

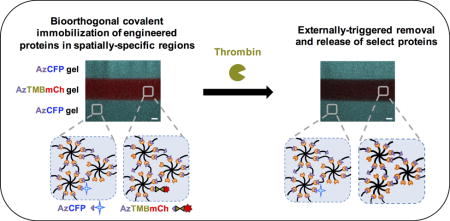
Graphical abstract
1. Introduction
Synthetic hydrogels are excellent modular platforms for use as sensors [1], drug delivery vehicles [2], and biomimetic scaffolds for tissue engineering applications [3,4]. Synthetic polymers such as poly(ethylene glycol) (PEG) are easily functionalized with a variety of reactive functionalities to provide a bioinert base for the creation of well-defined network architectures with high water content and controlled physical and biochemical properties [5]. In these materials, the incorporation and removal of biomolecules, such as proteins, is key for directing interactions with biological systems, including mammalian cells in culture and in the body for modulating their attachment, function, and fate [3]. A simple approach for protein incorporation into hydrogels is encapsulation and entrapment of full-length proteins; while effective for therapeutic delivery applications, use in cell culture typically is limited to only large proteins (e.g., whole laminins, fibronectin) that can be retained in select hydrogel compositions [6]. For more precise control of protein presentation from and within hydrogels, covalent modification of proteins can be broadly useful.
A variety of approaches with different levels of complexity have been developed for protein immobilization. These include i) orthogonal physical binding pairs (e.g., barnase-barstart and streptavidin-biotin) [7], ii) oxime ligation [8], and iii) thiol-ene reaction [9], where photo-uncaging or photo-initiating chemistries have been applied for spatial specificity of immobilization. While useful, these methods often rely on chemical modification of proteins by reacting with randomly-located amine or thiol groups on the protein backbone. Consequently, these modifications typically are not site-specific and the number of modifications on each protein cannot be easily controlled, which may cause potential undesired side reactions such as disulfide bond formation between proteins. In addition to these undesired side reactions, the modification can occur at an active site of the protein, with the potential to cause protein misfolding or dysfunction.
A few methods have emerged for enzyme-mediated covalent coupling of engineered proteins and protein fragments to pre-formed hydrogels with sortase A [10] and transglutaminase factor XIIIa (FXIIIa) [11], respectively. Additionally, single lysines have been incorporated within protein sequences designed for assembly to enable the subsequent introduction of reactive functionalities and covalent modification for crosslinking [12]. These approaches demonstrate the power of genetic modification for maintaining protein bioactivity during covalent coupling and immobilization. However, challenges remain in diffusion of enzymes and proteins into gels for homogenous reaction and in site-specific protein modification with reactive unnatural amino acids (UAAs) for maximizing coupling efficiency. Versatile methods that enable site-specific modification of proteins and bioorthogonal conjugation and immobilization are needed for the design of biomimetic materials with well-defined cell-matrix interactions.
The ability to control immobilization and removal of proteins from the hydrogel is of growing interest for a number of biological applications [8]. In the human body, gradients of soluble proteins, such as morphogens, growth factors, and cytokines, occurring over hundreds of micrometers at specific times are known to drive tissue morphogenesis in development and regeneration in wound healing [13,14]. For example, gradients of chemokines and matrix stiffness drive stem cell migration and condensation during cartilage development, ultimately leading to a shift from a fibronectin-rich to collagen-rich environment that facilitates stem cell differentiation and tissue development [15,16]. Materials that enable the release of proteins in time, as well as an anisotropic manner, are needed for studying and directing these processes within culture systems and implanted devices. One method to achieve this is through the cleavage and subsequent release of covalently-linked proteins. Cleavage of tethered proteins from hydrogel-based matrices has been accomplished through incorporation of photolabile groups [17,18] or protease cleavage sites [19]. For example, incorporation of an o-nitrobenzyl-ether group has been used to spatiotemporally control cleavage of integrin-binding proteins upon irradiation for directing MSC differentiation before and after protein cleavage [17].
To better mimic anisotropic, multipart tissues, patterned and more complex structures are required, such as intricate hydrogels containing multiple layers with different biochemical cues, various mechanical properties [20], or multiple cell types [21]. For example, to mimic the structure of cartilage which is composed of different zones, a multi-layer hydrogel composite was formed with different mechanical and biochemical properties within each layer to promote zone-specific chondrogenic differentiation of MSCs [20]. Additionally, co-culture of neural progenitor cells, astrocytes, and neurons each within a different layer of a multi-layer hydrogel accelerated neural migration and differentiation [21]. Methods that combine cleavage strategies with such layering techniques would provide a straightforward approach to controlling the presentation of proteins at times and locations of interest and facilitate bottom up design over multiple size scales, from the molecular to micro- and macro-scales.
Genetic engineering is a powerful tool for introducing unique and site-specific modifications to proteins that enable orthogonal coupling and cleavage reactions. Site-specific incorporation of UAAs for functionalizing proteins can be achieved using nonsense codon replacement [22–25]. Unlike sense codon replacement [26,27], nonsense codon replacement allows for the site-specific placement of UAAs in each protein molecule using either a stop codon or 4-base codon as the designator for the UAA. This strategy for placement of the UAA also minimizes protein dysfunction when, by design, the conjugation site is away from the active site of the protein. Furthermore, UAAs can be strategically placed alongside specific domains, such as protease cleavage sites, giving the benefits of unparalleled site-specific chemical conjugation and a specific biological release mechanism for the construction of well-defined and dynamic biomaterials.
Here, we report a modular approach for the controlled incorporation and removal of proteins in hydrogels using a combination of bio-orthogonal click chemistry and protease-responsive engineered proteins within defined geometries. Multi-arm PEG was functionalized with dibenzocyclooctyne (DBCO), which undergoes biocompatible and biorthogonal strain promoted alkyne-azide cycloaddition (SPAAC) click chemistry [28,29] with a di-azide functionalized PEG (PEG-2-Az) crosslinker for gel formation. The UAA p-azido-l-phenylalanine (pAzF), which provides azide functionality, was site-specifically incorporated into a cyan fluorescent protein (CFP), a mCherry fluorescent protein (mCh), and mCh decorated with a thrombin cut-site (AzTMBmCh). Gels containing layers with different protein compositions were created using a layering methodology, and in combination with the engineered proteins, permitted spatially-defined protein presentation initially and in time upon thrombin application. Immobilization of full-length proteins within hydrogels followed by controlled temporal enzymatic cleavage in defined regions is promising for use in a number of biological applications, including controlled cell culture, tissue engineering, diagnostics, and therapeutic delivery.
2. Materials and Methods
Eight-arm amine-functionalized poly(ethylene glycol) (PEG-8-NH2, 40 kDa) was purchased from JenKem Technology (Beijing, China). Bovine Serum Albumin (BSA) and dibenzocyclooctyne-acid (DBCO-Acid) were purchased from Sigma Aldrich (St. Louis, MO). HATU was purchased from ChemPep (Wellington, FL). N-N-Dimethylformamide (DMF, 99.8% anhydrous) was purchased from Acros Organics. PEG-bis-azide (PEG-2-Az, 3.4 kDa) was purchased from Creative PEGWorks. Dulbecco’s Phosphate Buffered Saline (DPBS, 1X) was purchased from Thermo Fisher Scientific (Grand Island, US). All primers were purchased from IDT (Coralville, IA). 4-Methylmorpholine (4-MMP), ethyl ether, and all reagents for culturing media were purchased from Fisher Scientific (Pittsburgh, PA). All reagents for SDS-PAGE were purchased from BIO-RAD (Hercules, CA). All enzymes related to DNA manipulation and cloning were purchased from New England Biolabs (Ipswich, MA). p-Azido-l-phenylalanine (4-azido-l-phenylalanine, ≥ 98% (HPLC)) was purchased from Chem-Impex International Inc. (Wood Dale, IL). All reagents purchased and received commercially were used without additional purification.
