

Abstract
We have used site-directed time-resolved fluorescence resonance energy transfer to determine the effect of a pathological mutation in the human ventricular essential light chain (hVELC) of myosin, on the structural dynamics of the actin-myosin complex. The hVELC modulates the function of actomyosin, through the interaction of its N-terminal extension with actin and its C-terminal lobe with the myosin heavy chain. Several mutations in hVELC are associated with hypertrophic cardiomyopathy (HCM). Some biochemical effects of these mutations are known, but further insight is needed about their effects on the structural dynamics of functioning actomyosin. Therefore, we introduced the HCM mutation E56G into a single-cysteine (C16) hVELC construct and substituted it for the VELC of bovine cardiac myosin subfragment 1. Using a donor fluorescent probe on actin (at C374) and an acceptor probe on C16 of hVELC, we performed time-resolved fluorescence resonance energy transfer, directly detecting structural changes within the bound actomyosin complex during function. The E56G mutation has no significant effect on actin-activated ATPase activity or actomyosin affinity in the presence of ATP, or on the structure of the strong-binding S complex in the absence of ATP. However, in the presence of saturating ATP, where both W (prepowerstroke) and S (postpowerstroke) structural states are observed, the mutant increases the mole fraction of the S complex (increasing the duty ratio), while shifting the structure of the remaining W complex toward that of S, indicating a structural redistribution toward the strongly bound (force-generating) complex. We propose that this effect is responsible for the hypercontractile phenotype induced by this HCM mutation in myosin.
Introduction
The structural transition of the actin-myosin complex from the weak (W, lever arm up) to the strong (S, lever arm down) binding states during the ATPase cycle generates the powerstroke that produces the force of muscle contraction (Fig. 1) (1). The myosin molecule is composed of three major structural domains: 1) the catalytic domain (CD) of the heavy chain, containing the actin and the nucleotide-binding sites; 2) the light-chain domain (LCD), which contains the essential and regulatory light chains (ELC, RLC) that are attached to two IQ domains of the heavy chain; and 3) the tail region of the heavy chain, which is responsible for thick filament formation (2, 3). The LCD is proposed to function as a lever arm that amplifies small structural changes in the CD into the larger motions by which myosin exerts force on actin. The light chains stabilize the lever arm and play an important role in regulating the actin-myosin interaction. In mammalian muscle, RLC potentiates force through modulation of the interaction of the myosin head with actin via phosphorylation of serine residues near its N terminus (4), whereas the removal of ELC from myosin results in a loss of actin filament motility (5) and a reduction in force (6).
Figure 1.
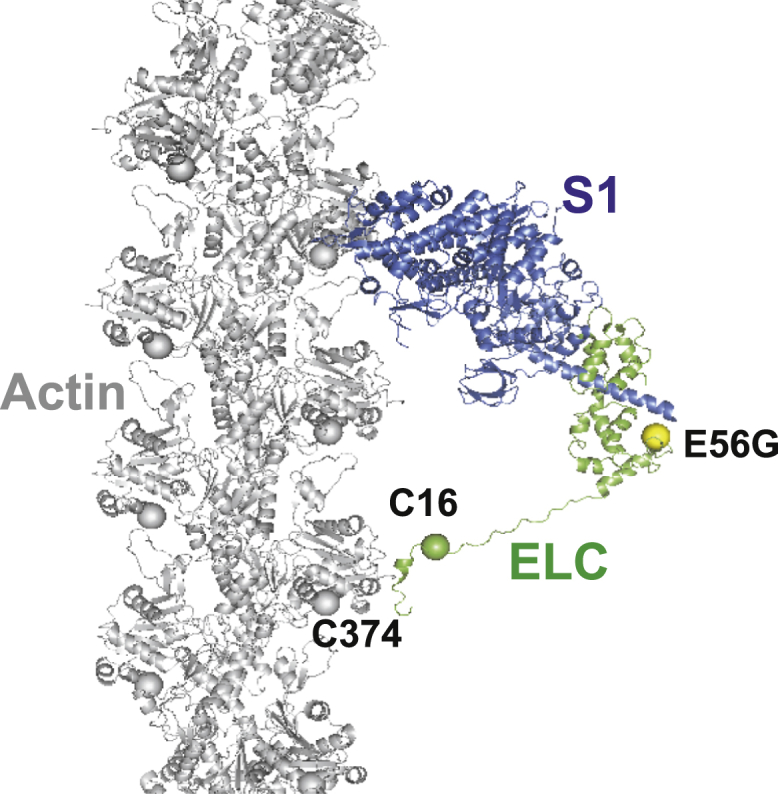
Given here is a model of an actin-myosin complex with skeletal myosin S1 (heavy chain, blue; ELC, green) in a strongly bound S state (lever arm down, postpowerstroke) on F-actin (gray). Spheres show the labeling sites on actin (C374, gray) and on myosin ELC (C16, green), and the HCM mutation (E56G, yellow). Not shown is the weakly bound W state (prepowerstroke), in which the ELC-containing lever arm is tilted up. The FRET sensor (donor at actin 374, acceptor at ELC C16) was designed to determine the effect of the E56G HCM mutation in hVELC on the W-to-S structural transition in cardiac actomyosin. The model structure was adapted from Aydt et al. (62). To see this figure in color, go online.
ELCs are highly conserved, and are expressed as two isoforms, A1 and A2. The principal difference between them is that A1 contains an N-terminal extension (NTE) of 40–45 additional amino acids. Smooth muscle and nonmuscle myosins express only the A2 isoform (7, 8). Fast skeletal muscle contains both A1 and A2 (9), whereas slow skeletal and cardiac muscles contain only A1 (10). In cardiac muscle, ventricular and atrial ELCs have NTEs of approximately the same length as their A1 counterparts in skeletal muscle, and are important regulators of cardiac contractility (11, 12). In skeletal muscle, A1 interacts with the negatively charged C terminus of actin, resulting in strongly actin-bound myosin heads with slower kinetics compared to A2 (12, 13, 14, 15, 16). Several point mutations in the human ventricular myosin essential light chain (hVELC) gene, MYL3, lead to hypertrophic cardiomyopathy (HCM), which is one of the most common and severe inherited cardiac disorders (13, 17). HCM results from mutations in 11 or more different sarcomeric genes (18) and is characterized by hypertrophy of the left ventricle and the interventricular septum, typically reducing ventricular chamber volume and causing myocyte and myofibrillar disarray. The clinical manifestations of the disease are quite variable (19), and the wide spectrum of functional perturbations induced by the different HCM mutations suggests that many different pathways lead to the HCM phenotype (18), so it is difficult to establish a prognosis based on the mutation (20).
Most of the HCM-linked mutations in hVELC are located in the EF-hand Ca2+ binding motifs, in highly conserved amino acid residues (21). In vitro studies on these mutations have shown a wide range of effects on force, actin sliding velocity, and actin-activated ATPase activity (22). However, the molecular mechanisms by which these mutations cause the HCM phenotype remain poorly understood. Thus, further insight is required to understand the structural and functional effects of these mutations on actomyosin.
