

Abstract
The unpredictability of actual physical, chemical, and biological experiments due to the multitude of environmental and procedural factors is well documented. What is systematically overlooked, however, is that computational biology algorithms are also affected by multiplicity of parameters and have no lesser volatility. The complexities of computation protocols and interpretation of outcomes is only a part of the challenge: There are also virtually no standardized and industry-accepted metadata schemas for reporting the computational objects that record the parameters used for computations together with the results of computations. Thus, it is often impossible to reproduce the results of a previously performed computation due to missing information on parameters, versions, arguments, conditions, and procedures of application launch. In this article we describe the concept of biocompute objects developed specifically to satisfy regulatory research needs for evaluation, validation, and verification of bioinformatics pipelines. We envision generalized versions of biocompute objects called biocompute templates that support a single class of analyses but can be adapted to meet unique needs. To make these templates widely usable, we outline a simple but powerful cross-platform implementation. We also discuss the reasoning and potential usability for such concept within the larger scientific community through the creation of a biocompute object database initially consisting of records relevant to the U.S. Food and Drug Administration. A biocompute object database record will be similar to a GenBank record in form; the difference being that instead of describing a sequence, the biocompute record will include information related to parameters, dependencies, usage, and other information related to specific computational instance. This mechanism will extend similar efforts and also serve as a collaborative ground to ensure interoperability between different platforms, industries, scientists, regulators, and other stakeholders interested in biocomputing.
Keywords: Biocompute object, NGS standardization, Computation reproducibility, Regulatory research, FDA
Background
The 20th and 21st centuries have witnessed many collaborative efforts resulting in such technological marvels as the Internet, microchips, telecommunications and wireless communications, Hadron particle collider, and space travel, among many others. These advances would have been impossible without the related scientific communities' willingness to standardize and harmonize their technologies. We hope that next-generation sequencing (NGS) (also known as high-throughput sequencing (HTS)), and standardization and bioinformatics harmonization efforts will yield another success story: a collaborative ground for ensuring future interoperability between platforms, industries, scientists, regulators, and developers.
NGS technology is still in the scientific research and innovation domain of its lifecycle, facing many challenges to its confident and appropriate use. Procedural differences in computational techniques and subject datasets can result in unreproducible results or false discoveries where computational artifacts are interpreted as scientific facts. For example, analysis tool choices and parameter settings can profoundly affect results from two common HTS analyses: transcriptome assembly and quantification [1] and somatic mutations identification [2]. Unless we establish highly scrutinized and consistent protocols for regulatory analytics, our conclusions will always be affected by the false impression of reliability. The need is urgent for validation procedures and computational protocols for results that will be used in regulatory sciences with a high health impact, and some progress is being made in this regard through the High-performance Integrated Virtual Environment (HIVE) project [3,4].
Regulatory scientists have already started receiving the front wave of HTS technology application reviews ranging across a wide spectrum of bioinformatics applications, including disease diagnosis, food safety, and infectious diseases. Although, it is difficult to regulate an industry still in its infancy, the promise of this particular technology and the immense potential of its applications are greatly accelerating the need for its regulation. Computation validation protocols used for analysis of large data are critical in the scientific conclusion-making process because the complexity and size of computation results can easily overwhelm regulatory and research scientists, rendering them unable to detect errors or validate inconsistencies by other means.
Significant amount of work has been done in sharing of workflows and experiments. The myExperiment project provides a virtual research environment for social depositing and sharing of bioinformatics workflows [5]. The concept of research objects has been explored within this context. It has been proposed that workflow-centric research objects with executable components can be considered as computational research objects [6,7]. Workflow creation and management software such as Taverna [8] and others [9] allows researchers to access different tools and resources and create complex analysis pipelines, and projects such as Open Science Framework, BioDBcore, and BioSharing provides the logical and conceptual backbone for reproducible science (https://osf.io/cdi38/) (https://biosharing.org/) [10,11].