2.1 Hydrogel components preparation
2.1.1 Cloning and Strains
Escherichia coli NEB5α (New England Biolabs, Ipswich, MA) [fhuA2 Δ(argF-lacZ)U169 phoA 23 glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17] was used as the host for plasmid construction and maintenance. E. coli strain BL21(DE3) (EMD Millipore, Madison, WI) [F- ompT hsdSB(rB- mB-) gal dcm (DE3) Δ(srl-recA)306::Tn10 (TetR)] was used for the production of all proteins. NEB5α grown in Luria-Bertani Broth (LB, 10.0 g/L tryptone, 5.0 g/L yeast extract, 5.0 sodium chloride) supplemented with either 50 μg/mL kanamycin or 100 μg/mL ampicillin during cloning. Co-transformants were grown in LB media supplemented with 100 μg/mL ampicillin and 50 μg/mL spectinomycin.
pET24a-w3CFP-his6, which bears the gene that encodes wild-type (WT) enhanced cyan fluorescent protein (ECFP), was cloned previously [26]. The w3CFP gene was inserted into pET22b to yield an ampicillin resistant plasmid bearing the same gene. The resulting plasmid pET22b-w3CFP-his6 served as the vector of choice for the UAA bearing proteins.
Gene fragment encoding ECFP was amplified via PCR using primers AzCFP-F and AzCFP-R (Table S1), through which an amber codon and NheI cut sites were genetically fused upstream and inframe to the ECFP gene fragment. The PCR amplicon was inserted into pET22b-w3CFP-his6 using SacII and XhoI sites to yield pET22b-w3-amber-CFP-his6. The plasmid was transformed into NEB5α and positive clones were subsequently co-transformed with pULTRA-CNF (a generous gift from Prof. Peter G. Schultz, [21]) for expression.
Gene fragment encoding mCherry was amplified via PCR using primers AzmCh-F and AzmCh-R (Table S1). The PCR product was then inserted into pET22b-w3-amber-CFP-his6 using NheI and XhoI sites to yield pET22b-w3-amber-mCherry-his6. Similarly, the thrombin cleavage site (Leu-Val-Pro-Arg-Gly-Ser) was incorporated via PCR using primers AzTMBmCh-F and AzmCh-R. The resulting PCR product was inserted into pET22b-w3-amber-CFP-his6 using NheI and XhoI to yield pET22b-w3-amber-thrombin cut site-mCherry-his6. Both mCherry gene bearing plasmids were transformed into NEB5α after ligation and positive clones were co-transformed with pULTRA-CNF into BL21(DE3).
2.1.2 Protein expression, purification, and characterization
w3CFP-his6 (CFP) was expressed as previously described [26]. BL21(DE3) bearing both the expression plasmids for w3-pAzF-CFP-his6 (AzCFP), w3-pAzF-mCherry-his6 (AzmCh), and w3-pAzF-thrombin cut site-mCherry-his6 (AzTMBmCh) and pULTRA-CNF plasmids were grown in terrific broth (TB) media (12 g/L tryptone, 24 g/L yeast extract, 0.4% (v/v) glycerol, 9.4 g/L potassium phosphate monobasic, 2.2 g/L potassium phosphate dibasic) supplemented with 100 μg/mL ampicillin and 50 μg/mL spectinomycin. Cultures were grown in shake flasks inoculated to an OD600 = 0.1 from an overnight pre-culture from a colony isolated from an LB agar plate. The culture was allowed to grow at 37°C until OD600 = 0.8, at which point the expression was induced with 1 mM IPTG and supplemented 1 mM pAzF. pAzF was stored previous to culturing as 1 M stock solutions in 1 M NaOH. The culture was further grown overnight (16 hrs) at 20°C.
All cultures were sedimented using centrifugation at 4,000×g for 10 min at 4°C. The supernatant was removed, and the pelleted cells were suspended in 1x phosphate buffered saline (PBS, pH 7.4) to an OD600 = 30. The cells were lysed using sonication, and the soluble portion was fractionated using centrifugation at 15,000 × g for 15 min at 4°C. All fluorescent proteins in this study have a C-terminal hexahistidine (his6) tag. Thus, all fluorescent proteins were purified using His-Bind Ni-NTA resin from Thermo Fisher (Pittsburgh, PA) gravity column used as instructed.
w3CFP-his6 (CFP) was produced with over 95% purity (based on densitometry) (Fig. S1) [30]. Similarly, full-length expression of w3-pAzF-CFP-his6 (AzCFP), an azide bearing CFP, w3-pAzF-mCherry-his6 (AzmCh), an azide bearing mCherry, and w3-pAzF-thrombin cut site-mCherry-his6 (AzTMBmCh) was observed only in cultures supplemented with p-azido-l-phenylalanine (pAzF) with over 90% purity for all azide bearing proteins (based on densitometry) (Fig. S1). After purification, all fluorescent proteins were stored in 1x PBS pH 7.4 with 250 mM imidazole (his elution buffer) at 4°C, and the samples were protected from light exposure until immobilization into hydrogels.
All fluorescent proteins had expected spectral properties (data not shown), and in all cases the identity of the fluorophore was determined easily by eye. Thrombin cleavage site incorporation (LVPRG↓S) into AzTMBmCh upstream of the mCherry domain was confirmed by binding ELP-2rSH3 [30,31], an ELP-SH3 fusion protein specific for the SH3 specific binding ligand on the N-terminus of AzTMBmCh (Fig. S2). When the complex is maintained, the entire complex will go through ELP transition cycling. Exposing this complex to thrombin releases the mCherry cargo, and this released cargo no longer goes through ELP transition cycling (Fig. S2).
2.1.3 Synthesis and purification of 8-arm 40 kDa PEG-DBCO (PEG-8-DBCO)
Eight-arm 40 kDa PEG-amine (1 equiv., JenKem, A8012) was dissolved in anhydrous DMF at a concentration of 0.25 g polymer/mL and subsequently was added to DMF solution of DBCO (2.2 equiv.), HATU (2 equiv.), and 4-MMP (4.5 equiv.) (Fig. S3). This reaction was stirred at room temperature overnight in the absence of light. Excess DBCO-acid must be removed before hydrogel formation because it can react with PEG-2-Az crosslinker, decreasing crosslink density and immobilization of proteins to the hydrogel. Ethyl ether precipitations were performed to remove excess DBCO-acid. The solution was diluted with DMF before the product was precipitated in cold ethyl ether (5×). The final product, a water-stable macromer PEG-8-DBCO where DBCO is linked to multi-arm PEG by amide bonds, was collected after two re-precipitations from DMF into cold ethyl ether, to increase removal of DBCO-acid, and dried in vacuo overnight. The degree of functionalization was characterized using a Bruker AV 400 NMR spectrometer (Bruker Daltonics, Billerica, MA). By averaging the results of four batches, 82% of NH2 groups were modified with DBCO with 5% standard deviation (approximately 6.6 DBCO per 8 arms of multiarm PEG), with no impurities or unreacted amine detected by 1H-NMR analysis (Figure S4). 1H NMR (DMSO-d6, 400 MHz). δ = 7.71(t, 1H), δ = 7.46 (m, 8H), 5.03 (d, 1H), 3.51 (m, PEG backbone), 3.12 (m, 2H), 2.17 (m, 1H), 1.84 (t, 2H), 1.75 (m, 1H), 1.20 (m, 4H)
2.2 Hydrogel formation and rheological characterization
All hydrogels were formed by mixing PEG-8-DBCO (10 mM final DBCO concentration, 1.5 mM final PEG-8-DBCO concentration) and PEG-2-Az (9.98 mM final azide concentration, 4.99 mM final PEG-2-Az concentration) in PBS containing 0.25% BSA (wash buffer); soluble BSA was included to reduce any nonspecific interactions between synthetic macromers and proteins. For protein incorporation into the hydrogel, including conjugation of AzCFP, AzmCh, and AzTMBmCh and encapsulation of CFP, all proteins were mixed and allowed to react overnight with PEG-8-DBCO (10 mM final DBCO concentration) in a microcentrifuge tube at 4°C. The next day, PEG-2-Az (9.98 mM final azide concentration) was added to the PEG-8-DBCO, starting the crosslinking reaction. This gel precursor solution (22 μL) was well mixed and immediately pipetted into a 1-mL syringe mold to form cylindrical hydrogels. Note, other mold geometries may be utilized to achieve gels of different shapes. Concentrations of protein incorporated within gels were 20 μM for AzCFP and CFP and 10 μM for AzmCh and AzTMBCh.