In this study, we focus on a mutation in hVELC, E56G, which causes HCM (22). We hypothesize that this mutation, located near the interface of heavy and light chains (Fig. 1), affects cardiac function by altering the structural states of the actin-myosin complex during the ATPase cycle. To test this hypothesis, we engineered a fluorescent biosensor to detect structural changes within hVELC bound to myosin. We expressed a single-cysteine (C16) construct of hVELC without (here designated “WT”) and with the E56G mutation (here designated “E56G”) (Fig. 2). Binding of the ELC N-terminus to actin is governed primarily by the first 11 residues of ELC (23). We avoided modifying this region by placing a probe at an engineered Cys at residue 16. We labeled C16 of hVELC with a nonfluorescent acceptor probe (DABCYL) for both WT and E56G, exchanged it with the native endogenous ELC in bovine cardiac myosin subfragment 1 (S1), and made homogenous preparations of WT and E56G cardiac S1. We labeled actin with a fluorescent donor probe (1–5 IAEDANS) at C374. Structural transitions, induced by addition of ATP, in the labeled acto-S1 complexes, were detected using time-resolved fluorescence resonance energy transfer (TR-FRET) from the probe on actin C374 to the probe on C16 of hVELC. TR-FRET from actin to myosin can resolve intermolecular distances within the bound actomyosin complex without interference from unbound S1. This is critical in the presence of ATP, where a large fraction of S1 is dissociated from actin because of its low duty ratio. Our previous study applied a similar approach to the skeletal muscle actomyosin complex, where we showed that the NTE of ELC has a large effect on the distribution of actomyosin structural states during ATPase activity (24). In addition, we showed that the movement of the LCD relative to actin probed by a FRET acceptor on the C-terminal lobe (C177) of ELC correlates well with the movement of an acceptor (C16) on the NTE (24). The latter acceptor site provides a much more robust FRET measurement, in which the resolved W and S structural complexes are well within the optimal range for resolution by FRET. Thus, in this study, we used the ELC-C16 acceptor to determine the effects of the E56G mutation on the structural dynamics of cardiac actomyosin during the ATPase cycle.
Figure 2.

Here is a schematic representation of the hVELCs used in this study. (A) Native endogenous hVELC has three cysteines. (B) WT hVELC has a single cysteine at residue 16. (C) E56G has a single cysteine (C16) with E56G. To see this figure in color, go online.
Materials and Methods
Protein preparations and labeling
Actin was prepared from rabbit skeletal muscle by extracting acetone powder in cold water, as described in Prochniewicz et al. (25). C374 in F-actin was labeled with the FRET donor as follows: 700 μM 1–5 IAEDANS, freshly dissolved in DMF, was mixed with 70 μM F-actin and incubated for 18 h at 4°C. Labeling was terminated by adding 10 mM DTT. After 30 min sedimentation at 350,000 × g, the F-actin pellet was suspended in G-Mg buffer (5 mM Tris, 0.5 mM ATP, 0.2 mM MgCl2, pH 7.5) followed by clarification at 300,000 × g for 10 min. Actin was again polymerized for 30 min at 25°C in the presence of 3 mM MgCl2, centrifuged at 300,000 × g for 30 min, and the pellet was suspended in F-Mg buffer (3 mM MgCl2, 10 mM Tris, pH 7.5) containing 0.2 mM ATP. The labeled F-actin was immediately stabilized against depolymerization and denaturation by adding one molar equivalent of phalloidin. For selected studies, F-actin was labeled with pyrene-iodoacetamide (producing pyrene-actin) as described in Guhathakurta et al. (26).
Bovine β-cardiac myosin was prepared as described in Lowey et al. (27) and Rohde et al. (28). β-cardiac S1 was prepared by α-chymotryptic digestion of bovine β-cardiac myosin. After overnight dialysis against 20 mM Imidazole pH 7, 120 mM KCl, and 1 mM EDTA, 2 mg/mL chymotrypsin was added dropwise to the myosin suspension at 25°C to a final concentration of 0.05 mg/mL and was stirred for 15 min. The digestion was stopped with 1 mM PMSF, and the digested myosin was centrifuged to pellet undigested myosin. The supernatant, containing mainly S1, was dialyzed overnight in 10 mM Tris, pH 7.5. S1 was flash frozen with 150 mM sucrose before storage at −80°C.
A pET15b vector encoding endogenous hVELC with an N-terminal 6× His tag and thrombin cleavage site was obtained from GenScript (GenScript, Piscataway, NJ) (Fig. 2 A). All three native cysteines (C67, C76, and C182) of hVELC were mutated to alanine, and a single cysteine at position A16 was engineered using a QuikChange kit (Invitrogen, Carlsbad, CA). This construct, containing a single cysteine, is designated wild-type (WT) (Fig. 2 B). The E56G mutation was engineered in the single-cysteine WT hVELC using a QuikChange kit (Stratagene, San Diego, CA), and is designated “E56G” (Fig. 2 C). All the sequences were confirmed by DNA sequencing. Both WT and E56G constructs were transformed into Escherichia coli BL21DE3-competent cells, grown in LB medium supplemented with 100 μg/mL ampicillin, and was induced with 1 mM IPTG. Both WT and E56G N-terminal His-tagged hVELCs were purified from bacterial cell pellets using Talon affinity resin (Clontech Laboratories, Mountain View, CA) as described in Guhathakurta et al. (24), then concentrated, dialyzed to 10 mM Tris pH 7.5, and stored frozen in −80°C in 150 mM sucrose.
Before labeling, hVELC was reduced with 5 mM DTT for 60 min on ice. Excess DTT was removed using Zeba spin columns (Pierce Biotechnology/Thermo Fisher Scientific, Waltham, MA) in 10 mM Tris pH 7.5. C16 of WT and E56G hVELC was labeled with the FRET acceptor as follows: 700 μM DABCYL Plus C2 maleimide (AnaSpec, Fremont, CA), was mixed with 70 μM hVELC in 6 M GuHCl, 5 mM EDTA, 10 mM Tris pH 7.5, and 1 mM TCEP, and incubated for 18 h at 23°C. Unbound dye was removed by extensive dialysis against 10 mM Imidazole pH 7 and Zeba Spin desalting columns (Pierce Biotechnology/Thermo Fisher Scientific). Protein was concentrated and stored frozen in 150 mM sucrose at −80°C. Labeling of C16 with the acceptor probe was complete and specific (>95%) (Fig. S7) as confirmed using electrospray ionization mass spectrometry (29).
Exchange of hVELCs in cardiac S1
Acceptor-labeled hVELC was exchanged into β-cardiac S1 as described in our previous study of skeletal S1 (24). A quantity of 50 μM cardiac S1 was incubated with 500-μM labeled hVELC in 100 mM imidazole pH 7, 2 mM DTT, 2 mM EDTA, and 4.7 M NH4Cl for 25 min on ice, followed by extensive dialysis in 10 mM Imidazole pH 7. The dialyzed sample was applied to a Talon affinity resin (Clontech Laboratories). S1 with endogenous ELC did not bind to the Talon resin. S1 containing labeled and exchanged His-tagged ELC together with free His-tagged ELC that binds to the resin were eluted in Talon elution buffer (200 mM Imidazole and 300 mM KCl, pH 7). Free His-tagged ELC was removed by pelleting S1 with actin (20 min for 300,000 × g) added at 1:1 molar ratio. The acto-S1 pellet was then suspended in the release buffer (3 mM MgCl2, 10 mM Tris pH 7.5, 0.1 M KCl, and 3 mM ATP) and was centrifuged at 300,000 × g for 20 min. This step separated actin from S1, and S1 with labeled hVELC was obtained in the supernatant, which was concentrated and dissolved in F-Mg buffer (3 mM MgCl2, 10 mM Tris pH 7.5) for experiments. A SDS-PAGE gel was used to observe the exchange S1 (Fig. S8 A). A native PAGE gel (4–25%) was used to detect the incorporation of labeled and bound ELC in final S1 (Fig. S8 B, lane 2). Absence of free ELC in the final S1 sample (Fig. S8 B, lane 2) indicates that the fluorescence signal is coming from bound ELC. Replacing endogenous ELC with completely labeled His-tagged ELC and utilizing His-tag for purification of ELC-bound S1 results in final cardiac S1 (used for experiment) with fully labeled ELC. Therefore, the S1 sample used for experiments was homogenous, with all S1 molecules containing a labeled ELC.