However, workflows are often very generic, and they cannot and should not be validated for all possible inputs and parameters. Instead, a workflow should be validated with a particular usage pattern, such as a limited set of input types and parameter settings. Running the workflow with a different usage pattern would fall outside the scope of validation and may produce valid or invalid results. To validate a workflow, then, one needs to strictly constrain applicability and parametric limits. An instance of a workflow executed at a particular point in time and space, applied to known subjects with known set of parameters using known experiment or research object or a pipeline, can be verified or invalidated through rigorous examination and made into valid biocompute object.
There exists an analogy between actual physical, chemical, or biological experiment and in-silico biocomputation; in this context we use the term generalized scientific experiment for both: bench experiments conducted at a wet lab, and computational experiments conducted either by computer software or through scientists' analytic derivations. Many judgments described in this concept article are applicable, not just to computations but to experiments in general.
To describe validation protocols in a generalized manner, we represent an experiment as a black box as shown in Figure 1 with certain predefined inputs, predetermined outputs types, and distinct parameters that control the experimental flow. For example, in the wet lab case of the black-box chemical experiment, the actual chemical reagents play the role of an input, yielded chemical compounds play the role of the results, and the conditions governing how the experiment is executed play the role of the parameters. Similarly, in a computational experiment aligning short high-throughput DNA sequencing reads, the alignment algorithm is the black box, the sequence files of the genome and sequence reads are the input, the alignment file that maps reads to genome is the result, and the set of algorithmic control arguments are the parameters driving the alignment procedure.
Figure 1.
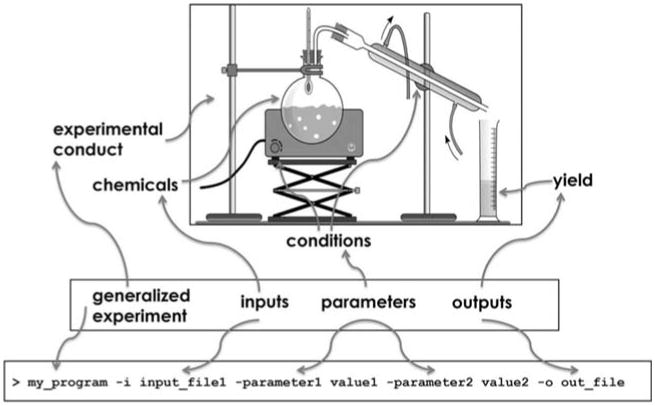
An experiment can be viewed as a black box that takes specific inputs under an established set or range of conditions and has predetermined outputs that are generated. A sample program command line is shown with generic terms covering the input, parameters, and output for the computation to emphasize the analogy of generalized experiment concept. Note: The image has been adapted from Pixabay released under Creative Commons CC0. No attribution or permission is required (https://pixabay.com/en/chemistry-distillationexperiment-161575/).
Well-designed and customizable experimental procedures are capable of producing different results for the same inputs depending on the given set of parameters. For validation, however, an instance of an experiment is complete if and only if the complete set of parameterization is well defined in the form of a protocol. This introduces a distinction between an experimental method, experimental protocol, and an actual experimental instance.
Figure 2 provides a visualization of an experimental procedure. Scientifically, the most fundamental aspect is an experimental method shown in the center of the illustration. Next is the experimental protocol, which includes details about the experimental method applicable in certain conditions within specific parameter/condition settings for running the experiment. Most specific is the experimental instance, which includes not only the parameter values of the experimental protocol and the general details of the experimental method, but also includes specific input files. A general experimental method might be an algorithm of an aligner such as BLAST [12] that is proven to produce mappings between subject and query sequences that meet specific algorithmic criteria. Expanding on this, an experimental protocol would be a BLAST analysis pipeline with certain parameters defined (i.e., gap penalty, etc.) in order to use the underlying algorithm for detecting viruses, bacteria, or human sequences. Finally, an experimental instance would be running a BLAST algorithm with a predefined set of parameters on a specific data at a specific point in time and place.
Figure 2.
