

Abstract
How can highly charged, cationic antimicrobial peptides (AMPs) translocate across hydrophobic lipid bilayers despite the prohibitive energetic penalty to do so? A common explanation has been the formation of peptide-lined channels. However, for most AMPs, no structures of membrane pores have been found despite clear evidence of membrane leakage and antimicrobial activity. The study here suggests an alternative and simple reason: for the AMP PGLa from Xenopus laevis (charge +5), such pores are not needed to explain both leakage and peptide translocation. Elevated-temperature multimicrosecond equilibrium simulations at all-atomistic level reveal that peptides spontaneously translocate across the membrane individually on a timescale of tens of microseconds, without forming pores. Both surface-bound peptides and lipids assist in the one-by-one translocation of the charged side chains. Single peptides can remain in a transmembrane orientation for many microseconds, snorkeling some charged residues to one interface and some to the opposite, but without inducing a water channel. Instead of stable pores, short-lived water bridges occur when two or three peptides connect at their termini, allowing both ion translocation and lipid flip-flop via a brushlike mechanism usually involving the C terminus of one peptide. The results here suggest that for some specific antimicrobial and other membrane active peptides, pore formation may not have to be invoked at all to explain peptide translocation and membrane permeabilization, which may explain why no channel structures for them have been determined experimentally.
Introduction
Antimicrobial peptides (AMPs) are an ancient, powerful, and ubiquitous component of the innate immune defense in all domains of life (1, 2, 3). AMPs are amphiphilic peptides that selectively target and kill a wide variety of microbial pathogens at low micromolar concentrations (4). These peptides vary greatly in size, sequence, and secondary structure, yet despite the discovery of thousands of AMPs over the last 20 years, the molecular basis of antimicrobial activity remains poorly understood (3, 5, 6). AMP are known to permeabilize membranes, and it is assumed many AMPs may form pores in cell membranes, but the detailed structures of these pores remain undetermined (7). The key difficulty for structure determination is that pores may not be stable under equilibrium conditions, but transient instead (8).
A typical highly charged AMP is PGLa, from the skin of the African frog Xenopus laevis (9, 10, 11, 12). It is part of the magainin family of peptides that exhibit broad antibacterial (13, 14), anticancer (15), antiviral (16), and antifungal activity (17). Detailed structural information is available on this AMP via recent solid-state NMR spectroscopy (18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30). These measurements have revealed that PGLa is fully helical when membrane-bound, and resides in a well-defined surface-bound S-state in dimyristoyl-phosphatidylcholine (DMPC) bilayers at peptide-to-lipid ratios of P/L = 1:200 and below. At higher peptide concentration, PGLa is thought to form pores in a transmembrane configuration (22, 31), and several tilted orientations have been found (19, 22, 25, 27, 29). Real membrane pores, however, have not been detected, presumably because they form transiently with lifetimes too short to be studied by NMR (27). However, in a mixture with Magainin 2, an almost upright (transmembrane) inserted I-state was found for PGLa, shedding some light on what such a pore could look like (22, 28, 29, 32, 33, 34, 35, 36). Unfortunately, for pure PGLa, no I-state is found under the same conditions.
If experimental pore detection is challenging, can they be predicted via computer simulations? Although some early molecular dynamics (MD) simulations using united-atom models indeed appeared to directly reveal AMP pore formation (37, 38), it has been recognized over the years that those models were unfortunately not accurate, with too-rapid (<100 ns) membrane insertion, too-strong membrane rupture, and even unfolded peptides in the membrane core—which is energetically unlikely, due to the high cost (4 kcal/mol) of breaking backbone hydrogen bonds in the membrane interior (39, 40). Other early attempts using coarse-grained MD models (41) can be considered only qualitative, as these force fields by construction cannot account for the complex interplay of interactions among protein, lipids, and water, which explains why the results have never been reproduced using more accurate all-atomistic models. Indeed, most recent MD simulations of AMPs using modern all-atom parameter sets, and resources such as ANTON, have not shown spontaneous channel-like pore formation for many microseconds, including PGLa (39, 42, 43, 44). Predicting pores has been limited to trying initial transmembrane arrangements as suitable pore candidates (44), but unbiased ab initio prediction has been challenging so far. We have, however, recently demonstrated this is achievable for the AMP maculatin, in which an ensemble of pore structures was found (45).