The concentrations of unlabeled proteins were determined by measuring absorbance at 280 nm, assuming the following molecular weights and absorption of 0.1% protein A280: S1 = 110,000 Da, A280 = 0.75; and A1 = 23,898 Da, A280 = 0.18. The concentrations of labeled S1, actin, and ELC were measured by the Bradford assay (Bio-Rad Laboratories, Hercules, CA) using corresponding unlabeled proteins of known concentration as standards.
Steady-state ATPase activity and cosedimentation assay
Actin-activated ATPase activity was measured at 25°C in F-Mg buffer (3 mM MgCl2, 10 mM Tris pH 7.5) containing 3 mM ATP, at a constant concentration of myosin S1 (0.1 μM) and increasing concentrations (10–50 μM) of nucleotide-free F-actin, using an NADH-coupled assay, with oxidation of NADH monitored by absorbance at 340 nm (26). Control experiments with donor-labeled actin showed that the label does not significantly affect Vmax (activity at saturating actin) or KATPase (actin concentration at half Vmax), which were determined as described in Guhathakurta et al. (24). The affinity of native and exchanged cardiac S1 for labeled actin in the presence of saturating ATP was measured at 25°C in F-Mg buffer containing 3 mM ATP by cosedimentation (quantitating the unbound S1 in the supernatant), and Kd was determined by fitting the data to a quadratic binding equation (24) (Fig. S3 B and Table 1).
Table 1.
Interaction of Cardiac S1 with Actin in the Presence of ATP
| S1 | Vmax (s−1) | KATPase (μM) | Catalytic Efficiencyb | Kd (μM)c |
|---|---|---|---|---|
| Nativea | 3.2 ± 0.4 | 15.9 ± 6.8 | 0.19 ± 0.05 | 11.1 ± 3.5 |
| WT | 3.0 ± 0.5 | 21.2 ± 6.8 | 0.14 ± 0.05 | 7.3 ± 0.9 |
| E56G | 3.6 ± 0.4 | 24.4 ±1.8 | 0.15 ± 03 | 5.9 ± 0.6 |
Mean ± SE for n = 3 to six preparations, p < 0.05.
Native tissue purified bovine cardiac S1 (unlabeled and unexchanged).
Vmax/KATPase (Vmax is the activity at saturating actin, and KATPase is the actin concentration needed for half-maximal activation V = Vmax/2).
Dissociation constant for actin and S1 in the presence of saturating ATP.
Interaction of myosin with pyrene-actin
The effect of the E56G mutation on the actin-binding surface of myosin was measured by the interaction of S1 with actin labeled with pyrene-iodoacetamide at C374 (pyrene-actin). Pyrene-actin (2 μM) fluorescence (excitation 350 nm, emission 407 nm) was measured at 25°C in F-Mg buffer in the absence and presence of S1 (10 μM) as described in Guhathakurta et al. (26). After addition of 3 mM ATP (which was saturating), pyrene-actin fluorescence was measured in the steady state, at its minimum value, before fluorescence began to recover. The measured fluorescence intensities of acto-S1 in the absence and in the presence of saturating ATP were normalized to the fluorescence intensity of actin alone, and results are plotted in Fig. S4.
Binding of MANT-ADP to myosin and actomyosin
Binding of MANT-ADP (Invitrogen) to myosin was detected by measuring sensitized emission of the MANT fluorophore (acceptor) at 440 nm using myosin tryptophans as donors with excitation at 290 nm (30). Increasing concentrations (0–20 μM) of MANT-ADP were added to 1 μM myosin with or without 2 μM nucleotide-free F-actin. Background fluorescence for each concentration of the added MANT-ADP was determined as the sum of emission intensity of free MANT-ADP and emission intensity of myosin ± actin in the absence of MANT-ADP. The fraction of the bound nucleotide (Fig. S5) was calculated as y = (F–F0)/Fmax, where F = fluorescence intensity of MANT-ADP in the presence of myosin construct, F0 = background fluorescence, and Fmax = fluorescence intensity corresponding to the saturating amount of bound MANT-ADP, obtained by fitting y = F–F0 to the Michaelis-Menten equation. The dissociation constant Kd of MANT-ADP for myosin was determined by fitting the fraction of bound nucleotide y to a quadratic equation.
Spectroscopic data acquisition and FRET analysis
Fluorescence waveforms were acquired using a high-performance time-resolved fluorescence spectrometer constructed in this laboratory, which uses direct waveform recording, providing 105 times higher throughput than the conventional method, time-correlated single-photon counting, while providing at least comparable performance in signal/noise, accuracy, and resolution of distinct components (31). Donor-labeled (at C374) actin (2 μM) was excited using a passively Q-switched microchip frequency-tripled YAG laser (NanoUV-532; JDS Uniphase, Milpitas, CA) at 355 nm with a pulse repetition frequency of 10 kHz. The high-energy (1 μJ/pulse), narrow (∼1 ns full width at half-maximum, FWHM) laser pulses are highly uniform in shape and intensity. Emitted photons were passed through a polarizer set at the magic angle 54.7°C, followed by an interference band-pass filter (470/20 nm; Semrock, Rochester, NY), and detected with a photomultiplier tube module (H5773-20; Hamamatsu Photonics, Hamamatsu, Japan), and digitizer (Acqiris DC252, time resolution, 0.125 ns; Agilent Technologies, Santa Clara, CA).
FRET experiments were performed at 25°C in F-Mg buffer. Acto-S1 complexes were formed by adding 1–10 μM of acceptor-labeled S1 (at C16 on ELC) (as needed to obtain a significant bound complex during the steady state) to 2 μM donor-labeled actin (at C374). Fluorescence emission decays were acquired first in the absence of ATP, then ATP was added at a final concentration of 1–3 mM (as needed to obtain a reliable steady state, assuring saturation by ATP), and samples were mixed quickly and measurements started typically within 5 s after addition of nucleotide. Consecutive decays were measured every ∼5 s for 10 min to demonstrate that the steady state was achieved.
TR-FRET waveforms were analyzed globally in model-independent and -dependent methods as described in the literature (24, 32, 33), and as elaborated in Supporting Material (Eqs. S1–S7). In the absence of ATP, a two-state model (Eq. S6, n = 1) was sufficient to fit the data: a mole fraction (1 − XB) of unbound donor (probe on actin), and a single-Gaussian distance distribution, presumably corresponding to the strongly bound S complex (lever arm down), characterized by a center distance RS and a full width at half-maximum ΓS (see Fig. 5 C, green). After addition of ATP, a three-state model (Eq. S6, n = 2) was necessary and sufficient (Supporting Material) to fit the data: a mole fraction (1 − XB) of unbound donor, a mole fraction XS of the S complex, and a mole fraction XW (= XB − XS) of the W complex (lever arm up), characterized by a center distance RW and FWHM ΓW (see Fig. 5 C, red). Later in Fig. 5, A and B, these mole fractions are plotted versus time after addition of ATP.
Figure 5.

Shown here are structural states of the actomyosin complex detected by TR-FRET. (A) Given here is the time dependence of FRET-detected mole fractions (X) after the addition of ATP to a mixture of donor-labeled actin and acceptor-labeled myosin S1 (top, WT and bottom, E56G). XB (squares, black) is the fraction of donor that has bound acceptor. XB = XW + XS, where XW (red) and XS (green) are the mole fractions of W and S complexes. (B) Given here is the time dependence of mole fractions (expanded view) in the first minute after the addition of ATP, demonstrating a true steady state. Steady-state duty ratio is shown in the box. (C) Given here is the interprobe distance distribution (best fit to a Gaussian function) corresponding to bound actin-S1 complex determined from S (ρS, green) and W (ρW, red) complexes (WT, top and E56G, bottom). Whereas the structure (distance distribution) of the S complex is not significantly affected by the mutation, the W complex is shifted to a shorter distance and a narrower width, so it is designated W′. Each curve is normalized to unit area, which is independent of the mole fraction XS or XW. Thus the distribution of actin-bound distances R at a given time after mixing is given by a linear combination ρ(R) = (XS/XB)ρS(R) + (XW/XB)ρW(R) (24) (Eq. S8). To see this figure in color, go online.