For rheological characterization, the gel precursor solution was prepared at a total volume of 10 μL and pipetted onto a Peltier plate on a rheometer with an 8 mm flat plate geometry (AR-G2, TA instruments, USA). Measurements were taken with a 150 μm gap at room temperature. Storage (G’) and loss (G”) moduli were recorded over time at 2% applied strain and 6 rad s−1 frequency, which was in the linear-viscoelastic regime for these hydrogels. Mineral oil was added to the space between the hydrogel outer surface and the lower plate once the G’ value reached 150 Pa to ensure that the gel remained hydrated throughout the time course of the measurement. The gel point was estimated with the crossover of G’ and G”, and the time to complete gelation was defined to be when the percent change in modulus between consecutive data points was less than 10% [28].
2.3 CFP and AzCFP hydrogels analyzed by typhoon scanner imaging and plate reader
Whole hydrogel images were recorded using a Typhoon Variable Mode Imager 9400 (Amersham Biosciences, Piscataway, NJ). AzCFP hydrogels, CFP encapsulating hydrogels, and blank PEG hydrogels (negative control) were imaged every hour for 9 hours after the start of swelling in the wash buffer (PBS with 0.25% BSA), followed by one long incubation overnight (~27 hours after the start of swelling). Each hydrogel was washed in a separate well of a 24-well plate, and the wash solutions were collected before each imaging step. The wash fractions were diluted (4x) and measured in triplicate on a Synergy 2 plate reader (BioTek, Winooski, VT). Samples were excited at a wavelength of 435 nm, and the fluorescence emission was measured at 485 nm. CFP fluorescence from the wash fractions was used to calculate CFP retention in the hydrogel at each time point. CFP retention was calculated by measuring the total amount of CFP in each wash fraction in triplicate. This experiment was repeated two more times, using fluorescence microscopy assessment of fluorescence within the hydrogels. Images of multiple positions of gels were captured to confirm retention of fluorescence associated with covalent immobilization of AzCFP and the loss of fluorescence associated with release of unattached CFP from the CFP gels (Fig. S6), which is in agreement with the typhoon scanner and plate reader analyses.
2.4 AzmCherry confocal imaging
AzmCh and AzTMBmCh hydrogels were made using 22 μL of solution at a concentration of 0.01 mM AzmCh and AzTMBmCh pre-reacted overnight in the gel precursor solution. After gel formation, the gels were swollen overnight in wash buffer. The AzmCh and AzTMBmCh hydrogels were placed side by side in a confocal chamber slide (Nunc™ Lab-Tek™ II Chamber Slide, Glass, 1 well). Thrombin solution (200 μL of 120 unit/mL) was added to the top of the hydrogels every two hours to ensure a source of thrombin and that gels remained hydrated without disturbing the gels over the course of the experiment. Z-direction images of the gels were taken (Zeiss 710 inverted confocal microscope with Zen software), and tiles were stitched with 20% overlay every hour over a period of twelve hours. A final image was taken after six more hours of incubation (18 hours total) with thrombin solution. Densitometry analysis of AzmCh and AzTMBmCh gels to assess mCherry cleavage over time was performed using Plot Profile in ImageJ. This experiment was performed in triplicate.
2.5 Layered hydrogel fabrication and temporal removal of AzTMBmCh
AzCFP gel precursor solution (20 μL) was pipetted into a syringe mold. The pre-gelled solution was flattened (e.g., gentle tapping the side of the syringe) and then 10 μL of it was pipetted out of the mold. After 30 minutes, sufficient time for gel formation based on rheometry, a second layer containing 10 μL of AzTMBmCh precursor solution was pipetted into the mold and reacted for 30 minutes. Finally, 10 μL of fresh AzCFP gel precursor solution was pipetted on top of the AzTMBmCh layer. After 30 minutes, the layered gel was removed from the mold and placed directly into a 24-well plate with 500 μL of PBS with 0.25% BSA (wash buffer) and placed on a rocker at room temperature to swell. The hydrogels were prepared, washed overnight to remove any unreacted components, and subsequently imaged in a confocal chamber slide. To cleave and release the second AzTMBmCh layer, the gel was placed in the 500 μL cleavage buffer (wash buffer with 1000 unit/mL thrombin) and placed on a rocker overnight at room temperature. After overnight exposure to the cleavage buffer, gels were rinsed with 500 μL wash buffer at room temperature for five minutes in triplicate before imaging (Zeiss 710 inverted confocal microscope). ImageJ analysis was used to measure CFP and mCherry fluorescence intensities in each layer before and after thrombin cleavage to determine the percentage loss in fluorescence (Fig. S8).
2.6 Cell encapsulation and viability
Human mesenchymal stem cells (hMSCs, passage 7, Lonza) were encapsulated in a PEG-8-DBCO gel (control), AzmCh gel, and a layered AzmCh/AzTMBmCh gel before and after thrombin cleavage. A live/dead cytotoxicity assay (ThermoFisher, L3224) was performed to determine cell viability in these hydrogels and assess any effects of the incorporation and removal of proteins and the gel layering procedure on cell viability. Precursor solutions were prepared for forming the PEG-8-DBCO control gel and the AzmCh gel, with 0.01 mM AzmCherry pre-reacted overnight within the gel precursor solution before gel formation, as previously described in section 2.2. Cells were encapsulated at 10 × 106/mL within 20 μL gels for 20 minutes, allowing sufficient time for gel formation based on measured gelation times (between 10–12 min, see Figure 1). After 1 day of culture, the live/dead assay was performed and z-stacks 300-μm thick were imaged by confocal microscopy (Zeiss LSM 800). All cell encapsulation images are orthogonal projections of these z-stacks (3 replicates per composition with 3 z-stacks taken per replicate), and cell counts of live (green) or dead (red) cells were performed using ImageJ to obtain the percentage of viable hMSCs (number of live cells/(number of live + dead cells) * 100%).
Figure 1. Hydrogel formation.
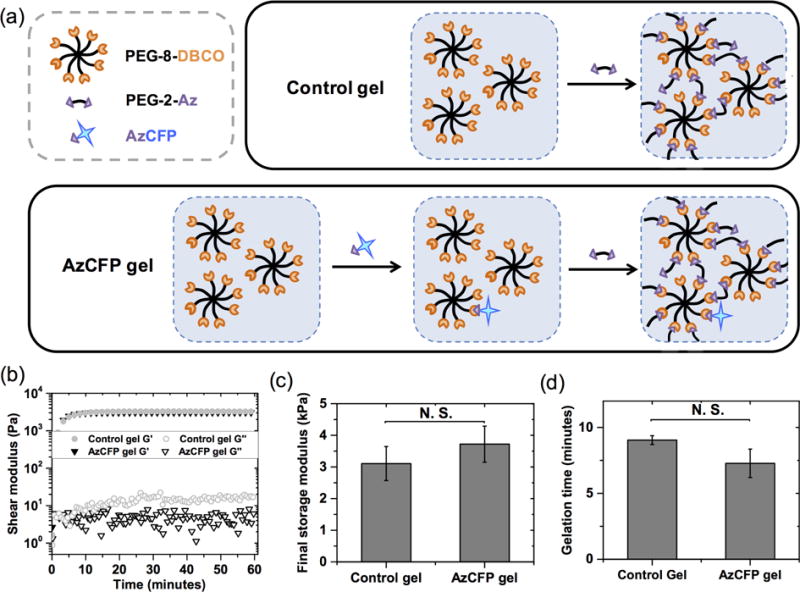
(a) Hydrogels were formed with 8-arm PEG functionalized with DBCO (PEG-8-DBCO) and PEG diazide (PEG-2-Az with or without pre-reacted AzCFP (AzCFP or control gel, respectively). The AzCFP gel was formed by pre-reacting AzCFP with PEG-8-DBCO overnight before polymerization with PEG-2-azide. (b) Rheometry was used to monitor gel formation. The control gel and the AzCFP gel have no significant difference in gelation kinetics. Further, no significant difference was observed for (c) final storage moduli and (d) gelation times. These data suggest that incorporation of an azid-bearing protein has little effect on the formation or properties of the hydrogels.