One possibility is that peptide-lined pores are not needed at all to explain how a highly charged peptide (+5) can translocate across a lipid bilayer, and how ion leakage through the membrane can be induced. Instead, as shown here using fully atomistic models, both appear to be relatively simple cooperative effects involving 2–3 peptides, occurring on the order of once every several microseconds per peptide. They are observed both in zwitterionic phosphatidylcholine (PC) bilayers as a model of the plasma membrane of mammalian cells, and in anionic membranes, involving PC and phosphatidylglycerol (PG) in a 3:1 mix, as a model for bacterial membranes. PGLa translocates across both membranes with similar efficiency. The results shown here demonstrate how a cationic peptide penetrates monomerically through a lipid bilayer as well as induces (some) leakage. Although it is certain that a large class of AMPs indeed forms pores, there may be some peptides for which channel-like pore models may not be needed to explain the action of membrane permeabilization. Indeed, translocation models without pores have been proposed for several cell-penetrating peptide sequences (46, 47, 48, 49). And recently, it has been shown that some cell-penetrating peptides with large fractions of cationic and hydrophobic residues are able to silently translocate membranes without causing too much leakage (50, 51, 52, 53, 54).
Materials and Methods
MD simulations
All simulations were performed and analyzed using GROMACS version 5.0.5 (http://www.gromacs.org) (55) and HIPPO β (http://www.biowerkzeug.com), using the CHARMM27 force field (56), and TIP3P water (57). CHARMM36 all-atom lipids parameters were used (58). Electrostatic interactions were computed using particle mesh Ewald, and a cutoff of 10 Å was used for the van der Waals interactions. Bonds involving hydrogen atoms were restrained using LINCS (59). Simulations were run with a 2 fs time-step, and neighbor lists were updated every 10 steps. All simulations were performed in the NPT ensemble, with water, lipids, and the protein coupled separately to a heat bath with T = 120–160°C and a time constant τT = 0.1 ps using the velocity rescaling method. An atmospheric pressure of 1 bar was maintained using weak semiisotropic pressure coupling with the compressibility κz = κxy = 4.6 · 10−5 bar–1 and a time constant τP = 1 ps.
The initial peptide setup was the surface-bound conformation, or S-state. This stable interfacial α-helical minimum can be rapidly predicted, as shown in Ulmschneider et al. (42). Simulations with 8/9 peptides were started with all peptides in the same membrane leaflet, by replicating the box of the single peptide simulation once it was stably inserted in the S-state. Typical bilayers consisted of 176–189 DMPC and DMPG lipids, and ∼36 water molecules per lipid and 0.1 M NaCl solution, as described in Ulmschneider et al. (42). Membranes were either pure DMPC, or DMPC/DMPG 3:1. The latter was used as a model membrane for an anionic bacterial membrane. Two to three lipids per peptide were initially removed in the leaflet containing the peptides, to reduce the mass imbalance of the bilayer and prevent biasing the results as a consequence of a highly nonequilibrium starting membrane. The resulting bilayers are very stable, and the first peptide translocation usually occurred only after many microseconds, indicating that peptide translocation is not driven by some initially prepared biased setup. Any concern regarding lipid mismatch of the bilayer was found to be unfounded as multiple lipid flip-flop events occur during all simulations, in both directions, allowing the membrane to adjust any potentially severe mass imbalance.
All simulations were performed on the PI supercomputer at Shanghai Jiao-Tong University and the MARCC supercomputing facility at Johns Hopkins University. Typical simulation length was between 10–40 μs for each trajectory, sufficient to capture at least one translocation event.
Results
Insertion mechanism
In all simulations, PGLa prefers to remain in the surface (S-) state. Insertion as well as translocation are rare events: in total, only 11 PGLa translocation events occur over a cumulative of 110 μs of seven individual simulations (Table 1). In the dominant insertion mechanism the peptide inserts with the C terminus first (Figs. 1 and 2). This is expected, as the N-terminus is charged, whereas the C terminus is amidated and therefore neutral, resulting in average tilt angles of τ > 90°, as shown in Ulmschneider et al. (42). In the absence of other peptides there is a limit on how strongly PGLa can tilt and insert into the membrane core. However, when assisted by one or two other surface peptides in mutual contact at their C termini, deep insertion can occur (Figs. 1 and 2): a transient water bridge is established through which Lys19 is translocated first. Translocation of Lys15 and Lys12 is then facilitated via cotransport of lipids and chloride anions. Other surface-bound peptides finally assist the translocation of the charged N-terminal groups (Lys5, Gly1) via deep insertion of their C termini. After <50 ns, the water bridge dissolves and all peptides return to the surface S-state, with one peptide translocated across the membrane. The same mechanism, and a similar timescale, is found for both pure DMPC (Fig. 1) and an anionic DMPC/DMPG 3:1 bilayer (Fig. 2). Again, the translocation of the Lys19, Lys15, and Lys12 side chains occurs first, this time assisted by numerous anionic lipids that shield the cationic side chains. A third event in pure DMPC follows the same pattern (Fig. S1). For C-terminal insertion, no long-lived transmembrane (TM) structures are formed, with peptides being rapidly translocated across, rather than inserted into, the membrane.