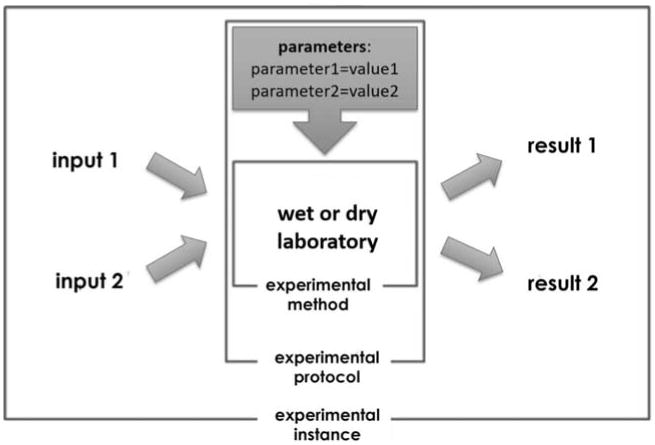
Visualization of an experimental procedure. The results are dependent on input, parameters and experimental methods, protocol, and instance.
Experimental Method
An experimental method is defined as application technique of underlying scientific theory to real-life objects with an expectation of gaining a particular result. To adhere to the conventional scientific methodology, an experimental method can be considered valid if it satisfies the following criteria:
Objectivity: observations made by one scientist should be consistent with those made by others following the same method.
Reproduction: the ability to conduct the protocol multiple times, each time returning the expected results within the acceptable and estimated range of errors and within the known limits of accuracy.
Deduction: a theoretical foundation explaining scientific facts and observations within an experimental method and accepted by the scientific community.
Prediction: observations that must conform to inductive theoretical hypotheses, allowing extraction of predictive knowledge about the experimental subjects under the assumption(s) of the applied theoretical model.
Experimental Protocol
Experimental protocol is defined as the specific set of procedures and conditions/parameters necessary to conduct an experiment within the limits of usability and scientific accuracy. Figure 3 displays the design of an experimental protocol that includes
Figure 3.
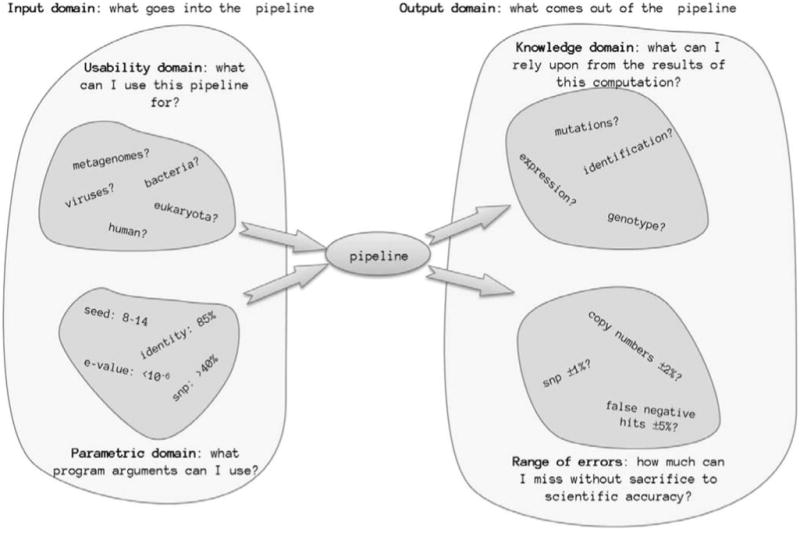
Illustration of an experimental protocol that will have a specific set of input and output domains.
Usability domain/domain of inputs: a set of inputs for which the protocol can be used and can produce scientifically valid outcomes within permissible error rates.
Parametric space: a set of parameters or conditions that are acceptable for the given experimental method to produce scientifically accurate results.
Knowledge domain/domain of outputs: a set of scientific results and/or conclusions extracted from observation of conducted experiments using experiment protocols; a particular protocol may reliably produce some scientific facts while unfit to produce others with requisite level of trustworthiness depending on domain of inputs and parametric space.
Range of errors: the accepted range of deviations from theoretically expected outcomes while maintaining the integrity of scientific methods employed in the protocol.
Figure 3 shows that an experimental protocol will have a specific set of input and output domains as illustrated. The input domains define what an analysis pipeline can validly accept, while the output domain defines what can be extracted from a pipeline's output. This covers both the range of what can be fed into the pipeline as well as what can be interpreted from the results.
