
The designed benchmark sets derived 1461 and 1557 SNVs for the training and testing datasets, respectively (Table 1). The design enables evaluation of SNVs with a broad range of allele frequencies and is also enriched in 0.5%–3% allele frequencies. As shown in Table 1, the percentage of SNVs with allele frequency no more than 3% is 64% in training benchmark and 68.9% in testing benchmark. Independent testing SNVs from individuals only in testing benchmark composed 38.6% of all testing benchmarks, in which >60% SNVs were with allele frequencies ≤3%.
Table 1.
Distribution of Single-Nucleotide Variants Across Designed Allele Frequency Ranges
| Training benchmark | Testing benchmark | |||||
|---|---|---|---|---|---|---|
| Designed AF (%) | Full training | Cumulative percentage | Full testing | Cumulative percentage | New testing | Cumulative percentage |
| 0.50 | 304 | 20.8 | 270 | 17.3 | 152 | 25.3 |
| 1 | 271 | 39.4 | 309 | 37.2 | 43 | 32.4 |
| 1.5–3 | 360 | 64.0 | 493 | 68.9 | 168 | 60.4 |
| 3.5–5 | 162 | 75.1 | 164 | 79.4 | 45 | 67.9 |
| 5.5–10 | 213 | 89.7 | 151 | 89.1 | 69 | 79.4 |
| 10.5–53 | 151 | 100.0 | 170 | 100.0 | 124 | 100.0 |
| All | 1461 | 100.0 | 1557 | 100.0 | 601 | 100.0 |
3.2. Position-specific error model
To evaluate the efficacy of the PSEM in identifying SNV candidates, we compared the performance of PSEM at 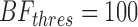 with Fisher's exact test-based VarScan2 (Koboldt et al., 2012), which tends to have higher recall. On both full and new testing sets, PSEM showed much higher overall recall and slightly lower precision (Table 2).
with Fisher's exact test-based VarScan2 (Koboldt et al., 2012), which tends to have higher recall. On both full and new testing sets, PSEM showed much higher overall recall and slightly lower precision (Table 2).
Table 2.
Overall Performances in Testing Benchmark by Methods
| All testing | New testing | |||||
|---|---|---|---|---|---|---|
| Methods | Recall | Precision | F1 score | Recall | Precision | F1 score |
| PSEM | 0.958 | 0.492 | 0.650 | 0.970 | 0.274 | 0.428 |
| VarScan2 | 0.830 | 0.533 | 0.649 | 0.812 | 0.301 | 0.439 |
| RareVar | 0.819 | 0.955 | 0.882 | 0.785 | 0.887 | 0.833 |
| Strelka | 0.360 | 0.899 | 0.514 | 0.429 | 0.804 | 0.560 |
| TVC | 0.638 | 0.934 | 0.759 | 0.644 | 0.843 | 0.730 |
| deepSNV | 0.165 | 0.959 | 0.282 | 0.235 | 0.928 | 0.375 |
| LoFreq | 0.528 | 0.956 | 0.680 | 0.567 | 0.900 | 0.696 |
3.3. Machine learning-based SNV calibration
Classification power of 29 features used in machine learning was ranked by IG. Sequencing-related features and alignment quality features ranked the highest, while sequence context feature—GC content—was removed from further analysis due to 0 IG (Supplementary Table S2).
The calibration effectively reduced false-positive SNVs identified by the PSEM. On the full testing benchmark dataset, overall precision increased from 0.492 to 0.955 after calibration, while on the new testing set, overall precision increased from 0.301 to 0.887 (Table 3).
Table 3.
Comparison of Precision and Recall Between Position-Specific Error Model (PSEM) and RareVar on Full Testing Set by Allele Frequencies
| (a) Precision comparison | ||||||
|---|---|---|---|---|---|---|
| PSEM | RareVar | |||||
| Observed frequency (%) | Predicted no. of SNVs | Recovered no. of SNVs | Precision | Predicted no. of SNVs | Recovered no. of SNVs | Precision |
| 0.25–0.75 | 1369 | 286 | 0.209 | 140 | 127 | 0.907 |
| 0.75–1.25 | 479 | 298 | 0.622 | 271 | 264 | 0.974 |
| 1.25–3 | 571 | 422 | 0.739 | 417 | 403 | 0.966 |
| 3–5 | 217 | 168 | 0.774 | 172 | 165 | 0.959 |
| 5–10 | 207 | 159 | 0.768 | 169 | 159 | 0.941 |
| 10–54 | 190 | 158 | 0.832 | 166 | 157 | 0.946 |
| All | 3033 | 1491 | 0.492 | 1335 | 1275 | 0.955 |
| (b) Recall comparison | ||||||
|---|---|---|---|---|---|---|
| PSEM | RareVar | |||||
| Expected frequency (%) | Expected no. of SNVs | Recovered no. of SNVs | Recall | Expected no. of SNVs | Recovered no. of SNVs | Recall |
| 0.5 | 270 | 230 | 0.852 | 270 | 96 | 0.356 |
| 1 | 309 | 296 | 0.958 | 309 | 251 | 0.812 |
| 1.5–3 | 493 | 483 | 0.980 | 493 | 458 | 0.929 |
| 3.5–5 | 164 | 162 | 0.988 | 164 | 156 | 0.951 |
| 5.5–10 | 151 | 150 | 0.993 | 151 | 149 | 0.987 |
| 10.5–54 | 170 | 170 | 1.000 | 170 | 165 | 0.971 |
| All | 1557 | 1491 | 0.958 | 1557 | 1275 | 0.819 |
3.3.1. Performance by allele frequencies
A closer examination of the RareVar performance by allele frequencies in full testing set showed that precision increased for all allele frequencies by at least 0.1 after calibration, with greater than 0.9 precision achieved for SNVs in all allele frequency ranges (Table 3). Lower frequencies showed higher increase, in which 0.70 and 0.35 increases in precision were achieved for 0.5% and 1%, resulting in >0.9 precision for both. The decrease in recall was mainly attributed to 0.5% and 1%, yet >0.8 recall was maintained for allele frequencies ≥1%. The ROC curve on different allele frequency ranges (Fig. 3a) showed that the model reaches relatively stable performance for >1% frequency. On the new testing set, the recall values are similar to the ones on the full set across all allele frequencies, while the precision values dropped noticeably (Table 4). Except for the drop caused by overfitting, part of the drop is due to the inflated false positives originated from other individuals in the full set and thus artificially lowered the precision.