Table 1.
List of Molecular Dynamics Simulations Performed
| Simulation | T [°C] | Length [μs] | Peptides [Number] | Peptides/Lipid | Lipid Composition | Number of Peptide Translocations |
|---|---|---|---|---|---|---|
| DMPC-393K | 120 | 37.2 | 8 | 1/22 | DMPC | 1 |
| DMPC-413K | 140 | 19.6 | 8 | 1/22 | DMPC | 1 |
| DMPC-433K-1 | 160 | 10.7 | 8 | 1/22 | DMPC | 1 |
| DMPC-433K-2 | 160 | 11.0 | 8 | 1/22 | DMPC | 3 |
| DMPC-453K | 180 | 5.5 | 8 | 1/22 | DMPC | 2 |
| DMPC-DMPG-413K-1 | 140 | 12.6 | 9 | 1/21 | DMPC/DMPG 3:1 | 1 |
| DMPC-DMPG-413K-2 | 140 | 13.0 | 9 | 1/21 | DMPC/DMPG 3:1 | 2 |
Bilayer were fully hydrated (∼36 waters/lipid) in a salt concentration of 0.1 M NaCl. The total simulation time was 110 μs.
Figure 1.

Rapid C-terminal-first peptide translocation mechanism, as observed in a pure DMPC bilayer. (a) Shown here is total peptide mass density. (b) Shown here are helix tilt angles of all PGLa peptides. (c) C-terminal insertion is facilitated by the lack of charge on this terminus, allowing the peptide to tilt and insert strongly (τ > 90°). All such translocation events involve three peptides in mutual contact at their C termini. Mutual deep insertion opens a transient water bridge through which Lys19 is translocated first. Translocation of Lys15 and Lys12 is facilitated via cotransport of two DMPC lipids and one chloride anion. Twenty-to-thirty nanoseconds later, the N-terminal charge groups (Lys5, Gly1) are translocated via assistance of the C termini of the other two strongly tilting peptides. The whole process occurs in 30 ns, after which the peptides return to their S-state and the water bridge dissolves. No stable pores are formed, but one peptide has been translocated across the membrane. To see this figure in color, go online.
Figure 2.

Rapid C-terminal-first peptide translocation mechanism, as observed in a DMPC/DMPG 3:1 bilayer. (a) Shown here is a total peptide mass density. (b) Shown here are the helix tilt angles of all PGLa peptides. (c) C-terminal insertion is facilitated by the lack of charge on this terminus, allowing the peptide to tilt and insert strongly (τ > 90°). The mechanism is almost identical to the pure DMPC simulation (Fig. 1). Here, translocation of Lys15 and Lys12 is assisted by numerous anionic lipids (one shown, red line). Again, no stable pores are formed, but one peptide translocates across the membrane in 40 ns. To see this figure in color, go online.
Longer-lived TM structures
There are, however, translocation events that can result in longer-lived TM orientations (Fig. 3). In one of these, the peptide inserts via its N-terminus. This is only possible via cotransport of anionic lipids, due to the positive charge on the N-terminus of Gly1. The peptide is arrested in a TM orientation for 4 μs before another surface-bound peptide assists in the translocation of the three remaining lysines. Of particular interest is the fact that the single-TM helix is dry—no persistent water channel is formed, with all lysines snorkeling to the interfacial region. Higher-order TM structures such as dimers and even trimers can also be observed in the same simulation, involving one peptide that has already translocated. However, these structures are extremely short-lived. They induce a temporary water bridge through which ions and lipids can translocate, but which then rapidly dissolves. Conformational clustering of all the orientational and oligomerization states in the bilayer reveals that over all DMPC/DMPG 3:1 simulations, the S-state clearly dominates (pS = 94%, ΔGS→TM = 2.3 kcal/mol), and the only TM state that is significantly populated is the single TM helix. In pure DMPC, the S-state is even more stabilized (pS = 98%, ΔGS→TM = 3.8 kcal/mol).