Statistics
Values are given as mean ± SE. Statistical differences between mean values were calculated using a paired t-test, with significance at p < 0.05.
Results
Effects of the E56G mutation on functional properties of cardiac myosin
We first examined the functional effects of labeling and exchange of hVELC onto cardiac S1 (Fig. S3, A and B and Table 1). Vmax and KATPase of actin-activated myosin S1 ATPase (Table 1) with the exchanged labeled hVELCs are similar to 1) those of native tissue-purified S1 containing endogenous VELC (Fig. S3 A and Table 1), which is used as a control; and to 2) those reported previously for native bovine cardiac S1 (34, 35). This indicates that the incorporation of the labeled hVELCs into S1 does not perturb its solution enzymatic properties. The E56G mutation has no significant effect on the actin-activated myosin ATPase (Fig. S3 A and Table 1). Cosedimentation binding assays of labeled actin and S1 show that the E56G mutation had no significant effect on the weak binding Kd (affinity in the presence of ATP) for S1 binding to actin (Fig. S3 B and Table 1).
In the absence of ATP, the quenching of pyrene-actin by E56G-S1 is identical (within 1%) to that of native and WT cardiac S1 (Fig. S4). The addition of saturating ATP relieved the quenching of pyrene-actin completely (within 1%) for all three acto-S1 complexes (Fig. S4). These data indicate that the interface of the strongly bound actomyosin complex is not altered by the presence of the E56G mutation hVELC, and the bound complex in the presence of saturating ATP shows no rigorlike component.
The intensity of sensitized fluorescence emission of MANT-ADP bound to myosin depends on the efficiency of fluorescence energy transfer from directly excited tryptophan donors. If all myosins have the same number of tryptophans located at the same distances from the MANT fluorophore, the intensity of sensitized fluorescence would be the same for all of them. The binding of MANT-ADP to S1 in the presence or absence of actin, measured using sensitized emission, is unaltered from WT to E56G S1 (Fig. S5). This shows that the conformation of the active site (nucleotide binding pocket) is not altered in E56G S1.
Actomyosin FRET in the absence of ATP
In the absence of ATP, the binding of acceptor-labeled WT and E56G S1 (labeled at C16 in the NTE) to donor-labeled actin (labeled at C374) leads to a faster decay (shorter lifetime) of the donor lifetime, indicating FRET (Fig. 3, black FD to green, FD+A = [1-XB]FD + XBFDA, where XB is the fraction of actin with bound S1). The FRET efficiency 〈E〉, as calculated from the average lifetime in a model-independent analysis (Eq. S3 and Fig. S1), was not affected by the E56G mutation (Fig. 4, green bars). Model-based analysis of FRET decays (FD+A) (Fig. S1) allows 1) resolution of the signals from bound (FDA) and free (FD) actin, and 2) direct structural determination of the bound actin-S1 complex by analyzing FDA and calculating the mean distance R and width of the distribution (FWHM = Γ) (Fig. 5 C). In the absence of ATP, this is relatively simple, because there is no free actin (XB = 1, FD+A = FDA), and there is no W complex. The mean distance RS and the distribution ΓS was essentially the same for WT (3.6 ± 0.2, 2.0 ± 0.2 nm) and E56G (3.5 ± 0.2, 2.0 ± 0.2 nm) acto-S1 complexes (Fig. 5 C, green).
Figure 3.
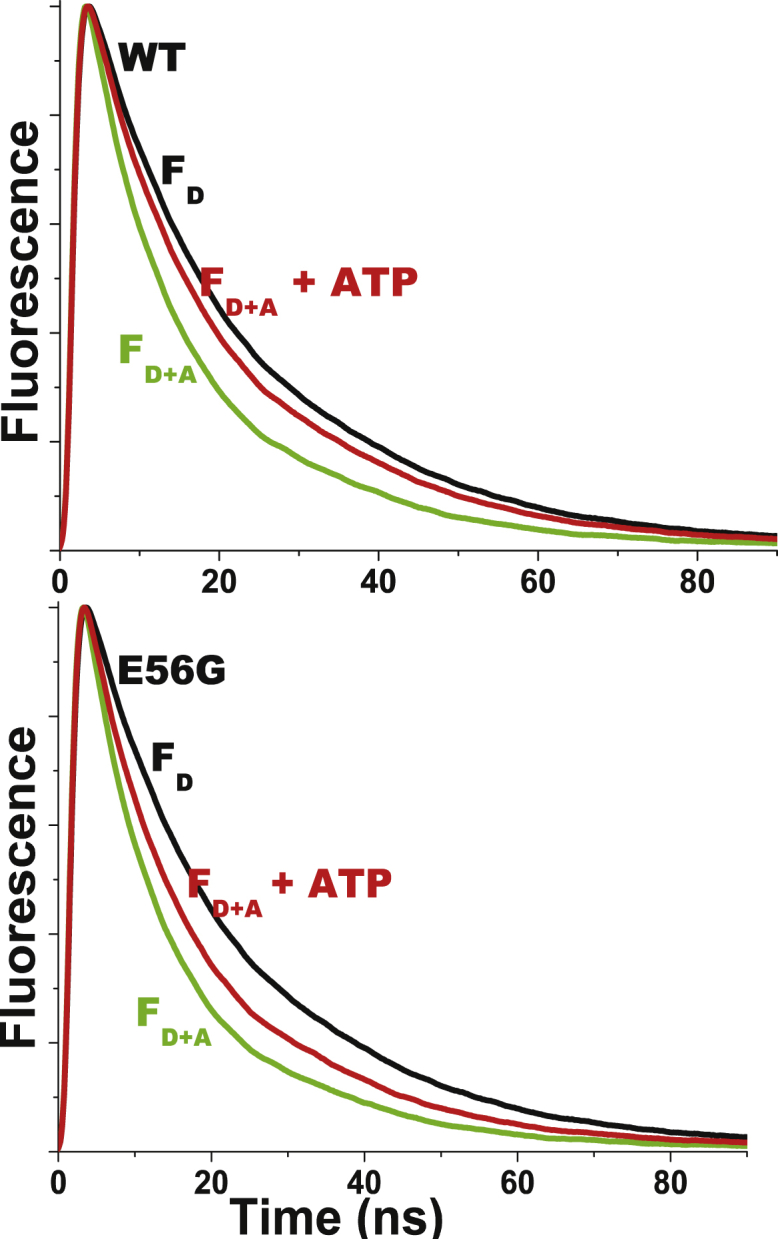
Given here are the representative fluorescence decays of 2 μM donor-labeled actin in the absence (black, FD, right) and presence (FD+A) of acceptor-labeled cardiac S1 (10 μM), and in the absence (green, left) and presence (red, middle) of saturating ATP (1 mM). (Top) WT-ELC S1. (Bottom) E56G-ELC S1. Unlabeled S1 had no effect on FD, so faster FDA decays indicate FRET. Signals are normalized to the peak intensity. To see this figure in color, go online.
Figure 4.