To determine cell viability after layering and thrombin cleavage, a two-layered gel was fabricated with a bottom layer of AzmCh gel and a top layer of AzTMBmCh gel (AzmCh/AzTMBmCh) with both layers containing encapsulated hMSCs. The layered gel was made as previously described in section 2.5, with AzmCherry and AzTMBmCherry whole proteins, respectively, pre-reacted overnight in the gel precursor solutions followed by layer-by-layer molding of gels in syringes. To encapsulate cells in a layered gel, a 10-μL AzmCherry gel-forming solution containing hMSCs (10 × 106 cells/mL) was added to the syringe mold and allowed to gel for 20 minutes (bottom layer); subsequently, 10 μL of AzTMBmCherry gel-forming solution containing hMSCs (10 × 106 cells/mL) was added on top, which was allowed to gel for 20 minutes (top layer). After one day of culture, the live/dead cytotoxicity assay was used to determine the viability of hMSCs before the addition of thrombin for cleaving the AzTMBmCherry layer. Thrombin (1000 U/mL) subsequently was added to remaining replicates and incubated overnight in the culture medium of the AzmCh/AzTMBmCh two-layered gels, and viability of hMSCs on day 2 was analyzed by live/dead cytotoxicity assay. Z-stacks (500-μm thick) of cells within these gels were imaged with confocal microscopy, and orthogonal projections of these stacks were used to determine cell viability using ImageJ as described above.
2.7 Statistical analysis
Results are reported as mean ± standard error (SE). Specific numbers of replicates (n ≥ 3) are noted for each experiment within the Results and Discussion. Statistical significance was determined by performing a two-sided Student’s T-test with significance accepted at p < 0.05.
3. Results and Discussion
In this study, we demonstrate the rapid functionalization and externally triggered removal of whole proteins from synthetic hydrogels. Azide functional groups and an enzymatically cleavable group were site-specifically, genetically encoded within the proteins of interest, demonstrated here with model fluorescent proteins CFP and mCh. In combination of SPAAC chemistry, this tool enabled the facile construction of hydrogel layers with different compositions for spatially-defined presentation of proteins and temporal cleavage of a specified protein with the application of a biological trigger, thrombin, to cleave the incorporated thrombin cut-site. This approach provides multiple handles for tuning of synthetic matrix properties in vitro, or potentially for responsive release in vivo, which may prove useful in studying and directing a variety of biological processes.
3.1 Site-specific modification of proteins with azides allows facile incorporation within hydrogels using bioorthogonal click chemistry
Bioorthogonal azide functional groups were site-specifically positioned within recombinant proteins by genetic incorporation of pAzF, starting with AzCFP. Rather than substitute an amino acid within the fluorescent protein, the amber codon, the designated codon for pAzF incorporation, is upstream of all functional components. This placement is unlikely to affect protein folding or function, even after conjugation. In addition to selective purification of proteins with the correctly placed azido UAA (Fig. S1 and Fig. S2), retention of AzCFP fluorescence within the hydrogel over time (Fig. 1) supports that the placement of pAzF did not hinder protein folding and enabled conjugation to the hydrogel. During expression, we expected two major products to form: the full-length Az-fluorescent protein and a truncated product. Full-length expression only occurs if the amber codon, which natively designates the termination of translation, is suppressed and the UAA is incorporated. For this reason, all of the azide-bearing proteins have C-terminal hexahistidine tags that enabled the selective purification of the azide-modified full-length product (Fig. S1).
Hydrogels decorated with whole proteins were formed using bioorthogonal SPAAC chemistry by reacting multi-arm PEG functionalized with cyclooctyne groups (PEG-8-DBCO) with azide-bearing proteins and PEG-2-Az, covalently crosslinking the multi-arm PEG (Fig. 1 and Fig. S3). For this, PEG-8-DBCO was synthesized by reacting PEG-8-amine with excess DBCO-acid, followed by ethyl ether purification, resulting in DBCO functionality of 82 ± 5% (n=4; the error indicates standard deviation, Fig. S3 and Fig. S4). To ensure complete immobilization of protein, AzCFP was pre-reacted with PEG-8-DBCO overnight before mixing with the PEG-2-Az crosslinker, which immediately commences gel formation under physiological pH. With an approximate rate constant on the order of 10−1 M−1 S−1 for SPAAC reactions [32,33], addition of a low concentration of azide protein (on the order of 0.01 mM) results in an overall reaction rate that requires day(s) for completion of alkyne-azide cycloaddition. Because of this, the relatively small ratio of AzCFP (0.02 mM) to PEG-2-Az crosslinker (9.98 mM) would lead to a lower yield of immobilized protein if these azide groups were simultaneously mixed with PEG-8-DBCO in the gel-forming solution. Pre-reacting PEG-8-DBCO with azido protein thus allows for more consistent conjugation of protein within the hydrogel structure (Fig. S7).
Mechanical properties and gelation times were measured for PEG-8-DBCO SPAAC hydrogels with and without AzCFP (Fig. 1). Gel formation was characterized using rheology, as described in section 2.2. The gelation times for non-functionalized CFP (control) and AzCFP gels were both under 10 minutes and statistically similar at 9.04 ± 0.33 minutes and 7.28 ± 1.08 minutes, respectively. The crossover point (G’ = G”) was difficult to measure consistently: the short time allowed for mixing and handling of the pre-gelled solution (~ 30 seconds) before measurements on the rheometer could be commenced frequently led to crossover already having occurred by the time the first measurement was acquired. With the variance in handling time, we did not observe the crossover point in some of the samples from both the control and the AzCFP gel formulations. Based on our qualitative observations during sample handling (adding in crosslinker, mixing the prep solution in the tube, and placing the solution on the rheometer) we estimate the crossover point occurs within approximately one minute of mixing. The mechanical properties of the resulting gels were consistent, making the SPAAC formulation easy to mold and handle, and final shear moduli for control and AzCFP hydrogels were statistically similar at 2900 ± 470 Pa and 3200 ± 420 Pa, respectively.
SPAAC occurred at ambient temperature, in the absence of any potentially harmful catalysts, UV-light, and free-radicals often used in hydrogel formation, and allowed effective conjugation of AzCFP. Further, hydrogels formed consistently and robustly with or without biofunctionalization. Both the observed time to complete gelation and storage moduli of the resulting gels were the same order of magnitudes as those of similar hydrogel systems fabricated using SPAAC click chemistry [28]. Of note, the model protein concentrations tested here (10 μM and 20 μM) are relevant for many bioactive proteins, including growth factors and cytokines. For example, growth factors, such as PDGF-BB (~ 0.7 μM) [3] and EGF (~ 2 to 20 μM) [34], have been immobilized within hydrogel-based matrices at similar concentrations to modulate cellular functions and regenerative processes in vitro and in vivo, such as proliferation, differentiation, and protein secretion. Further, large receptor-binding whole proteins or related peptide mimics have been incorporated within hydrogel-based matrices at similar or higher concentrations (~ 50 to 2000 μM) to enable cell adhesion and promote specific cellular activities: these include 1) integrin-binding peptides RGDS (fibronectin/vitronectin-derived mimic), IKVAV and YIGSR (laminin-derived), or (POG)3POGFOGER(POG)4 (collagen-derived) to promote adhesion and viability and facilitate differentiation of induce pluripotent stem cells (iPSCs) [35] and human mesenchymal stem cells (hMSCs) [36]; 2) recombinant, integrin-binding protein fragments fibronectin type III repeat 9–10 [37] or E8 derived from laminin isoforms (on plates) to promote hMSC adhesion or iPSC self-renewal [38], respectively; and 3) whole proteins E-cadherin and EpCAM to mimic cell-cell contact and facilitate reprogramming of somatic cells into iPSCs [37]. While not demonstrated here, the material system affords the capacity for incorporating higher concentrations (e.g., 2000 μM) by reacting an increased fraction of DBCO-functionalized PEG arms with azide-functionalized proteins. Theoretically, only 3 arms of PEG-8-DBCO are required for use as crosslinks to achieve gel formation (i.e., >2 crosslinks per PEG-8-DBCO required for gelation with the difunctional linear PEG-2-Az) [39,40], and the remaining fraction of DBCO-functionalized PEG arms could be used for protein immobilization, up to ~ 3.6 arms per PEG-8-DBCO with an average of 6.6 out of 8 arms based on NMR analysis (~ 5400 μM DBCO in current gel formulation). With this capacity, the material system and approach should have broad utility for the immobilization and controlled presentation of a variety of bioactive receptor-binding whole proteins or their fragments, such as those noted above, toward modulating cell function and fate.