Figure 3.

Simulation in a mixed DMPC/DMPG 3:1 bilayer, where two peptides translocate via forming longer-lived transmembrane orientations. (a) Shown here is total peptide mass density (red line = peptide 1, black line = peptide 2). (b) Shown here are the helix tilt angles. (c) Shown here is population occupancy during the simulation, clustering the peptides into their main states (S, surface; TM1, single TM; TM2, TM dimer; TM3 = TM trimer). (e) N-terminal insertion is a rare event due to the positive charge on the N-terminus (requiring assisting lipids), and observed only once over the 110 μs simulation time. The peptide is trapped in a TM configuration for 4 μs, with three lysines snorkeling to one interface, whereas Lys5 snorkels to the opposite interface (TM1). The TM orientation allows numerous lipid flip-flops (red lines), as well as Na+ ions to translocate (green lines), but is dry most of the time, with no water pore. (f) The same simulation shows how previously translocated peptides facilitate translocation events by other peptides via formation of TM dimers or trimers. All TM structures other than a single TM helix are extremely short lived and rare, with PGLa strongly preferring the S-state (ΔGS→TM = 2.3 kcal/mol). (g) The overall population distribution reveals the dominance of the surface states over any TM structures. To see this figure in color, go online.
Membrane permeabilization
At present, there is no quantitative link between the potency of AMP activity with the degree of vesicle leakage. However, it is clear that AMPs somehow permeabilize bacterial membranes (7, 60, 61). This could, in principle, lead to cell death via leakage of ions (H+, Na+, K+), ATP, or larger metabolites via membrane disruption, by depolarizing the membrane, or by osmotic lysis via increased water influx. Indeed, numerous ion leakage events are observed (7 Na+, 2 Cl−) through short-lived membrane water bridges during the simulations, including those formed during peptide translocation (Fig. 4). Ion transitions occur despite the fact that no applied membrane voltage, which would drive such events, is applied. The most frequent mechanism involves one surface peptide and one TM inserted peptide, as shown in Fig. 4. Initially, the membrane is completely dry. However, if an S-state peptide touches the TM helix at its polar side with its C terminus, it can tilt strongly and initiate a short-lived water-filled bridge. The terminus acts like a brush, shuttling both lipids and ions in both directions. The lifetime of these water bridges is very short (5–50 ns), and they do not resemble any of the classical pore models suggested for AMPs. In other cases, no TM-inserted AMP is needed, as the transient water bridge is generated by the three peptides touching each other at their termini via strong tilting and deep insertion (as shown in Fig. 3). Again, the lifetime of these water bridges is very short, as the snorkeling lysines rapidly pull the peptides back to a less tilted orientation.
Figure 4.

Mechanism of Na+ ion leakage and lipid flip-flop via spontaneous formation of short-lived membrane water bridges. A total of seven Na+ translocation events are found over all simulations. (a) The most common mechanism (i.e., brushing) involves one S-state peptide and one already TM inserted peptide. The charged lysines of the TM inserted peptide snorkel to both membrane leaflets, with the center of the membrane being completely dry (i.e., no water pore exists). The spontaneous formation of a temporary water channel is initiated by an S-state peptide, which tilts to place its polar C terminus toward the charged face of the TM peptide. Facilitated by anionic or zwitterionic lipids, small cations can translocate the channel, with the lipids cotransported, and the S-state peptide C terminus acts like a polar brush. (b) Such events can be very fast (5 ns) (green sphere, Na+; red sphere, DMPG phosphate; orange sphere, DMPC phosphate). (c) Given here is an example of a larger event (40 ns), in which two Na+, one DMPC, and one DMPG lipid translocate. (d) Short-lived water bridges can also spontaneously form via synchronized tilting of three S-state peptides, either all from the same interface, as well as (shown here) if one AMP has already translocated. (e) Similarly, the bridge is very short lived (∼50 ns) and immediately dissolves. To see this figure in color, go online.