Thus, an experimental protocol is the procedural basis of the experimentation process. It is important to recognize that a single experiment can have multiple usability domains and parametric subspaces where validity can be confirmed independently. The simple example shown in Figure 4 demonstrates the variable validity of a single pipeline with respect to distinct usability domains. For instance, short-read aligners are practical and mostly accurate for divergent viruses and bacteria with smaller k-mer seed size and large mismatch rates. However, the same arguments might be not practical when applied to large eukaryotic subjects like humans where heuristic-alignment algorithms cannot exhaust all potential alignment locations. Instead, for human subjects, the same programs are useful when considering much larger seed sizes and allowing significantly smaller mismatch rates.
Figure 4.
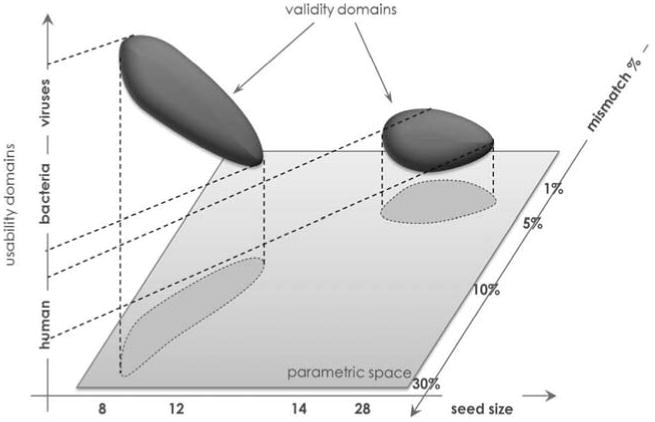
Illustration of validity domains of a pipeline in three dimensions. The parametric space (3 axis) in this case is expansive, but only a small subset represented by the dark blue shapes are valid in the pipeline. Parameters that fall outside of these areas are not within the scope of the pipeline and not guaranteed to result in usable outputs.
Unlike the image, however, real-world scenarios involve much larger parametric spaces and can have as many dimensions as potential arguments available for customization in the bioinformatics application.
Figure 4 illustrates the validity domains of a pipeline in three dimensions. The parametric space in this case is expansive, but only small subsets represented by the dark blue shapes are valid in the pipeline. Parameters that fall outside these areas are not within the scope of the pipeline validity and cannot guarantee validation of results as scientifically meritable. With complex computations using heuristic algorithmic methods, sometimes the operating system and the execution platform can affect outcomes as much as data and the parameters. When a validation question is raised on platform-dependent bioinformatics, there should be a clear measure of minimal system requirements for which the application is expected to perform accurately. A platform's requirement specifications should be reasonably justifiable, and an attempt should be made to avoid narrowly defined, heavily customized, specific computing platforms with unique requirements.
Experimental Instance
Experimental instance is defined as the actual physical execution of an experiment at a given time and location where the protocol is applied to a specific set of inputs within both its usability domain and parametric space. The resulting outcomes are then used as observations from within the knowledge domain with accepted error rates.
Biocompute Object (Experimental Instance)
To address traceability and reproducibility issues of the bioinformatics protocols, it is proposed (and implemented in the HIVE platform) [4] to record all values from parametric space of a validated bioinformatics application in the form of a biocompute metadata record that can be stored in a database. Such records store all arguments of the underlying software application together with the version information of the executable program used to run the bioinformatics computations. If a particular pipeline is validated to run with an exact set of arguments, strict constraints can be used to limit the computation scope to a single point in parametric space. If a pipeline is validated for a range of different values for some arguments, a set of flexible constraints can be used to limit the volume of accessible parametric space. The popular Galaxy platform (http://galaxyproject.org) [13] has implemented a concept similar to biocompute objects in analysis histories. Galaxy histories maintain a complete record of each analysis tool run in a multi-step analysis, including its version, inputs, parameter settings, and outputs. All of this information is stored in Galaxy's internal database. A history downloaded from a Galaxy server includes all analysis information in a simple text file as well as all input and output datasets in the history. A downloaded history can be stored for archival purposes or uploaded to any Galaxy server. When a history is uploaded to a server, it is recreated in its entirety with all analysis details automatically populated in the Galaxy database. Galaxy also has limited forms of validation during tool and workflow execution.