3.2 Conjugation enables retention of pAzF functionalized protein and its activity over time
For successful biofunctionalization of PEG hydrogels using SPAAC, we wanted a system that specifically and homogeneously immobilized proteins without affecting hydrogel mechanical properties. AzCFP covalent coupling to hydrogels for immobilization was confirmed by comparing CFP retention over time for hydrogels formed with AzCFP or control CFP (i.e., no azide functionality) (Fig. 2). AzCFP hydrogels and control CFP hydrogels were synthesized using purified AzCFP and CFP proteins, respectively (purity > 90%, Fig. S1). Fluorescence was tracked for the entire hydrogel through fluorescence imaging on a Typhoon Variable Mode Imager, as discussed in section 2.3. Hydrogel fluorescence was imaged over approximately 1 day with constant replacement of the wash solution every hour until the 10th hour, at which time little change in fluorescence was observed (Fig. 2a).
Figure 2. AzCFP incorporation and activity within the hydrogel.
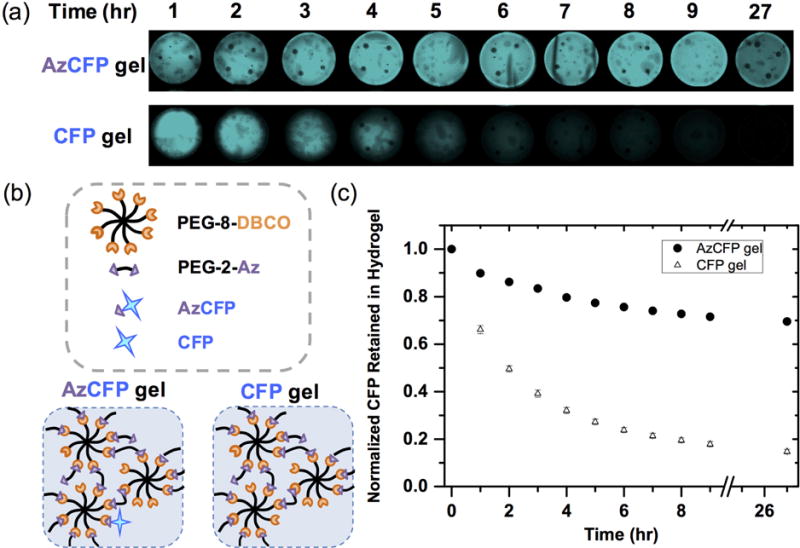
(a) Images were taken of AzCFP and CFP hydrogels on the Typhoon scanner at various time points over 27 hours. The AzCFP gel maintained similar fluorescence, whereas the control CFP gel exhibited decreased fluorescence. Without covalent modification, encapsulated CFP diffused out of the gel until there was no noticeable fluorescence in the hydrogel. (b) Comparison of AzCFP gel (CFP covalently immobilized to the gel) and CFP gel (encapsulation of CFP into the gel) allows assessment of the fraction of AzCFP covalently incorporated within the hydrogel, as the encapsulated CFP diffuses out of hydrogel network due to a larger mesh size of the hydrogel compared to the CFP hydrodynamic diameter. (c) Quantitative plot of the CFP retained in the hydrogel as a function of time shows that only ~ 20% CFP was retained in the control CFP gel after eight hours of incubation in wash buffer, whereas the AzCFP gel still retained ~ 75% AzCFP, supporting the covalent immobilization of AzCFP.
Diffusion of any unconjugated CFP out of the gel and into the bulk wash buffer also was confirmed by measuring CFP fluorescence in wash fractions (Fig. 2c). The cumulative CFP loss was subtracted from the initial CFP amount loaded within the hydrogel and normalized to the initial CFP amount to yield CFP retention. About 80% of the initial CFP was lost from the control CFP gel within the first six hours, indicating ~ 20% retention of CFP by encapsulation alone, whereas AzCFP hydrogels retained more than 70% of the initial CFP fluorescence over 27 hours of washing. These data support that the AzCFP protein was covalently bound to the hydrogel with an overall minimal loss of fluorescence. Retention of a small percentage of the control CFP within the hydrogel after extensive washing likely was due to nonspecific interactions between the hydrogel backbone and CFP, where inclusion of BSA as a blocking agent in the gel and wash buffer decreased the amount of residual CFP (Fig. S5). Homogenous immobilization of AzCFP was further confirmed using fluorescence microscopy before and after 3 days of incubation in the wash buffer (Fig. S6).
Diffusion of encapsulated proteins is dependent on hydrogel mesh size, the molecular-level pore size, where void spaces allow free to hindered diffusion within the hydrogel. Here, the mesh size of the hydrogel formulation used in all studies shown here (10 mM DBCO concentration from 1.5 mM PEG-8-DBCO) was estimated to be ξ = 19 nm (calculated by the Peppas Merrill equation [41], Fig. S9), whereas CFP, as variant of GFP, has a barrel-shaped structure with a diameter of 2.4 nm and a height of 4.2 nm [42]. Because the mesh size was roughly an order of magnitude larger than the diameter of CFP, unconjugated CFP diffused out of the 20 μL hydrogels on the order of hours (Fig. 2c). The SPAAC crosslinked PEG hydrogels created here thus allowed not only immobilization of large protein cargo during gel formation, but also diffusion of such large biomolecules through the structure for removal of unattached protein and with implications for controlled release.
3.3 Enzyme triggered cleavage of immobilized azide-mCherry with thrombin cut-site (AzTMBmCh)
In addition to the facile method for immobilizing full-length proteins, we wanted to establish a complementary approach for externally-triggered cleavage of proteins from these hydrogels, enabling dynamic tuning of matrix properties and protein release. Specifically, controlled release of a model protein, mCherry, from hydrogels by a proteolytic trigger, thrombin, was demonstrated by genetically incorporating a thrombin cleavage site (TMB) between pAzF and mCherry domains in AzTMBmCh. Before testing cleavage of mCherry from the hydrogel using thrombin, the functionality of a thrombin cleavage sequence in AzTMBmCh and the relative position of the cleavage site within the protein was assayed in solution by using ELP-2rSH3; ELP-2rSH3 is an elastin-like polypeptide domain fused to two repeats of an SH3, which provides a reversible phase transition for purification of protein-ELP fusions when heated above the lower critical solution temperature of ELP [30]. The N-terminal of all proteins consisted of a specific SH3-binding ligand (w3) and was fused in-frame to pAzF, which contains the azide conjugation site for SPAAC, followed by the thrombin cleavage site and mCherry (w3-pAzF-thrombin cleavage site-mCherry). Release of w3-pAzF by thrombin cleavage prevented the cleaved mCherry from complexing with ELP-2rSH3, resulting in a soluble mCherry even above the LCST of ELP. SDS-PAGE analysis of this experiment confirmed the location and accessibility of the thrombin cleavage site (Fig. S2).
To test enzymatic cleavage of the thrombin cut-site in hydrogels, non-cleavable AzmCh hydrogels and cleavable AzTMBmCh hydrogels were synthesized (Fig. 3a). These gels were swollen overnight and washed to remove unreacted AzmCh/AzTMBmCh from their respective hydrogels. Thrombin (120 U/mL) was added to the AzmCh and AzTMBmCh hydrogels over time. Resulting release of mCherry from the hydrogels was examined using confocal microscopy to image cross-sections of both hydrogels. Release of mCherry was observed from the hydrogel with the thrombin incorporated cut-site protein, whereas retention of mCherry was observed in the non-cleavable control gel (Fig. 3b). Fluorescence densitometry of the confocal images for the AzmCh and AzTMBmCh hydrogels were used to analyze the release profile of mCherry in response to the addition of thrombin (Fig. 3c), where an approximately linear trend was observed for triggered removal and release of mCherry over time.
Figure 3. Controlled presentation and release of mCherry.