Because the short-lived water bridges are tiny and partly filled with positively charged lysine residues from at least one participating peptide, cation leakage is observed to be tied to the cotransport of one or several shielding anionic DMPG lipids. All Na+ leakage events are therefore found in the DMPC/DMPG 3:1 simulations, and none in the pure DMPC bilayer studies. Thus, in marked contrast to the peptide translocation mechanism, which appears more or less similar for both membranes, PGLa appears selective toward anionic membranes, at least when the topic is leakage of cations. However, there are too few ion leakage events during all simulations to consider this a converged observation, and longer simulations will be needed in the future, when hardware performance increases will make this feasible.
Translocation kinetics
The simulation protocol used here is based on the stability of membrane-bound peptides against thermal denaturation, as shown experimentally in numerous previous studies (62, 63, 64, 65). This allows MD simulations to be carried out at elevated temperatures, which greatly speeds up sampling (62, 63, 64). This has no effect on partitioning thermodynamics (62), and the simulations here demonstrate that this holds true also for cationic AMPs such as PGLa. No helical restraints were used, yet PGLa remains fully helical up to 140°C in the simulations. Some slight terminal fraying of the helix is observed for T ≥ 160°C, therefore these simulations were only used in kinetic estimates, not in the analysis of the translocation mechanism as described. Similarly to what was observed for hydrophobic sequences, the translocation rates were found to fit extremely well to an Arrhenius equation (Fig. 5), with an activation energy of 16 kcal/mol, and alternatively to an Eyring equation with ΔH‡ = 15 kcal/mol and ΔG‡ = 19 kcal/mol. This is in line with what has been reported for the translocation barrier of charged lysine (66). Extrapolation to physiological temperatures indicates that translocation of one peptide would approximately occur every ∼10 ms at 37°C (green circle), albeit at a significant expected error. In previous studies on hydrophobic sequences, it was possible to directly measure the kinetics at these temperatures and demonstrate that such an extrapolation is indeed reliable (62). Thus, it would be expected that ∼40 ms is required to distribute half of the eight peptides in the simulation box across both bilayer leaflets at 37°C.
Figure 5.
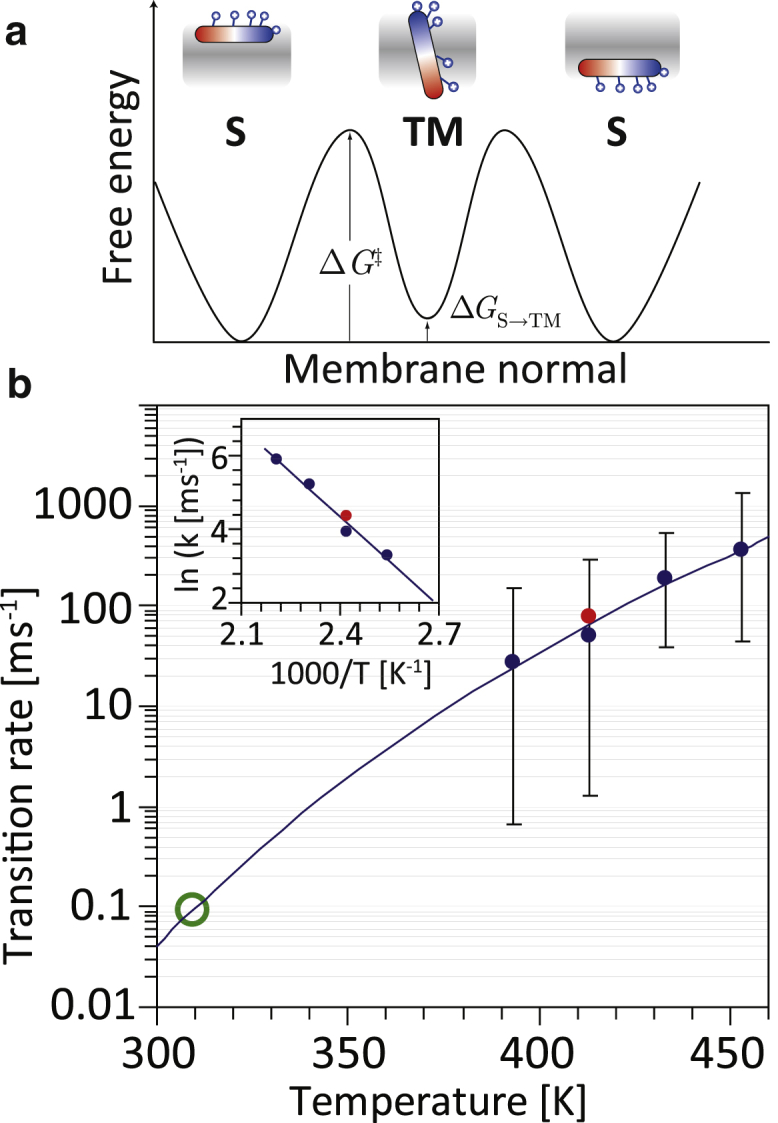
Energetics and kinetics of peptide translocation. (a) Given here is a schematic illustration of the presumed free energy profile for PGLa, for the simulation containing the long-lived monomeric TM state. A converged free energy profile is not shown, as it would require orders-of-magnitude longer simulations. The results suggest a three-state system: the surface S-state is lower in free energy than the monomeric TM state by ΔGS→TM = 2.3 (DMPC/DMPG 3:1) and 3.8 kcal/mol (pure DMPC). Thus, instead of forming pores, the peptides strongly prefer to reside at the interface. The values for ΔGS→TM are only rough estimates, as they depend on the overall converged population of the metastable TM state, which was infrequently visited. (b) The measured peptide translocation rates (blue dots, DMPC; red dots, DMPC/DMPG 3:1) fit well both to an Arrhenius equation (dashed line; inset, Arrhenius plot) with an activation energy of 16 kcal/mol, and to an Eyring equation with ΔH‡ = 15 kcal/mol and ΔG‡ = 19 kcal/mol, demonstrating the large barrier for lysine translocation. Extrapolation to physiological temperatures indicates that translocation of one peptide would (very approximately) occur every ∼10 ms at 37°C (green circle), or ∼40 ms required for the distribution of half of the eight peptides in the simulation box across both bilayer leaflets. Error bars are 95% confidence intervals by assuming a Poisson process. To see this figure in color, go online.