Having validated biocompute types in a computation universe would be equivalent to having standardized data types in the data universe. Correspondingly, just like databases that contain metadata records, one could create (and such exist in the HIVE platform) [3,4]) a database of biocompute objects containing the set of uniquely identifiable computation instances. References to these may then be used in publications and for review processes in the same manner as for data: genomes, reads, etc. In Galaxy, Pages—interactive Web-based research supplements with embedded datasets, histories, workflows, and visualizations—enable reviewers and readers to inspect and reproduce analyses performed in a paper [14–17].
Biocompute objects can be used in two distinct scenarios: federated and integrated systems. In the scenario where computation is conducted in a high-performance computational environment such as HIVE, the input data are already integrated into the execution environment and addressable with unique internal references (identifiers) and, as such, biocompute objects do not need to include a copy of input or output data to be complete (self-sufficient) and provide a level of reproducibility. In federated environments, such as in the Galaxy ecosystem with many public (http://bit.ly/gxyservers) and private servers where input data or outputs are detached in either location or time, there is a need to specify concise and uniquely identifiable external references or include a copy of input and output data in order to provide comprehensive provenance to computation and become complete. Whatever a particular requirement, a biocompute object is valid only if it is complete.
Different experimental instances may have distinct types of inputs and outputs. One can therefore assemble multiple experiments into complex experimental pipelines by joining appropriate nodes of internal inputs and outputs into a workflow graph. From the validation perspective, however, a complex experimental pipeline is still an experimental instance where the collection of all external inputs, produced results, and parameters play similar roles. Thus, reference to bioinformatics applications are extended to all potential components or combinations thereof: algorithms, standalone tools, integrated applications, pipelines, and workflows.
Biocompute Template (Experimental Protocol) and Template Library
It is conceivable to use well-identified, characterized, and validated biocompute objects as a basis for other computations with the same protocol and alter only a few of the conceivable elements of parametric and input spaces. This process is called templating and allows modification of all parameters or only a limited subset of an existing biocompute object. Such templated biocompute objects turn into re-useable constructs for computational protocols. Templated objects can then be transformed into analysis pipelines or workflows and used for batch execution of computations with different inputs in a series of experiments to provide analytical consistency in large-scale studies.
To ensure that biocompute templates are widely usable, tools that use these templates could be implemented within Docker (https://www.docker.com/) software containers. Docker containers make it simple to run software across a wide variety of computing platforms, including different flavors of Linux, Mac OS, and cloud computing frameworks. With a biocompute template in a Docker container, using the template becomes as simple as downloading its container and filling in the template for a particular analysis. This simplicity is maintained no matter how complex the analysis and how many software tools it uses. Because the container would work across platforms, the same template could be used on a local workstation to analyze one sample and then used again in the cloud to analyze thousands of samples.
Validation Schema
Validation of a scientific experimental method extends outside the scope of this perspective: It is generally assumed that the peer review–publication process serves as a reliable tool to determine a method's scientific validity. In addition, benchmark datasets such as those from the Genome in a Bottle consortium are simplifying scientific validation [18] (http://jimb.stanford.edu/giab/). Here we are talking about the concept of computational authentication of experimental protocols from a biocompute object viewpoint. More precisely, testing an experimental protocol as applied to regulatory bioinformatics is the subject of this discussion. A well-defined validation schema for an experimental protocol needs to include the test sets of inputs, parameterizations, expected results, and limits of allowed errors. The actual validation happens by comparing the expected with actual results and accepting or disqualifying the protocol based on their similarity within the acceptable range of errors. Creating and maintaining certified test and validation kits for each type of experimental protocol facilitates the ability to test the scientific accuracy of a method at any given time. Thus, in order to validate a protocol, one must provide enough information to satisfy the experimental method criteria (discussed above) and determine the experimental protocol design characteristics (above) using one of the standardized biocomputation metadata objects appropriate to the particular experiment.
With respect to inputs for the testing protocol, one may use data generated from actual well-characterized biological specimens or may use a simulated, artificially synthesized dataset that mimics a particular behavior of a biological system.