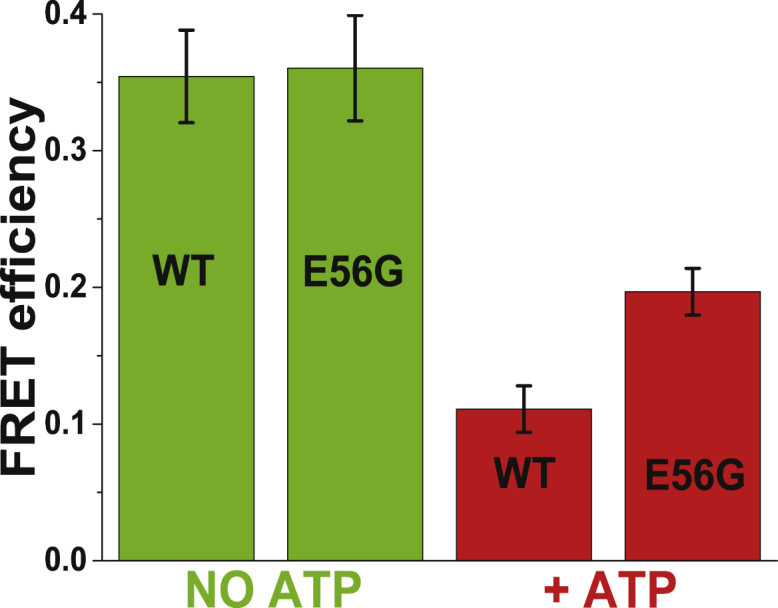
Given here is the model-independent ensemble-average FRET efficiency 〈E〉, (Eq. S3) between actin and cardiac S1 in the absence (green) and presence (red) of ATP. To see this figure in color, go online.
The analysis of FRET measurements performed in the presence of substoichiometric or excess acceptor S1 (Fig. S2) showed that the mean distance RS between donor and acceptor probes in the S complex is independent of the fraction of bound acceptor-labeled S1. This indicates that FRET is not affected by the proximity of multiple acceptors and that the unbound S1 does not contribute to FRET. Thus, the distance between probes on actin and cardiac S1 in the S complex is not affected by the E56G mutation. This distance RS is ∼0.5 nm longer than the distance previously measured between equivalent sites in the skeletal acto-S1 complex (24). We conclude that the NTE of cardiac hVELC is in close proximity to actin (Fig. 1) as previously observed for skeletal A1 (24), and that the structure of the S complex, as detected by our FRET assay in the absence of ATP, is not affected by the E56G mutation.
Actomyosin FRET in the presence of saturating ATP
Resolution of the structural states in the presence of ATP required acquisition of TR-FRET decays during the brief steady state of the actomyosin ATPase cycle. Acceptor-labeled S1 (5–10 μM final concentration) was added to donor-labeled actin (2 μM final concentration), followed by adding saturating ATP (1–3 mM). ATP decreased the rate of decay compared to the S complex, indicating a decrease in FRET (Fig. 3, green, FD+A to red, FD+A + ATP). FRET efficiency 〈E〉, as calculated from the average lifetime in a model-independent analysis (Eq. S3), was significantly greater for the E56G mutant (Fig. 4, red bars). These results were obtained at 6 s intervals from 6 to 60 s, varying ATP from 1 to 3 mM, with no significant change, indicating that a true steady state existed and that ATP was saturating.
In the presence of ATP, actomyosin is best described by a three-state model: a mole fraction (1 − XB) of unbound donor, with the bound state (mole fraction XB), which is resolved into a two-Gaussian distance distribution characterized by a mole fraction XS of the bound S state with a mean distance Rs and FWHM of ΓS, and a mole fraction XW (= XB − XS) of the bound W state, with a mean distance RW and FWHM of ΓW (Supporting Material). No improvement in the fit (decrease in χ2) was obtained by allowing the S-state parameters (RS, ΓS) to vary from the values observed in the absence of ATP (Fig. S6), so these parameters were fixed to those values in the analysis shown in Fig. 5. This indicates that the structure of the S complex that populates during the steady state is similar to that of the rigor complex. This is consistent with previous EPR results indicating that the orientation of the strong-binding structural state of myosin LCD during contraction is indistinguishable from that observed in the absence of ATP (36). For both WT and E56G S1, the steady state of ATP hydrolysis persisted for ∼2 min after addition of ATP, as reflected in the constant values of bound mole fractions (XB) of both proteins (Fig. 5 B). Acquisition of high signal/noise waveforms at intervals of several seconds, as required to accurately capture the steady-state signal (Fig. 5, A and B, red), was made possible by our direct waveform recording technology (31). The steady-state value of XB was 0.662 ± 0.022 and 0.643 ± 0.015, for WT and E56G, respectively, indicating that the mutation does not significantly affect actin-S1 affinity, consistent with our results in the cosedimentation assays (Fig. S3).
In contrast, the mole fractions XS and XW, corresponding to bound actin-myosin structural states S and W (Fig. 5, A and B), are significantly different for WT and E56G. The duty ratio (DR) is typically defined as the fraction of time a myosin head spends in a strong-binding state (equivalent to the fraction of heads XS in the S state during the steady state) (6, 37). It is usually measured under conditions where the fraction of bound heads XB is <1 (e.g., in isometric muscle contraction, in a motility assay, or in solution). In solution, the most meaningful value is obtained by extrapolating to infinite [actin], so that DR is equal to the fraction of bound heads that are in the S state, DR = XS/XB = XS/(XS + XW) (38). In this study, we achieve this directly in a single time-resolved FRET experiment that resolves and quantitates the bound actomyosin structural states with high resolution and precision. The steady-state DR (averaged over the first minutes of Fig. 5 B) is significantly higher for E56G (0.308 ± 0.012) than for WT (0.231 ± 0.018) (Fig. 5 B), indicating a 33 ± 11% increase in the fraction of attached heads that are in the S state due to the mutation. With increasing time of data acquisition, leading to exhaustion of ATP, in all cases XB and XS gradually increased toward 1, as observed in the absence of ATP (XB = 1) (Fig. 5 A). Control experiments showed that the dissociation of the weakly bound actin-S1 complex by adding 0.1 M KCl completely eliminated FRET, indicating that all S1 is capable of binding ATP, thus weakening the interaction with actin (data not shown).
The increased FRET caused by the mutation in Fig. 4 (red) is not simply due to increased duty ratio (increase in XS), because model-based analysis shows that the structure of the bound W complex is clearly affected by the mutation (Fig. 5 C, red). The mutation decreases the mean distance between probes from 5.2 ± 0.2 nm in WT (Fig. 5 C, red, top) to 4.2 ± 0.2 nm (Fig. 5 C, red, bottom), although also decreasing the width ΓW (corresponding to structural disorder) from 3.8 ± 0.2 to 2.1 ± 0.2 nm (Fig. 5 C, red). Thus, the mutation shifts the structure of the W complex toward that of the S complex, as measured by both the mean distance RW and the structural disorder ΓW. We designate this new structural state W′ to distinguish it from the W state in WT S1. In addition, the W and S structural states induced by the E56G mutation in cardiac S1 (Fig. 5 C, bottom) are found to be remarkably similar to those observed with the same labeling sites in WT skeletal S1A1 (Fig. 4 in Guhathakurta et al. (24). This suggests that the W structural state of E56G S1 is shifting toward that of a faster, stronger myosin, consistent with the HCM phenotype (37).
As observed in the S complex in the absence of ATP, FRET analysis as a function of added S1 shows that the bound structural states are independent of the fraction of bound myosin, indicating that there are no artifacts due to multiple acceptors and no cooperative effects of bound S1 on the structures of nearby actin-S1 complexes.
Our results show that the W-S structural transition in actomyosin, as detected by FRET between actin and the NTE of hVELC, is modulated by the E56G mutation: the population of the S state is increased, and the structure of the W state is shifted toward that of the S state.