Discussion
Despite thousands of known sequences and two decades of systematic studies, the mechanisms of AMP membrane targeting and poration are poorly understood, and the role of membrane permeabilization in antibacterial activity is still debated (7). Although it is certain that AMPs target and permeabilize bacterial membranes (7, 60, 61), detailed pore structures have only been reported in a few cases (67). Possible reasons are that many AMPs may form transient pores, are too unstable to observe in equilibrium, or that only a small proportion of peptides might be involved in pore formation.
Although it is certain that many AMPs form pores, the results here indicate that for some AMPs like PGLa, structured channels may not be required to explain both translocation and permeabilization. This could explain why defined oligomeric channels have not been experimentally detected before for this AMP and many related ones. In all simulations, peptide translocation occurs, but no pore formation. In addition, a different model of AMP action—the formation of large surface aggregates (“carpets”) that dissolve the membrane in a concerted action—is not observed for PGLa in any of the simulations. PGLa does not aggregate at all, even at the chosen high P/L ratio of 1:22. This is not surprising, given its charge of +5. Instead, PGLa appears to behave more like a series of recently described cell-penetrating and antimicrobial sequences that appear to be able to stealthily, or silently, sneak across membranes, without large pore formation and dye influx (50, 51, 52, 53, 54).
The lack of stable pores has a relatively easy explanation: in the simulation model, TM states (any TM state) have a significantly higher free energy compared to the S-state (Fig. 5). Due to the four charged lysines, the surface-bound S-state constitutes the global free energy minimum of PGLa in the membrane, with ΔGS→TM = 2.3 (DMPC/DMPG 3:1) and 3.8 kcal/mol (pure DMPC) calculated over the 110-μs simulation time via ΔGS→TM = −kT ln(pTM/pS). These values are poorly converged because they depend strongly on the population of the TM state, which was encountered only briefly. In addition, the trapped TM state, which is dry, differs from the short-lived TM states where a peptide translocates in ∼40 ns, so the TM population is a mix of states, sharing only the upright position of the peptide. Spontaneous water bridges involving 2–3 peptides are extremely short lived because the strongly tilting participating peptides force the attached lysines to snorkel, ultimately pulling the peptides back to the S-state. The only reasonably populated TM state is a monomeric inserted peptide, which is kinetically trapped by lysines snorkeling to both interfaces (Fig. 3), but actually ΔGS→TM >2.3 kcal/mol higher than the S-state. This free energy penalty is for a thin and leaky DMPC and DMPC/DMPG bilayer. The energy penalty will be even higher in the case of a more realistic thicker bacterial bilayer.