However, use of an experimentally generated data that is not well characterized as a test input might result not only in false validation, but also the false perspective of what a “validated” bioinformatics pipeline, to which future tools would be compared, should be. It is, therefore, important to ensure only the highest quality test input data are being used for this goal.
It has been argued that mathematical validity of underlying applications is better tested using simulated test data. Robustness, however, is better validated with actual biological data where the unexpected variability of input data may be a better filter for imperfections in computational approaches.
Based on the discussion above, the outlined procedure could be followed when validating scientific merit and interpretation unambiguity of biocompute objects:
Provide references to publications where underlying scientific method is discussed.
Describe experimental protocol by clearly defining usability domain, parametric space, knowledge domain, error rates (if applicable), prerequisite datasets, and minimal requirements for an execution platform.
Generate or synthesize in-silico well-characterized input test sets.
Execute application and accumulate results.
Analyze results in detail to ensure outputs' validity.
Create and register a biocompute metadata record.
Save all valid outcomes associated with the biocompute object.
Template a biocompute object for further uses.
Ensure availability of dependencies, such as software versions and databases.
The validation can proceed as follows:
A mechanism similar to Bankit can be set up for people to enter biocompute objects (https://submit.ncbi.nlm.nih.gov/subs/genbank/SUB1125119/submitter/) (it is possible to go one step further and enable direct submission from HIVE and/or Galaxy or other platforms).
Submitters will submit information for generating a flat/xml or other format file (JSON, YAML, etc.) for human or machine reading.
This database will have two sections: a validated and reviewed section of biocompute objects and an un-reviewed and/or partially validated section. The validation can proceed through automatic and/or semi-automatic methods and can be either crowd-sourced or through dedicated curation.
Initially, authors propose to create biocompute objects for pipelines most appropriate for FDA HTS regulatory projects and research projects supporting scientific evaluation of regulated medical products. Efforts such as Common Workflow Language (CWL) (http://www.commonwl.org/) and Workflow Definition Language (WDL) (https://software.broadinstitute.org/wdl/) that provides portability of data analysis workflows can be adopted or adapted to provide workflow and biocompute templates that are of interest to the FDA but are currently being validated by other communities. It is important to note that biocompute objects have an important unique feature that differentiate them from CWL/WDL: Biocompute objects are concrete instantiations of a workflow with results in a database (see below). CWL and WDL do not explicitly have a notion of computational instances, archiving workflow templates and storing validated results in a database. It is possible that workflows available in myExperiment [5] can form the basis for the creation of new biocompute objects.
Biocompute Database
Biocompute object runs and biocompute template library elements can be submitted and stored in a database after going through a validation procedure as described in earlier sections. All entries (objects) can be versioned, and old objects can be archived. Mechanisms to identify duplicate objects and to refer to other objects within the database can be inherent in the database entry format. Users can browse, search, download via a web-browser, or programmatically retrieve individual entries or the entire database and use the objects to run computations within their software platforms.
Users will be able to upload or type in biocompute objects that have metadata (information not needed for the computation), information on workflow, pipeline with all internal arguments, parametric domain instance, input domain, and output domain (Figure 5).
Figure 5.
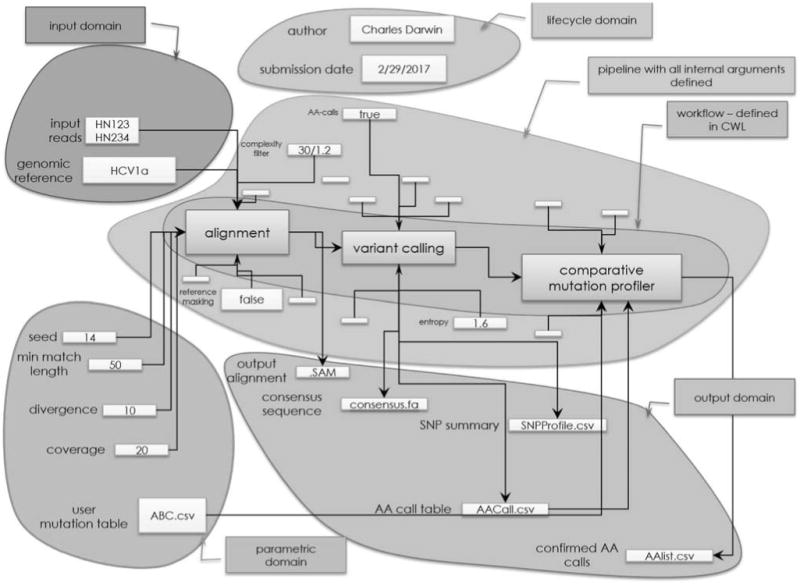
A visual description of the biocompute object that a user would create and upload into the Biocompute Database. The figure provides a view of how a biocompute object encapsulates workflow, pipeline, parametric domain instance, input domain, and output domain.