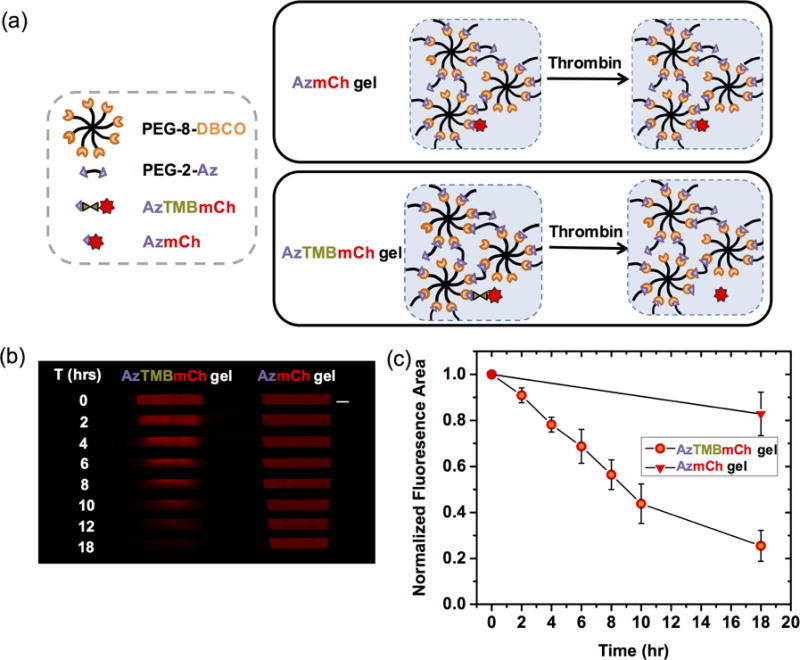
(a) AzTMBmCh was covalently immobilized to the hydrogels for temporally triggered release upon addition of thrombin. Hydrogels with noncleavable AzmCh were created as enzyme-stable, noncleavable controls. (b) Selective release of AzTMBmCh is observed with confocal microscopy (confocal tile images stitched together) over time after thrombin addition, whereas minimal loss of fluorescence from the control AzmCh hydrogel was observed in the presence of thrombin (Scale bar, 1 mm). (c) Fluorescence of these hydrogels was quantified using ImageJ and normalized to t=0 values.
Nearly all of the mCherry was released from the hydrogel by 10 hours after thrombin addition. Compared to strictly the release of unconjugated CFP by diffusion out of the hydrogel (complete release within ~ 8 h, Fig. 2), mCherry release was slightly slower. Since the kinetics of thrombin cleavage is relatively fast, the release of mCherry is almost exclusively dependent on the diffusion of thrombin in (~36 kDa, effective hydrodynamic diameter ~ 4.7 nm [43]) and subsequently mCherry out of the hydrogel. Despite the dependence on the diffusion of thrombin, the release of mCherry was still on the order of hours much like the release of unconjugated CFP. These observations support that the relatively large void spaces afforded by the mesh size of these hydrogels allowed thrombin diffusion to be relatively unhindered.
Note, when thinking more broadly in terms of design principles for future applications, the rate for diffusion and release will depend on the mesh size as well as the dimensions of the gel. For example, increasing the total concentration of PEG in the gel precursor solution (while keeping the ratio of DBCO and Az from PEG constant) will decrease the mesh size, which can cause slower diffusion of protein as the mesh size approaches the size of the protein. Additionally, a thicker or wider gel will result in a more pronounced protein gradient and a longer protein diffusion time. Here, we selected a hydrogel composition with modulus similar to various soft tissues (E ~ 10 kPa) [15] and with a large enough mesh size (ξ ~ 19 nm) for relatively unhindered diffusion of our model proteins (CFP and mCherry), which are similar in size (or larger than) many growth factors, cytokines, and integrin-binding protein fragments (e.g., recombinant fibronectin type III repeat 9–10) [44]. Further, while response to thrombin was demonstrated here, this approach is amenable to use with other biological triggers where associated cleavage sites can be incorporated into similar proteins, including other protease cleavage motifs and protein domains that self-cleave based on external stimuli, such as pH sensitive self-cleaving inteins [45].
Responsive release of proteins from hydrogels has been achieved by a variety of approaches [46]. However, the overwhelming majority of studies encapsulating proteins utilize hydrogels as merely delivery vehicles rather than matrices for promoting interactions with biological systems (e.g., mammalian cells), limiting the scope of these hydrogels to largely drug delivery applications. For instance, the required mesh size for encapsulation is dependent on the size of the encapsulated protein, limiting the range of hydrogel crosslink densities and moduli that can be used. Furthermore, encapsulated proteins are prone to premature leakage, as shown by CFP diffusion from the gel, and the hydrogel structure can be compromised after the release of proteins is triggered (e.g., gel is degraded), precluding studies where the hydrogel structure must be maintained during release. The successful incorporation of a thrombin responsive tether for mCherry protein release is an important function to our study, whereby we can control the release of the protein while maintaining a highly crosslinked hydrogel network with robust mechanical properties. This independent control of properties is promising for not only drug delivery applications, but also controlled cell culture and tissue engineering for the dynamic tuning of protein presentation to probe and direct cell fate. Our approach, incorporating a site-specific reactive functional group and protease cut-site, adds to the growing suite of tools for the controlled presentation and release of proteins from hydrogel-based materials using different cleavable chemistries, such as photolabile (e.g., o-nitrobenzyl ether and coumarin groups [17,18]) and other enzyme responsive (e.g., sortase and MMPs [10,19]) groups.
3.4 Layered gel formation and triggered protein release allow the creation of anisotropic and dynamic gel architectures
Spatial presentation of proteins was demonstrated by creating an AzCFP/AzTMBmCh/AzCFP multi-layer hydrogel (Fig. 4). A syringe mold was used for layering the hydrogels, allowing control of the height of the gel (300–400 μm) on size scales relevant for mammalian cells and tissues. Each new gel layer was conjugated sequentially to its adjacent layer utilizing unreacted DBCO and azides on the gel surface. To prevent any mixing of protein between layers and to make gels with discrete boundaries between layers, each gel layer was allowed to polymerize for 30 minutes prior to adding the next layer to allow enough time for substantial crosslinking to occur.
Figure 4. Spatial control and release of proteins from layered hydrogels.
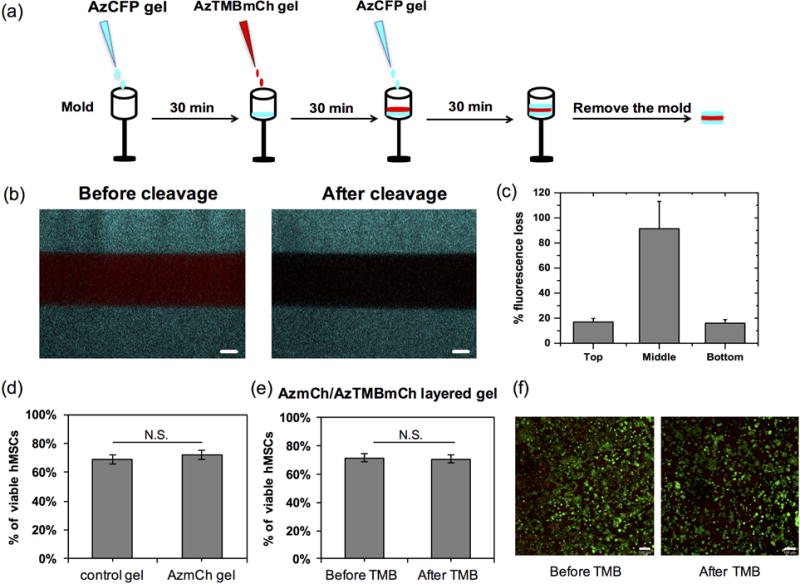
(a) Layers of gels with different proteins were created. Using a syringe mold, 10 μL of gel precursor solution was added and allowed to polymerize for 30 minutes before the addition of the next layer. With this method, a hydrogel with layers of AzCFP, AzTMBmCh, and AzCFP was created. (b) Confocal images of the layered gel show the three separate fluorescent layers before and after mCherry cleavage using thrombin. Thrombin enzyme was added overnight (adding 100 μL of thrombin solution every 2 hours), followed by three washes before imaging the next day. Release of mCherry was observed (lack of fluorescence in the middle layer after incubation with thrombin) (Scale bars, 100 μm). (c) Select removal of mCherry from layered hydrogels was quantified: the top and bottom AzCFP layers exhibit minimal (~15%) loss in fluorescence, whereas the middle AzTMBmCh layer that contains the thrombin cut-site exhibits a 90% loss in fluorescence after the addition of thrombin. (d) hMSC viability in PEG-8-DBCO (control) and AzmCh gels indicate no statistical difference in viability with incorporation of a covalently-immobilized whole protein. (e) and (f) hMSC viability in two-layered AzmCh/AzTMBmCh gels before and after thrombin addition, followed by mCherry cleavage and release, indicate no statistical difference in cell viability (right, example confocal projections for live (green)/dead (red) staining of hMSCs encapsulated in AzmCh/AzTMB layered gel before and after thrombin addition. Scale bar = 100 μm.