The value for ΔGS→TM depends strongly on the sequence: in a recent study on maculatin 1.1, a value of ΔGS→TM close to zero was found, with numerous pores found and TM states occupied half of the time (45). The much higher ΔGS→TM for PGLa is due to the many more charged lysines, which render any TM orientation highly unfavorable. It is difficult to envision how this penalty could be compensated by any form of oligomerization. One could imagine a putative pore made of several TM inserted peptides, with the lysine residues lining the sides of a membrane-spanning water channel (44). However, the simulations here reveal that the establishment of any water bridge catalyzes rapid translocation of the lysine side chains of PGLa to the opposing membrane leaflet, thus allowing the peptides to return to the energetically preferred S-state (Figs. 1 and 2). Essentially, once a water pore enables lysine movement, nothing would stop the whole peptide from transitioning back to the surface. Thus, such a pore appears inherently unstable compared to the S-state. In a recent study of the AMP Piscidin 1, a similar instability of preformed pores, and strong preference for the S-state, were found (68). However, this observation is correct only for a highly charged peptide like PGLa, and not—as shown—for maculatin (45). There is also the possibility that current MD force fields get ΔGS→TM significantly wrong, which will require further investigation.
Can the observed monomeric translocation events explain membrane leakage? Although not forming durable channels, the membrane is frequently short-circuited via short-lived water bridges during peptide translocation that allow for the movement of lipids, anions, and cations. In addition, peptides can remain monomerically inserted in a TM state for considerable time, allowing formation of water bridges via contact of an additional S-state peptide. No voltages are applied in the simulation, mimicking the situation in a typical leakage assay. Under these conditions, as many as seven Na+ ions were seen to translocate in one 13-μs simulation in a bilayer patch of 80 nm2 containing nine peptides. However, it is difficult to compare these numbers directly with experimental leakage rates: first, there are too few ion conduction events for reliable kinetics. Second, the extrapolation to 37°C is problematic, because ion leakage may also occur when peptides simply cause some membrane defect, without translocation, as seen in the simulations. If the same kinetic speedup factor of ∼250 is assumed, this would indicate a rate of one ion translocation every ∼0.5 ms at 37°C, for a surface patch of 80 nm2 at 0.11 peptides/nm2, at P/L = 1:22. This extrapolation is expected to have a huge error, so a direct comparison with experimental leakage rates is reserved for future studies with longer simulation timescales, and when the temperature dependency for ion leakage has been determined. It remains to be seen whether larger cargo, such as ATP, or fluorescent dyes used in experimental leakage assays, can be cotranslocated in a similar way as ions and lipids.
The results here may provide an explanation for the frequent experimental observation that leakage from dye-filled liposomes occurs in bursts immediately after addition of the peptide, and then stops, only to restart by addition of even more peptides. This suggests that peptides translocate until both bilayer leaflets are equally populated, lowering the driving force for further translocation and thus leakage through the short-lived defects during translocation. Simulations are currently under way to ascertain whether the translocation rate is lower when peptides are initially placed in both bilayer leaflets, as compared to starting with all peptide in the same bilayer interface.
Does the mechanism found here shed light on how PGLa kills bacteria, or why it is selective for bacterial over eukaryotic membranes? Although it can be expected that ion leakage would be much larger if a membrane potential were present—as for the membrane of a living bacterium—more investigations are needed to determine if the leakage mechanism observed here is sufficient to overwhelm the bacterial metabolism maintaining the transmembrane voltage. Also, membrane leakage might not be the sole mechanism driving antimicrobial activity. Other nonpore mechanisms could involve AMP interference with cell-wall biosynthesis, cell division, or acting on periplasmic or cytoplasmic targets after translocation (7, 61, 69). It has been suggested that pore formation may merely be a by-product of getting peptides efficiently across membranes so that the peptides can cause cell-death by modulating key components of the cellular machinery (70). However, the simulations here reveal that PGLa does not detach from membranes after translocation, thus excluding it as candidate for intracellular interactions, although it might still interfere with membrane-specific proteins. The simulations here, however, can only test for membrane leakage.
Interestingly, although lipid and anion translocations are possible in all simulations, cation leakage is only observed for the anionic DMPC/DMPG 3:1 membrane simulations, and never for pure DMPC. Na+ can only traverse the short-lived water defects when associated with one or several negatively charged PG lipid headgroups. This is understandable, as the water bridge is frequently lined with the lysine side chains, which would block all cations from passing unless screened by anionic groups. Thus, the observed cation leakage is indeed selective for anionic membranes, whereas peptide translocation is not. However, much longer simulations, featuring many more ion translocations and/or also including fluorescent dyes, other cargoes or molecules used in typical leakage assays, are needed to determine whether the rare leakage events shown here are indeed representative of the presumed mechanism of membrane permeabilization.