Discussion
Properties not affected by the mutation
We start this discussion by summarizing the properties of the actin-myosin system that the E56G mutation does not affect. We found that it has no significant effect (p < 0.05) on the standard steady-state biochemical properties of the actomyosin system, such as the actin-activated myosin ATPase (Fig. S3 A; and Vmax and KATPase in Table 1) or the actin-binding affinity of cardiac S1 in the presence of saturating ATP (Fig. S3 B; and Kd in Table 1). These results are consistent with those determined for several HCM mutations on the myosin heavy and light chains. Previously studied hVELC mutations, such as A57G (39), and three RLC mutations, A13T, F18L, and E22K (40), also showed no significant changes in solution ATPase activity. Recent studies on converter domain mutations in human β-cardiac myosin, R719W and R723G, showed little or no effect on ATPase activity (41, 42). The interaction of S1 with pyrene-actin is also unaffected by the E56G mutation, in the presence or absence of saturating ATP (Fig. S4), so the effects of the mutation cannot be attributed to structural changes at the actin-myosin CD interface, as previously observed for other mutations in myosin light chains (39, 40). Similarly, MANT-ADP binding and fluorescence is unaffected by the mutation (Fig. S5), so there is no evidence that this mutation alters the conformation of the nucleotide binding site. Nucleotide binding studies on an HCM mutation in the myosin heavy chain (R453C) also showed no change in ADP binding (43). All the biochemical and structural measurements have been performed at low ionic strength, as required to detect significant binding of myosin to actin in solution. Our results are consistent with studies on skeletal and cardiac muscle fibers, which indicate that the NTE of ELC interacts with actin similarly at physiological and low ionic strength (14, 39, 44).
In the absence of ATP, the distance between probes on actin and myosin was not significantly affected by the E56G mutation (Figs. 4 and 5). The FRET-detected distance RS = 3.6 ± 0.2 nm between C374 of actin and C16 of hVELC (Fig. 5 C, green) is in good agreement (3.6 nm) with a reported structural model of skeletal acto-S1A1 (Fig. 1). Both the mean distance RS values and the width of the distribution Γ-values are essentially the same for both WT and E56G actomyosin (Fig. 5 C, green), indicating an ordered structural state in both cases.
Properties affected by the mutation
Significant mutation-dependent differences in TR-FRET were detected only in the presence of ATP. TR-FRET provides quantitative information about both the populations of the W and S states (Fig. 5, A and B) and their structural properties (Fig. 5 C). From the populations, we determine the duty ratio (DR), defined as XS/XB, the fraction of actin-bound heads in the S structural state. DR was increased by 33% in the steady state of saturating ATP (Fig. 5 B). Our approach offers a key advantage, unique in the actin-myosin literature: the location of the donor on actin, coupled with time-resolved FRET detection, allows us to detect directly the bound actomyosin complexes in the presence of ATP. The duty ratio is typically calculated from several separate measurements, usually requiring extrapolation to infinite actin (6, 37), but TR-FRET directly detects and resolves the populations of the W and S structural states of actin-bound myosin heads with high precision in a single measurement, thus determining DR directly. A recent study in which β-cardiac myosin data was analyzed comprehensively (45), calculated a duty ratio of 0.20; this is in excellent agreement with our finding of 0.23, when the actin concentration is 20 Km (approaching our conditions).
The direct detection of TR-FRET in the bound actin-myosin complex also provides a direct measurement of the structural properties of the resolved W and S states. In the presence of ATP, we found no FRET evidence for an effect of the mutation on the structural properties of the S complex, but those of the W complex were clearly affected. In WT, the mean distance Rw between actin and C16 was 5.2 ± 0.2 nm, whereas in E56G, this distance decreased to 4.2 ± 0.2 nm (Fig. 5 C, red). The effect of the mutation on structural disorder is even greater: in WT, the W complex is nearly twice as disordered as the S complex, but this difference is negligible for the E56G mutation (Fig. 5 C, red). Thus, in the presence of the E56G mutation, the structure of the W′ complex is intermediate between those of the W and S complexes of WT cardiac S1.
Our structural measurements do not provide direct information about the correlation of existing structural states (W and S) with the biochemical identities of bound ligands (ATP, ADP.P, ADP, etc.), so the correlation of structural and biochemical states remains to be fully determined, but transient kinetics studies provide useful context (Fig. 6). A recent transient structural kinetics study on β-cardiac myosin (28), in which TR-FRET (using a FRET biosensor within myosin to detect the power stroke) resolved the lever-arm-up (W) and the lever-arm-down (S) structural states of myosin, showed that the powerstroke is only slightly faster than phosphate release (28), probably following both pathways 1 and 2 in Fig. 6. This explains why we observe both W (lever arm up, prepowerstroke) and S (lever arm down, postpowerstroke) structural states for actin-bound myosin in the steady state in the presence of saturating ATP. We observe no pyrene quenching (indicated by blue actin in Fig. 6) in the presence of saturating ATP (Fig. S4), despite our observation that both W and S structural states are populated; this is consistent with studies of pyrene-actin quenching in various myosin isoforms (46, 47). The E56G mutation shifts the W-S transition toward the S state, probably by shifting the system in the direction of pathway 1 in Fig. 6.
Figure 6.

Shown here is the actin-activated myosin ATPase cycle. In this cycle, changes in the ligand at myosin’s nucleotide site are coupled to changes in actin-binding affinity and conformation between W (lever arm up, red) and S (lever arm down, green). After ATP hydrolysis, release of phosphate (Pi) is associated with the W-to-S transition (power stroke), producing force and movement. Based on kinetics (45) and transient FRET (28) data, the W-S transition for skeletal actomyosin proceeds primarily by pathway 1 (power-stroke before Pi release), whereas β-cardiac actomyosin proceeds by both pathways 1 and 2. The E56G mutation Increases the fraction of the S state, presumably by favoring pathway 1. The color of actin distinguishes those actomyosin states that quench pyrene-actin (blue, leftmost) from those that do not (yellow, all of the rest). To see this figure in color, go online.
Thus, TR-FRET shows that the E56G mutation alters the distribution of structural states of the actin-myosin complex, perturbing the W-S transition, which is a primary component of force generation (1, 48, 49). In the presence of saturating ATP, the mutation causes 1) an increase in the fraction of actin-bound myosin molecules that are in the S state, interacting strongly with actin (the duty ratio); and 2) a shift in the structure of the W state toward that of the S state, as illustrated by the model in Fig. 7. How might these effects be related to the hypercontractile HCM phenotype? 1) The increased duty ratio is consistent with greater force production (41). 2) Similarly, the perturbation of the W structural state in the direction of the S state is consistent with insufficient relaxation (diastole), which has also been proposed to characterize some HCM phenotypes (50). These results are consistent with a proposed mechanism of HCM mutations, in which there is an increase in the number and/or unitary force of force-generating actin-attached myosin heads, producing a hypercontractile heart (51, 52).
Figure 7.

Given here is the proposed model for the effect of the E56G mutation on the structural complex of actin (yellow) and cardiac S1 (blue) during the ATPase cycle. Based on TR-FRET (gray arrows) measured from probes attached to actin C374 (blue spheres) to C16 (magenta spheres) on the NTE of hVELC, two conformations of the complex (W and S) are observed during the steady state. The effect of the HCM mutation is primarily on the W complex, which decreases in population (indicated by black straight arrows), while decreasing its difference from the S complex in distance (gray arrows and numbers) and disorder (curved arrows) to produce a new state, W′. LA, lever arm (light-chain domain). To see this figure in color, go online.