Utility and Evolving Perspective
The utility of the biocompute object database is manifold. The biocompute database will have entries describing computations for targeted purposes. For example, a biocompute object submitted by a user could capture the details about software version and parameters used to identify a foodborne pathogen in an instance of an outbreak from fresh spinach. After such biocompute object validation and template creation it can be re-applied to analyze future outbreaks in spinach and other similar food items.
Analogous to a GenBank record (which has information related to a specific sequence), this object can be made into human-readable text, xml, or any other format. Further, it can be used programmatically, can be browsed, searched, compared, versioned, and so on for a variety of purposes. Submission of such biocompute objects can be made as easy as a click of a button from Galaxy [20], HIVE [4], or other workflow-centric platforms and resources.
The usability of biocompute objects can be extended far beyond the HTS perspective and offer a mechanism for improving the reliability of treatments based on biomedical computations for patient-centric outcomes in a clinical setting. And it has critical importance for regulatory organizations such as the FDA, where the validity of the underlying computational algorithm can affect decisions on cancer diagnostics, vaccine and biosimilar safety, tissue and blood analysis, adverse event and outbreak analysis, and so forth.
Several evolving and ongoing efforts are geared towards standardization of analysis methods, results interpretation, and data sharing [21,22] (https://biocaddie.org, https://github.com/common-workflow-language/commonworkflow-language, ga4gh.org, https://pebourne.word press.com/2014/10/07/the-commons/). Such evolution of the methodology and workflows should be reflected in novel validation schemas. All changes to implicated validation protocols should be reassessed from the perspective of inductivity within the new framework of understanding at the time such a change occurs.
Acknowledgments
We would like to mention with gratitude the following people for providing valuable comments regarding research and regulatory aspects to HTS: Jean et Danielle Thierry-Mieg, Konstantin Chumakov, Carolyn Wilson, Danica Marinac-Dabic, Khaled Bouri, Mark Walderhaug, Charles Hadley King, Robel Kahsay, Amanda Bell, and Benoit Mandelbrot. This work has been funded in part by FDA (HHSF223201510129C).
Footnotes
Disclaimer: The contributions of the authors are an informal communication and represent their own views. These comments do not bind or obligate the FDA or George Washington University.
Competing Interests: The authors declare that they have no competing interests.
Authors' Contributions: VS and RM initiated the discussions that led to this paper. VS, JG, and RM contributed to the writing of the manuscript. All authors read and approved the final manuscript.