FIG. 3.

Performances by allele frequencies and effect of depth on recall on full testing benchmark. (a) The precision and recall are evaluated at classification probabilities from 0 to 0.95 in steps of 0.05. Probabilities of 0.50 points are highlighted. (b) The depth is sampled from 10% to 100% of the original in steps of 5%.
Table 4.
RareVar Performance Comparison Between Full Testing and New Testing Sets Across All Frequency Ranges
| (a) Recall comparison | ||||||
|---|---|---|---|---|---|---|
| Full testing | New testing | |||||
| Expected frequency (%) | Expected no. of SNVs | Recovered no. of SNVs | Recall | Expected no. of SNVs | Recovered no. of SNVs | Recall |
| 0.5 | 270 | 96 | 0.356 | 152 | 55 | 0.362 |
| 1 | 309 | 251 | 0.812 | 43 | 34 | 0.791 |
| 1.5–3 | 493 | 458 | 0.929 | 168 | 154 | 0.917 |
| 3.5–5 | 164 | 156 | 0.951 | 45 | 40 | 0.889 |
| 5.5–10 | 151 | 149 | 0.987 | 69 | 68 | 0.986 |
| 10.5–54 | 170 | 165 | 0.971 | 124 | 121 | 0.976 |
| All | 1557 | 1275 | 0.819 | 601 | 472 | 0.785 |
| (b) Precision comparison | ||||||
|---|---|---|---|---|---|---|
| Full testing | New testing | |||||
| Observed frequency (%) | Predicted no. of SNVs | Recovered no. of SNVs | Precision | Predicted no. of SNVs | Recovered no. of SNVs | Precision |
| 0.25–0.75 | 140 | 127 | 0.907 | 57 | 44 | 0.772 |
| 0.75–1.25 | 271 | 264 | 0.974 | 83 | 76 | 0.916 |
| 1.25–3 | 417 | 403 | 0.966 | 137 | 123 | 0.898 |
| 3–5 | 172 | 165 | 0.959 | 53 | 46 | 0.868 |
| 510 | 169 | 159 | 0.941 | 78 | 68 | 0.872 |
| 1054 | 166 | 157 | 0.946 | 124 | 115 | 0.927 |
| All | 1335 | 1275 | 0.955 | 532 | 472 | 0.887 |
3.3.2 Depth effect on performance by allele frequencies
The average depth for the testing dataset was 3973. To evaluate its influence on performances, we gradually downsampled the testing benchmark sequencing data by randomly selecting a fixed percentage of reads from the original data. The precision is not affected, but the recall steadily decreases with reducing average depths for all frequencies (Fig. 3b). The decreasing trend is more severe when the depth is less than 1000 × . This result suggests that depth should be predetermined when detecting SNVs at a specific frequency range. In addition, for variants >0.5%, the recall curve reaches a plateau when average depth is greater than 2000 × . Thus, further increasing the sequencing depth will not improve the detection sensitivity.
3.3. Performance comparison with existing methods
We compared RareVar with established variant detection tools, including VarScan2, TVC from Torrent Suite™ software, Strelka, deepSNV, and LoFreq. The default settings for these tools aim at SNVs with higher frequencies, thus customized parameters were selected by optimizing for rare variants (Supplementary Table S3). On both full and new testing sets, RareVar was the best method in overall performance by F1 score (Table 2), with the advantage over the second and third best methods, TVC and LoFreq, most evident for 1% and 0.5% frequencies (Fig. 4). At 1%, precision is similar for these three methods, while RareVar achieved 0.812 recall, compared with <0.4 for the other 2. Even at 0.5% allele frequency, RareVar maintained 0.907 precision and 0.356 recall. It is expected that TVC also demonstrates good performance for >1% since it is specifically designed for Ion Proton™ technology. Overall, RareVar shows the best performance among all the tools tested, in particular for the SNVs with low allele frequencies (0.5%–3%). However, all methods, RareVar included, showed decreased recall, which is partially due to inflated false positives.