E56 is predicted to be in a helix that connects the NTE to the core of ELC. The importance of this helix is confirmed by the presence of an adjacent mutation (A57G) in hVELC that causes HCM (53). Transgenic mice with the A57G mutation showed increased Ca2+ sensitivity of force, decreased maximal tension, and increased passive tension of muscle fibers to induce myocardial stiffness (54). E56G is also in close proximity to myosin’s converter domain (55), which plays an important role in force generation. Previous studies on the HCM mutations in the converter region indicate that this region is a major element of cross-bridge compliance (56). Our results indicate that the structure of myosin’s actin-binding surface and nucleotide-binding site is unaltered by the E56G mutation, as detected by pyrene-actin fluorescence (Fig. S4) and MANT-ADP binding (Fig. S5), so it is likely that the mutation alters the interface between the heavy and light chains, interfering with the structural states of the myosin lever arm (Fig. 7).
Clarification of the mechanistic significance of these results awaits further structural and functional analysis of this and other HCM mutations in hVELC in the β-MHC background. In addition, extension of this TR-FRET approach to two-headed myosin and/or skinned muscle fibers would provide a more direct correlation between structural dynamics and contractility.
Conclusions
The high resolution, in both time and structure, of our TR-FRET technology, coupled with structure-based design of a fluorescent biosensor pair on actin and the NTE of the hVELC light chain, has provided direct insight into a mechanism for the perturbation of the actin-myosin structural interactions by an HCM mutation in the LCD. Despite a lack of effect on the steady-state ATPase kinetics, actin-myosin binding affinity, or actomyosin structure in the absence of ATP, our structural dynamics-based approach reveals that the E56G mutation in hVELC affects the structure of the actin-myosin complex in the presence of ATP. The mutation increases the population in the S structural state (increasing the duty ratio), and changes the structure of the W state, so that it more closely resembles the S state. Future FRET and functional studies on other HCM mutations in hVELC are needed to test and refine this model (Fig. 7).
Immediately, however, the structural model in Fig. 7 suggests that TR-FRET can be a potential tool to 1) characterize other mutations that cause HCM and 2) screen for compounds that can rescue HCM-related effects. I.e., our FRET construct can serve as a biosensor, which can be a powerful tool for therapeutic discovery, coupled to our recently developed direct-waveform recording high-throughput fluorescence lifetime detection technology (57, 58).
Author Contributions
P.G. and D.D.T designed the research. P.G. performed most of the experiments. P.G., E.P., O.R., J.A.R., and D.D.T. analyzed the data and wrote the paper.
Acknowledgments
Fluorescence experiments were performed at the Biophysical Technology Center, University of Minnesota, directed by Dr. J. Michael Autry. We thank Dr. Joseph M. Muretta for comments on data interpretation, and Kurt C. Petersen for technical advice.
This study was supported by grants from the National Institutes of Health (NIH) to D.D.T. (AR032961 and AG26160).
Editor: David Warshaw.
Footnotes
Supporting Materials and Methods and eight figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30566-0.
Supporting Citations
References (59, 60, 61) appear in the Supporting Material.
Supporting Material
References
- 1.Thomas D.D., Muretta J.M., Kast D. Spectroscopic probes of muscle proteins. In: Egelman E.H., editor. Comprehensive Biophysics. Elsevier; Amsterdam, the Netherlands: 2012. pp. 226–250. [Google Scholar]
- 2.Rayment I., Rypniewski W.R., Holden H.M. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 3.Houdusse A., Cohen C. Structure of the regulatory domain of scallop myosin at 2 Å resolution: implications for regulation. Structure. 1996;4:21–32. doi: 10.1016/s0969-2126(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 4.Sweeney H.L., Bowman B.F., Stull J.T. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am. J. Physiol. 1993;264:C1085–C1095. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 5.Lowey S., Waller G.S., Trybus K.M. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature. 1993;365:454–456. doi: 10.1038/365454a0. [DOI] [PubMed] [Google Scholar]
- 6.VanBuren P., Waller G.S., Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc. Natl. Acad. Sci. USA. 1994;91:12403–12407. doi: 10.1073/pnas.91.26.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trybus K.M. Regulation of expressed truncated smooth muscle myosins. Role of the essential light chain and tail length. J. Biol. Chem. 1994;269:20819–20822. [PubMed] [Google Scholar]
- 8.Chen T.L., Kowalczyk P.A., Chisholm R.L. Targeted disruption of the Dictyostelium myosin essential light chain gene produces cells defective in cytokinesis and morphogenesis. J. Cell Sci. 1995;108:3207–3218. doi: 10.1242/jcs.108.10.3207. [DOI] [PubMed] [Google Scholar]
- 9.Frank G., Weeds A.G. The amino-acid sequence of the alkali light chains of rabbit skeletal-muscle myosin. Eur. J. Biochem. 1974;44:317–334. doi: 10.1111/j.1432-1033.1974.tb03489.x. [DOI] [PubMed] [Google Scholar]
- 10.Lowey S., Risby D. Light chains from fast and slow muscle myosins. Nature. 1971;234:81–85. doi: 10.1038/234081a0. [DOI] [PubMed] [Google Scholar]
- 11.Morano I., Ritter O., Michel G. Myosin light chain-actin interaction regulates cardiac contractility. Circ. Res. 1995;76:720–725. doi: 10.1161/01.res.76.5.720. [DOI] [PubMed] [Google Scholar]
- 12.Morano I. Tuning the human heart molecular motors by myosin light chains. J. Mol. Med. (Berl) 1999;77:544–555. doi: 10.1007/s001099900031. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez O.M., Jones M., Szczesna-Cordary D. Myosin essential light chain in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1643–H1654. doi: 10.1152/ajpheart.00931.2006. [DOI] [PubMed] [Google Scholar]
- 14.Sweeney H.L. Function of the N terminus of the myosin essential light chain of vertebrate striated muscle. Biophys J. 1995;68:112S–118S. discussion 118S–119S. [PMC free article] [PubMed] [Google Scholar]
- 15.Morano I., Haase H. Different actin affinities of human cardiac essential myosin light chain isoforms. FEBS Lett. 1997;408:71–74. doi: 10.1016/s0014-5793(97)00390-6. [DOI] [PubMed] [Google Scholar]
- 16.Morano M., Zacharzowski U., Morano I. Regulation of human heart contractility by essential myosin light chain isoforms. J. Clin. Invest. 1996;98:467–473. doi: 10.1172/JCI118813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramaraj R. Hypertrophic cardiomyopathy: etiology, diagnosis, and treatment. Cardiol. Rev. 2008;16:172–180. doi: 10.1097/CRD.0b013e318178e525. [DOI] [PubMed] [Google Scholar]
- 18.Refaat M.M., Fahed A.C., Nemer G. The muscle-bound heart. Card. Electrophysiol. Clin. 2016;8:223–231. doi: 10.1016/j.ccep.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arad M., Seidman J.G., Seidman C.E. Phenotypic diversity in hypertrophic cardiomyopathy. Hum. Mol. Genet. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 20.Marsiglia J.D., Pereira A.C. Hypertrophic cardiomyopathy: how do mutations lead to disease? Arq. Bras. Cardiol. 2014;102:295–304. doi: 10.5935/abc.20140022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonne G., Carrier L., Schwartz K. Familial hypertrophic cardiomyopathy: from mutations to functional defects. Circ. Res. 1998;83:580–593. doi: 10.1161/01.res.83.6.580. [DOI] [PubMed] [Google Scholar]
- 22.Huang W., Szczesna-Cordary D. Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 2015;36:433–445. doi: 10.1007/s10974-015-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timson D.J., Trayer H.R., Trayer I.P. The N-terminus of A1-type myosin essential light chains binds actin and modulates myosin motor function. Eur. J. Biochem. 1998;255:654–662. doi: 10.1046/j.1432-1327.1998.2550654.x. [DOI] [PubMed] [Google Scholar]