Selective release of the middle AzTMBmCh layer was demonstrated by introducing thrombin to the multi-layer gel. The AzCFP/AzTMBmCh/AzCFP multi-layered hydrogel was imaged using confocal microscopy before and after the addition of thrombin (Fig. 4b). After 1 day of incubation with thrombin, which was expected to be sufficient for mCherry cleavage and release (see Fig. 3), the AzTMBmCh middle layer showed a 90% loss in fluorescence, which was statistically significant at a p-value < 0.05 (Fig. S8), while the bottom and top AzCFP layers had minimal to no loss in fluorescence (Fig. 4c). Taken together, these results demonstrate the utility of this materials strategy for user-controlled immobilization and removal of proteins within hydrogels from spatially-defined regions and at times of interest.
Viability of hMSCs encapsulated in single- or multi-layered gels with and without protein immobilization/removal were examined to establish the effectiveness of the materials and procedures for cell culture. First, the effect of whole protein incorporation on hMSC viability was assessed by encapsulating hMSCs in a PEG-8-DBCO gel (control) and an AzmCh gel. After encapsulation and 1 day of culture, hMSC viability was statistically the same for SPAAC-formed hydrogels with and without AzmCherry protein (68 ± 3% and 71 ± 3%, respectively) (Fig. 4d). These data indicate that the addition of azide-functionalized proteins to the hydrogel does not affect cell viability. Note, DeForest et al. observed ~90% viability of hMSCs encapsulated within similar PEG hydrogels formed by SPAAC and presenting covalently-immobilized vitronectin to facilitate cell integrin binding and adhesion. We hypothesize the addition of such integrin-binding proteins or peptides enhances cell viability within these otherwise bioinert matrices; however, even absent such integrin-binding ligands, our data support sufficient hMSC viability for cell culture in SPAAC-formed hydrogels in addition to no effects of protein immobilization on cell viability. To examine the effect of gel layering and protein removal on hMSC viability, a two-layered AzmCh/AzTMBmCh gel was fabricated. After one day of culture and before thrombin addition, hMSC viability in the two-layered gel was 71 ± 3% (Figs. 4e and 4f). Thrombin was added to the culture medium of the layered gels on day 1 and incubated overnight, and viability of hMSCs on day 2 (after TMB addition and mCh removal) was determined to be 71 ± 3%. Again, good viability of hMSCs was observed in the two-layered gel, before and after thrombin cleavage of mCherry in the top layer, and viability was statistically similar for all conditions. This study demonstrates the relevance of the material system for cell culture, including use of layering for 3D culture and thrombin addition for triggered protein removal.
This layering technique enables customizable and spatially separated layers with broad implications for biomedical applications. Theoretically, not only could layers harbor different proteins with their own distinct triggers, but each layer could also have significantly different mechanical properties. While not explored here, the backbone of the polymer network comprising each layer also can be engineered to degrade for further evolution of matrix properties (e.g., cleavage of crosslinks in response to cell secreted enzymes to facilitate matrix remodeling [47] or in response to applied light for user-directed property control) [28,48]. The ability to form multi-layer structures is an important step toward recapturing the different properties found within layered tissue architectures, as well as enabling the co-culture of different cell types. Toward this, we have demonstrated that the materials and approach presented enable the three-dimensional culture of hMSCs: good cell viability is maintained during protein immobilization/removal and gel layering and is statistically similar to SPAAC control gels (Fig. 4d–f), which have been shown useful by others for the culture of a variety of cell types [28,29,49,8]. More importantly, the ability to temporally control protein presentation within these layers by bio-orthogonal triggers could enable a variety of investigations for probing and directing cell response to anisotropic matrix properties, such as stem differentiation down specific lineages associated with tissue interfaces (e.g., bone, cartilage, and ligament cells at the bone-ligament interface) in response to temporally and spatially-defined presentation of growth factors and integrin-binding ligands [50].
4. Conclusions
We have demonstrated the use of SPAAC chemistry to control immobilization and release of bio-orthogonal azide-functionalized proteins to a hydrogel network. Genetic engineering allowed the site-specific attachment of an azide and a thrombin cleavage site to model fluorescent proteins without any loss of protein function. Long term fluorescence of AzCFP indicated covalent conjugation to the hydrogel, and the loss of mCherry fluorescence after thrombin cleavage demonstrated the triggered, spatially-specific release of a protein. Lastly, a method was developed for creating layered hydrogels with defined compositions using biorthogonal chemistry; to our knowledge, this is the first time a layered hydrogel was formed using SPAAC chemistry. The formation of the AzCFP/AzTMBmCh/AzCFP layered gel followed by protein cleavage from the middle layer, all while maintaining the structure of the layered hydrogel, demonstrates the versatility of this approach, affording numerous possible applications for protein release and studying biological processes.
Supplementary Material
Statement of Significance.
Controlling protein presentation within biomaterials is important for modulating interactions with biological systems. For example, native tissues are composed of subunits with different matrix compositions (proteins, stiffness) that dynamically interact with cells, influencing function and fate. Toward mimicking such temporally-regulated and spatially-defined microenvironments, we utilize bio-orthogonal click chemistry and protein engineering to create hydrogels with distinct regions of proteins and modify them over time. Through nonsense codon replacement, we site-specifically functionalize large proteins with i) azides for covalent conjugation and ii) an enzymatic cleavage site for user-defined release from hydrogels. Our results exemplify not only the ability to create unique bio-functionalized hydrogels with controlled mechanical properties, but also the potential for creating interesting interfaces for cell culture and tissue engineering applications.
Acknowledgments
The authors would like to thank the Bio-Imaging Center at the Delaware Biotechnology Institute, especially Dr. Michael Moore, and the NMR laboratory at the University of Delaware for use of their instruments and training. The authors would like to acknowledge support, for related work in their laboratories, from the National Science Foundation (NSF) CAREER Award (DMR-1253906) and NSF Grant (CBET-1510817), the Delaware COBRE program with a grant from the National Institute of General Medical Sciences (NIGMS P20GM104316 and 5 P30 GM110758-02) from the NIH, the Pew Charitable Trusts (00026178), the Burroughs Wellcome Fund (1006787), and the University of Delaware Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no conflict of interests to report.