FIG. 4.

Precision and recall at ≤3% allele frequencies. Benchmark performance-optimized parameters were applied for VarScan2, TVC, LoFreq, deepSNV, and Strelka. Upper panels show the performance evaluated on full testing set, while lower panels show the performances evaluated on new testing variants not used in training.
4. Discussion
The framework of RareVar provides guidance for low-frequency SNV identification from both experimental and algorithmic aspects. Many components in the next-generation sequencing pipeline, including library preparation, target enrichment assay, and sequencing technology, affect the sensitivity and fidelity of SNV detection. The comparison of RareVar with other algorithms underscores the necessity of modeling frequency detection limits and the significance of a model tailored for each technology. It is impractical to have a universal parameter or threshold setting scheme that fits all sequencing platforms and experimental protocols. To solve this problem, we construct a benchmark sample containing variants with desired allele frequencies. The distribution of nucleotide mismatch patterns around the positive and negative variant loci in the benchmark sample provides a valuable guideline for optimizing the parameter and threshold settings during the variant identification process. In addition, the benchmark sample also enables fair evaluation on the performance of the detection.
The two-stage computational modeling, position-specific error model (PSEM) and machine-learning-based calibration, was designed to take the advantages of sequencing signals on the invariant loci and designed variant loci, respectively. The PSEM step intends to model how genomic sequence contexts impact the sequencing error profiles that are associated with the experimental protocol. The derived model serves as an important base for accurately estimating background error signal that is specific to any particular nucleotide position. This step is critical in improving the detection accuracy of SNVs with extreme low frequency, as opposed to using a universal background error rate for all the genomic loci.
The machine-learning-based variant recalibration considered experiment-related features, such as strand bias and mapping quality. This design effectively avoids using a series of filters that often involves multiple ad hoc thresholds. We demonstrated that the variant calibration step significantly increased the specificity of variant identification and further improved overall accuracy.
Supplementary Material
Acknowledgments
This study is supported by funds from the US National Institutes of Health U10AA008401 (Collaborative Study on the Genetics of Alcoholism to H.J.E.), Pilot funds from the breast cancer program of IUSCC (Indiana University Simon Cancer Center), and Zeta Tau Sorority and Susan G. Komen for the Cure grant SAC110025 (to Y.L. and H.N.). The sequencing was performed in the Center for Medical Genomics (CMG) sequencing core at Indiana University School of Medicine.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.1000 Genomes Project Consortium, Abecasis G.R., Auton A., et al. . 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda C., Sausen M., Leary R.J., et al. . 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg L.M., Stone G., Butler M.K., et al. . 2013. Shining a light on dark sequencing: Characterising errors in Ion Torrent PGM data. PLoS Comput. Biol. 9, e1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. 2001. Random forests. Machine Learning 45, 5–32 [Google Scholar]
- Carter S.L., Cibulskis K., Helman E., et al. . 2012. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., et al. . 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E., Di Nicolantonio F., Loupakis F., et al. . 2013. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484 [DOI] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., et al. . 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F., Li M., Dressman D., et al. . 2005. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. U. S. A. 102, 16368–16373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Ley T.J., Larson D.E., et al. . 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew T., Murtaza M., Parkinson C., et al. . 2012. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 4, 136ra168. [DOI] [PubMed] [Google Scholar]
- Gerstung M., Beisek C., Rechsteiner M., et al. . 2012. Reliable detection of subclonal single-nucleotide variants in tumor cell populations. Nat. Commun. 3, 811. [DOI] [PubMed] [Google Scholar]
- Hao Y., Zhang P., Xuei X., et al. . 2016. Statistical modeling for sensitive detection of low-frequency single nucleotide variants. BMC Genomics 17, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O., Schwab R.B., Bao L., et al. . 2011. Detection of low prevalence somatic mutations in solid tumors with ultra-deep targeted sequencing. Genome Biol. 12, R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda M.D., Bonavia R., Mukasa A., et al. . 2010. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., et al. . 2012. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker M.L. 2010. Sequencing technologies—The next generation. Nat. Rev. Genet. 11, 31–46 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S., Van Loo P., Wedge D.C., et al. . 2012. The life history of 21 breast cancers. Cell 149, 994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rawe J., Jiang T., Sun G., et al. . 2013. Low concordance of multiple variant-calling pipelines: Practical implications for exome and genome sequencing. Genome Med. 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M.G., Russ C., Costello M., et al. . 2013. Characterizing and measuring bias in sequence data. Genome Biol. 14, R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C.T., Wong W.S., Swamy S., et al. . 2012. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 [DOI] [PubMed] [Google Scholar]
- Walter M.J., Shen D., Ding L., et al. . 2012. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 366, 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A., Aw P.P., Bertrand D., et al. . 2012. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.