- 24.Guhathakurta P., Prochniewicz E., Thomas D.D. Amplitude of the actomyosin power stroke depends strongly on the isoform of the myosin essential light chain. Proc. Natl. Acad. Sci. USA. 2015;112:4660–4665. doi: 10.1073/pnas.1420101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prochniewicz E., Walseth T.F., Thomas D.D. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43:10642–10652. doi: 10.1021/bi049914e. [DOI] [PubMed] [Google Scholar]
- 26.Guhathakurta P., Prochniewicz E., Thomas D.D. Allosteric communication in Dictyostelium myosin II. J. Muscle Res. Cell Motil. 2012;33:305–312. doi: 10.1007/s10974-012-9304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowey S., Lesko L.M., Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α- or β-myosin heavy chain backbone. J. Biol. Chem. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohde J.A., Thomas D.D., Muretta J.M. Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc. Natl. Acad. Sci. USA. 2017;114:E1796–E1804. doi: 10.1073/pnas.1611698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agafonov R.V., Nesmelov Y.E., Thomas D.D. Muscle and nonmuscle myosins probed by a spin label at equivalent sites in the force-generating domain. Proc. Natl. Acad. Sci. USA. 2008;105:13397–13402. doi: 10.1073/pnas.0801342105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De La Cruz E.M., Ostap E.M. Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 2009;455:157–192. doi: 10.1016/S0076-6879(08)04206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muretta J.M., Kyrychenko A., Thomas D.D. High-performance time-resolved fluorescence by direct waveform recording. Rev. Sci. Instrum. 2010;81:103101. doi: 10.1063/1.3480647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J., James Z.M., Thomas D.D. Structural and functional dynamics of an integral membrane protein complex modulated by lipid headgroup charge. J. Mol. Biol. 2012;418:379–389. doi: 10.1016/j.jmb.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muretta J.M., Petersen K.J., Thomas D.D. Direct real-time detection of the actin-activated power stroke within the myosin catalytic domain. Proc. Natl. Acad. Sci. USA. 2013;110:7211–7216. doi: 10.1073/pnas.1222257110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor R.S., Weeds A.G. The magnesium-ion-dependent adenosine triphosphatase of bovine cardiac myosin and its subfragment-1. Biochem. J. 1976;159:301–315. doi: 10.1042/bj1590301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith S.J., Cusanovich M.A. Bovine cardiac myosin subfragment 1. Transient kinetics of ATP hydrolysis. J. Biol. Chem. 1984;259:9365–9368. [PubMed] [Google Scholar]
- 36.Baker J.E., Brust-Mascher I., Thomas D.D. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc. Natl. Acad. Sci. USA. 1998;95:2944–2949. doi: 10.1073/pnas.95.6.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spudich J.A. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deacon J.C., Bloemink M.J., Leinwand L.A. Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell. Mol. Life Sci. 2012;69:2261–2277. doi: 10.1007/s00018-012-0927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kazmierczak K., Paulino E.C., Szczesna-Cordary D. Discrete effects of A57G-myosin essential light chain mutation associated with familial hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H575–H589. doi: 10.1152/ajpheart.00107.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farman G.P., Muthu P., Moore J.R. Impact of familial hypertrophic cardiomyopathy-linked mutations in the NH2 terminus of the RLC on β-myosin cross-bridge mechanics. J. Appl. Physiol. 2014;117:1471–1477. doi: 10.1152/japplphysiol.00798.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spudich J.A., Aksel T., Ruppel K.M. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J. Exp. Biol. 2016;219:161–167. doi: 10.1242/jeb.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawana M., Sarkar S.S., Spudich J.A. Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci. Adv. 2017;3:e1601959. doi: 10.1126/sciadv.1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bloemink M., Deacon J., Geeves M.A. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin. J. Biol. Chem. 2014;289:5158–5167. doi: 10.1074/jbc.M113.511204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muthu P., Wang L., Szczesna-Cordary D. Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J. 2011;25:4394–4405. doi: 10.1096/fj.11-191973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mijailovich S.M., Nedic D., Geeves M.A. Modeling the actin.myosin ATPase cross-bridge cycle for skeletal and cardiac muscle myosin isoforms. Biophys. J. 2017;112:984–996. doi: 10.1016/j.bpj.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenfeld S.S., Sweeney H.L. A model of myosin V processivity. J. Biol. Chem. 2004;279:40100–40111. doi: 10.1074/jbc.M402583200. [DOI] [PubMed] [Google Scholar]
- 47.Geeves M.A. Dynamic interaction between actin and myosin subfragment 1 in the presence of ADP. Biochemistry. 1989;28:5864–5871. doi: 10.1021/bi00440a024. [DOI] [PubMed] [Google Scholar]
- 48.Thomas D.D., Ramachandran S., Ostap E.M. The mechanism of force generation in myosin: a disorder-to-order transition, coupled to internal structural changes. Biophys. J. 1995;68(4, Suppl):135S–141S. [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas D.D., Kast D., Korman V.L. Site-directed spectroscopic probes of actomyosin structural dynamics. Annu. Rev. Biophys. 2009;38:347–369. doi: 10.1146/annurev.biophys.35.040405.102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonow R.O. Left ventricular diastolic function in hypertrophic cardiomyopathy. Herz. 1991;16:13–21. [PubMed] [Google Scholar]
- 51.Spudich J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015;43:64–72. doi: 10.1042/BST20140324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adhikari A.S., Kooiker K.B., Ruppel K.M. Early-onset hypertrophic cardiomyopathy mutations significantly increase the velocity, force, and actin-activated ATPase activity of human β-cardiac myosin. Cell Rep. 2016;17:2857–2864. doi: 10.1016/j.celrep.2016.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee W., Hwang T.H., Park J.E. Different expressivity of a ventricular essential myosin light chain gene Ala57Gly mutation in familial hypertrophic cardiomyopathy. Am. Heart J. 2001;141:184–189. doi: 10.1067/mhj.2001.112487. [DOI] [PubMed] [Google Scholar]
- 54.Kazmierczak K., Yuan C.C., Szczesna-Cordary D. Remodeling of the heart in hypertrophy in animal models with myosin essential light chain mutations. Front. Physiol. 2014;5:353. doi: 10.3389/fphys.2014.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rayment I., Holden H.M., Milligan R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 56.Seebohm B., Matinmehr F., Kraft T. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys. J. 2009;97:806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gruber S.J., Cornea R.L., Thomas D.D. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 2014;19:215–222. doi: 10.1177/1087057113510740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muretta J.M., Rohde J.A., Thomas D.D. Direct real-time detection of the structural and biochemical events in the myosin power stroke. Proc. Natl. Acad. Sci. USA. 2015;112:14272–14277. doi: 10.1073/pnas.1514859112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kast D., Espinoza-Fonseca L.M., Thomas D.D. Phosphorylation-induced structural changes in smooth muscle myosin regulatory light chain. Proc. Natl. Acad. Sci. USA. 2010;107:8207–8212. doi: 10.1073/pnas.1001941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lakowicz J.R. Kluwer Academic/Plenum Press; New York: 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 61.Agafonov R.V., Negrashov I.V., Nesmelov Y.E. Structural dynamics of the myosin relay helix by time-resolved EPR and FRET. Proc. Natl. Acad. Sci. USA. 2009;106:21625–21630. doi: 10.1073/pnas.0909757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aydt E.M., Wolff G., Morano I. Molecular modeling of the myosin-S1(A1) isoform. J. Struct. Biol. 2007;159:158–163. doi: 10.1016/j.jsb.2007.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.