References
- 1.Russell RJ, Pishko MV, Gefrides CC, McShane MJ, Coté GL. A Fluorescence-Based Glucose Biosensor Using Concanavalin A and Dextran Encapsulated in a Poly(ethylene glycol) Hydrogel. Anal Chem. 1999;71:3126–3132. doi: 10.1021/ac990060r. [DOI] [PubMed] [Google Scholar]
- 2.Lei Y, Segura T. DNA delivery from matrix metalloproteinase degradable poly(ethylene glycol) hydrogels to mouse cloned mesenchymal stem cells. Biomaterials. 2009;30:254–265. doi: 10.1016/j.biomaterials.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saik JE, Gould DJ, Watkins EM, Dickinson ME, West JL. Covalently immobilized platelet-derived growth factor-BB promotes angiogenesis in biomimetic poly(ethylene glycol) hydrogels. Acta Biomater. 2011;7:133–143. doi: 10.1016/j.actbio.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shendi D, Dede A, Yin Y, Wang C, Valmikinathan C, Jain A. Tunable, bioactive protein conjugated hyaluronic acid hydrogel for neural engineering applications. J Mater Chem B. 2016;4:23–26. doi: 10.1039/C5TB02235E. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen MK, Alsberg E. Bioactive factor delivery strategies from engineered polymer hydrogels for therapeutic medicine. Prog Polym Sci. 2014;39:1235–1265. doi: 10.1016/j.progpolymsci.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kloxin AM, Benton JA, Anseth KS. In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels. Nat Mater. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 8.DeForest CA, Tirrell DA. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 9.Lin C, Anseth KS. Cell – cell communication mimicry with poly (ethylene glycol) hydrogels for enhancing β-cell function - Supporting Information. Pnas. 2011;108:1–6. doi: 10.1073/pnas.1014026108/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1014026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cambria E, Renggli K, Ahrens CC, Cook CD, Kroll C, Krueger AT, Imperiali B, Griffith LG. Covalent Modification of Synthetic Hydrogels with Bioactive Proteins via Sortase-Mediated Ligation. Biomacromolecules. 2015;16:2316–2326. doi: 10.1021/acs.biomac.5b00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP. In situ cell manipulation through enzymatic hydrogel photopatterning. Nat Mater. 2013;12:1072–1078. doi: 10.1038/nmat3766. [DOI] [PubMed] [Google Scholar]
- 12.Madl CM, Katz LM, Heilshorn SC. Bio-Orthogonally Crosslinked, Engineered Protein Hydrogels with Tunable Mechanics and Biochemistry for Cell Encapsulation. Adv Funct Mater. 2016;26:3612–3620. doi: 10.1002/adfm.201505329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smithmyer ME, Sawicki La, Kloxin AM. Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease. Biomater Sci. 2014;2:634. doi: 10.1039/c3bm60319a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rehmann MS, Kloxin AM. Tunable and dynamic soft materials for three-dimensional cell culture. Soft Matter. 2013;9:6737–6746. doi: 10.1039/C3SM50217A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science (80-) 2009;59:1–6. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeForest CA, Tirrell DA. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 18.Azagarsamy MA, Anseth KS. Wavelength-controlled photocleavage for the orthogonal and sequential release of multiple proteins. Angew Chemie - Int Ed. 2013;52:13803–13807. doi: 10.1002/anie.201308174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metzger S, Blache U, Lienemann PS, Karlsson M, Weber FE, Weber W, Ehrbar M. Cell-Mediated Proteolytic Release of Growth Factors from Poly(Ethylene Glycol) Matrices. Macromol Biosci. 2016:1–11. doi: 10.1002/mabi.201600223. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen LH, Kudva AK, Saxena NS, Roy K. Engineering articular cartilage with spatially-varying matrix composition and mechanical properties from a single stem cell population using a multi-layered hydrogel. Biomaterials. 2011;32:6946–6952. doi: 10.1016/j.biomaterials.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z-N, Freitas BC, Qian H, Lux J, Acab A, Trujillo CA, Herai RH, Nguyen Huu VA, Wen JH, Joshi-Barr S, Karpiak JV, Engler AJ, Fu X-D, Muotri AR, Almutairi A. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc Natl Acad Sci. 2016;113:201521255. doi: 10.1073/pnas.1521255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG. A versatile platform for single-and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry. 2013;52:1828–1837. doi: 10.1021/bi4000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–7. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 24.Deiters A, Cropp TA, Summerer D, Mukherji M, Schultz PG. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg Med Chem Lett. 2004;14:5743–5. doi: 10.1016/j.bmcl.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Brock a, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 26.Kiick KL, van Hest JCM, Tirrell DA. Expanding the scope of protein biosynthesis by altering the methionyl-tRNA synthetase activity of a bacterial expression host. Angew Chemie Int Ed English. 2000;39:2148–2152. doi: 10.1002/1521-3757(20000616)112:12<2232::AID-ANGE2232>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Link AJ, Tirrell DA. Reassignment of sense codons in vivo. Methods. 2005;36:291–298. doi: 10.1016/j.ymeth.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 28.McKinnon DD, Brown TE, Kyburz Ka, Kiyotake E, Anseth KS. Design and characterization of a synthetically accessible, photodegradable hydrogel for user-directed formation of neural networks. Biomacromolecules. 2014;15:2808–2816. doi: 10.1021/bm500731b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J, Callahan LA Smith, Hao J, Guo K, Wesdemiotis C, Weiss RA, Becker ML. Strain-Promoted Crosslinking of PEG-based Hydrogels via Copper-Free Cycloaddition. ACS Macro Lett. 2012;1:1071–1073. doi: 10.1021/mz3003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H, Chen W. A non-chromatographic protein purification strategy using Src 3 homology domains as generalized capture domains. J Biotechnol. 2016;234:27–34. doi: 10.1016/j.jbiotec.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 31.Waugh DS. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80:283–93. doi: 10.1016/j.pep.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih HW, Kamber DN, Prescher JA. Building better bioorthogonal reactions. Curr Opin Chem Biol. 2014;21:103–111. doi: 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Patterson DM, Nazarova LA, Prescher JA. Finding the right (bioorthogonal) chemistry. ACS Chem Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 34.Cambria E, Renggli K, Ahrens CC, Cook CD, Kroll C, Krueger AT, Imperiali B, Griffith LG. Covalent Modification of Synthetic Hydrogels with Bioactive Proteins via Sortase-Mediated Ligation. Biomacromolecules. 2015;16:2316–2326. doi: 10.1021/acs.biomac.5b00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam J, Carmichael ST, Lowry WE, Segura T. Hydrogel Design of Experiments Methodology to Optimize Hydrogel for iPSC-NPC Culture. Adv Healthc Mater. 2014:n/a–n/a. doi: 10.1002/adhm.201400410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rehmann MS, Luna JI, Maverakis E, Kloxin AM. Tuning microenvironment modulus and biochemical composition promotes human mesenchymal stem cell tenogenic differentiation. J Biomed Mater Res Part A. 2016;104:1162–1174. doi: 10.1002/jbm.a.35650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caiazzo M, Okawa Y, Ranga A, Piersigilli A, Tabata Y, Lutolf MP. Defined three-dimensional microenvironments boost induction of pluripotency. Nat Mater. 2016;15:344–352. doi: 10.1038/nmat4536. [DOI] [PubMed] [Google Scholar]
- 38.Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M, Kawasaki M, Hayashi M, Kumagai H, Nakatsuji N, Sekiguchi K, Kawase E. Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat Commun. 2012;3:1236. doi: 10.1038/ncomms2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubinstein M, Colby RH. Polymer Physics. Oxford University Press; 2003. [Google Scholar]
- 40.Paul TPL, Hiemenz C. Polymer Chemistry. Second. CRC Press; 2007. [Google Scholar]
- 41.Peppas NA, Merrill EW. Crosslinked Poly(Vinyl Alcohol) Hydrogels As Swollen Elastic Networks. J Appl Polym Sci. 1977;21:1763–1770. doi: 10.1002/app.1977.070210704. [DOI] [Google Scholar]
- 42.Hink MA, Griep RA, Borst JW, Van Hoek A, Eppink MHM, Schots A, Visser AJWG. Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J Biol Chem. 2000;275:17556–17560. doi: 10.1074/jbc.M001348200. [DOI] [PubMed] [Google Scholar]
- 43.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 A crystal structure of human alpha-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.2210/pdb1ppb/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials. 2009;30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wood DW, Wu W, Belfort G, Derbyshire V, Belfort M. A genetic system yields self-cleaving inteins for bioseparations. Nat Biotechnol. 1999;17:889–892. doi: 10.1038/12879. [DOI] [PubMed] [Google Scholar]
- 46.Kharkar PM, Kiick KL, Kloxin AM. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem Soc Rev. 2013;42:7335–72. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat Mater. 2013;12:458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kharkar PM, Kiick KL, Kloxin AM. Design of thiol- and light-sensitive degradable hydrogels using Michael-type addition reactions. Polym Chem. 2015;6:5565–5574. doi: 10.1039/C5PY00750J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamura M, Yanagawa F, Sugiura S, Takagi T, Sumaru K, Kanamori T. Click-crosslinkable and photodegradable gelatin hydrogels for cytocompatible optical cell manipulation in natural environment. Sci Rep. 2015;5:15060. doi: 10.1038/srep15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee N, Robinson J, Lu H. Biomimetic strategies for engineering composite tissues. Curr Opin Biotechnol. 2016;40:64–74. doi: 10.1016/j.copbio.2016.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.