References
- 1.Goecks J, Coraor N, Nekrutenko A, Taylor J. NGS Analyses by Visualization with Trackster. Nat Biotechnol. 2012;30(11):1036–1039. doi: 10.1038/nbt.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alioto TS, Derdak S, Beck TA, Boutros PC, Bower L, et al. A Comprehensive Assessment of Somatic Mutation Calling in Cancer Genomes. bioRxiv. 2014:012997. [Google Scholar]
- 3.Wilson CA, Simonyan V. FDA's Activities Supporting Regulatory Application of “Next Gen” Sequencing Technologies. PDA J Pharm Sci Technol. 2014;68(6):626–630. doi: 10.5731/pdajpst.2014.01024. [DOI] [PubMed] [Google Scholar]
- 4.Simonyan V, Mazumder R. High-Performance Integrated Virtual Environment (HIVE) Tools and Applications for Big Data Analysis. Genes (Basel) 2014;5(4):957–981. doi: 10.3390/genes5040957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goble CA, Bhagat J, Aleksejevs S, Cruickshank D, Michaelides D, et al. myExperiment: A Repository and Social Network for the Sharing of Bioinformatics Workflows. Nucleic Acids Res. 2010;38(Suppl 2):W677–W682. doi: 10.1093/nar/gkq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roure D. Towards Computational Research Objects. DPRMA ′13 Proceedings of the 1st International Workshop on Digital Preservation of Research Methods and Artefacts; New York, NY: ACM; 2013. pp. 16–19. [Google Scholar]
- 7.Hettne KM, Dharuri H, Zhao J, Wolstencroft K, Belhajjame K, et al. Structuring Research Methods and Data with the Research Object Model: Genomics Workflows as a Case Study. J Biomed Semantics. 2014;5(1):41. doi: 10.1186/2041-1480-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolstencroft K, Haines R, Fellows D, Williams A, Withers D, et al. The Taverna Workflow Suite: Designing and Executing Workflows of Web Services on the Desktop, Web or in the Cloud. Nucleic Acids Res. 2013;41:W557–W561. doi: 10.1093/nar/gkt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiwari A, Sekhar AK. Workflow Based Framework for Life Science Informatics. Comput Biol Chem. 2007;31(5):305–319. doi: 10.1016/j.compbiolchem.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Destro Bisol G, Anagnostou P, Capocasa M, Bencivelli S, Cerroni A, et al. Perspectives on Open Science and Scientific Data Sharing: An Interdisciplinary Workshop. J Anthropol Sci. 2014;92:179–200. doi: 10.4436/JASS.92006. [DOI] [PubMed] [Google Scholar]
- 11.Gaudet P, Bairoch A, Field D, Sansone SA, Taylor C, et al. Towards BioDBcore: A Community-defined Information Specification for Biological Databases. Database (Oxford) 2011:baq027. doi: 10.1093/database/baq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 13.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch VJ, Bedoya-Reina OC, Ratan A, Sulak M, Drautz-Moses DI, et al. Elephantid Genomes Reveal the Molecular Bases of Woolly Mammoth Adaptations to the Arctic. Cell Rep. 2015;12(2):217–228. doi: 10.1016/j.celrep.2015.06.027. [DOI] [PubMed] [Google Scholar]
- 15.Heydarian M, Luperchio TR, Cutler J, Mitchell CJ, Kim MS, et al. Prediction of Gene Activity in Early B Cell Development Based on an Integrative Multi-Omics Analysis. J Proteomics Bioinform. 2014;7:50–63. doi: 10.4172/jpb.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pond SK, Wadhawan S, Chiaromonte F, Ananda G, Chung WY, et al. Windshield Splatter Analysis with the Galaxy Metagenomic Pipeline. Genome Res. 2009;19(11):2144–2153. doi: 10.1101/gr.094508.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, et al. Polar and Brown Bear Genomes Reveal Ancient Admixture and Demographic Footprints of Past Climate Change. Proc Natl Acad Sci U S A. 2012;109(36):E2382–E2390. doi: 10.1073/pnas.1210506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zook JM, Catoe D, McDaniel J, Vang L, Spies N, et al. Extensive Sequencing of Seven Human Genomes To Characterize Benchmark Reference Materials. Sci Data. 2016;3:160025. doi: 10.1038/sdata.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2007;35:D21–D25. doi: 10.1093/nar/gkl986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goecks J, Nekrutenko A, Taylor J. Galaxy: A Comprehensive Approach for Supporting Accessible, Reproducible, and Transparent Computational Research in the Life Sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terry SF. The Global Alliance for Genomics & Health. Genet Test Mol Biomarkers. 2014;18(6):375–376. doi: 10.1089/gtmb.2014.1555. [DOI] [PubMed] [Google Scholar]
- 22.Brownstein CA, Beggs AH, Homer N, Merriman B, Yu TW, et al. An International Effort towards Developing Standards for Best Practices in Analysis, Interpretation and Reporting of Clinical Genome Sequencing Results in the CLARITY Challenge. Genome Biol. 2014;15(1):R53. doi: 10.1186/gb-2014-15-3-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]