Author Contributions
J.P.U. designed the research, performed the simulations, developed software, analyzed the data, and wrote the article.
Acknowledgments
The author thanks Erik Strandberg and Anne Ulrich for helpful discussions. Simulation resources were provided by the PI supercomputer facility at Shanghai Jiao Tong University and the MARCC facility at Johns Hopkins University.
This research was supported by a grant from China 1000 Plan’s Program for Young Talents (13Z127060001) to J.P.U.
Editor: D. Peter Tieleman.
Footnotes
One figure is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30623-9.
Supporting Material
References
- 1.Epand R.M., Vogel H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. (BBA) [DOI] [PubMed] [Google Scholar]
- 2.van ’t Hoff W., Veerman E.C., Amerongen A.V. Antimicrobial peptides: properties and applicability. Biol. Chem. 2001;382:597–619. doi: 10.1515/BC.2001.072. [DOI] [PubMed] [Google Scholar]
- 3.Phoenix D.A., Dennison S.R., Harris F. Wiley; Berlin, Germany: 2013. Antimicrobial Peptides. [Google Scholar]
- 4.Fernandez D.I., Sani M.-A., Separovic F. Interactions of the antimicrobial peptide maculatin 1.1 and analogues with phospholipid bilayers. Aust. J. Chem. 2011;64:798–805. [Google Scholar]
- 5.Wang G., Li X., Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44(Suppl D1):D1087–D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasupuleti M., Schmidtchen A., Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit. Rev. Biotechnol. 2012;32:143–171. doi: 10.3109/07388551.2011.594423. [DOI] [PubMed] [Google Scholar]
- 7.Wimley W.C., Hristova K. Antimicrobial peptides: successes, challenges and unanswered questions. J. Membr. Biol. 2011;239:27–34. doi: 10.1007/s00232-011-9343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strandberg E., Ulrich A.S. AMPs and OMPs: is the folding and bilayer insertion of β-stranded outer membrane proteins governed by the same biophysical principles as for α-helical antimicrobial peptides? Biochim. Biophys. Acta. 2015;1848:1944–1954. doi: 10.1016/j.bbamem.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann W., Richter K., Kreil G. A novel peptide designated PYLa and its precursor as predicted from cloned mRNA of Xenopus laevis skin. EMBO J. 1983;2:711–714. doi: 10.1002/j.1460-2075.1983.tb01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter K., Aschauer H., Kreil G. Biosynthesis of peptides in the skin of Xenopus laevis: isolation of novel peptides predicted from the sequence of cloned cDNAs. Peptides. 1985;6(Suppl 3):17–21. doi: 10.1016/0196-9781(85)90345-6. [DOI] [PubMed] [Google Scholar]
- 11.Andreu D., Aschauer H., Merrifield R.B. Solid-phase synthesis of PYLa and isolation of its natural counterpart, PGLa [PYLa-(4-24)] from skin secretion of Xenopus laevis. Eur. J. Biochem. 1985;149:531–535. doi: 10.1111/j.1432-1033.1985.tb08957.x. [DOI] [PubMed] [Google Scholar]
- 12.Lohner K., Prossnigg F. Biological activity and structural aspects of PGLa interaction with membrane mimetic systems. Biochim. Biophys. Acta. 2009;1788:1656–1666. doi: 10.1016/j.bbamem.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Maloy W.L., Kari U.P. Structure-activity studies on magainins and other host defense peptides. Biopolymers. 1995;37:105–122. doi: 10.1002/bip.360370206. [DOI] [PubMed] [Google Scholar]
- 14.Soravia E., Martini G., Zasloff M. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 1988;228:337–340. doi: 10.1016/0014-5793(88)80027-9. [DOI] [PubMed] [Google Scholar]
- 15.Hoskin D.W., Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinchar V.G., Bryan L., Rollins-Smith L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology. 2004;323:268–275. doi: 10.1016/j.virol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 17.Helmerhorst E.J., Reijnders I.M., Nieuw Amerongen A.V. A critical comparison of the hemolytic and fungicidal activities of cationic antimicrobial peptides. FEBS Lett. 1999;449:105–110. doi: 10.1016/s0014-5793(99)00411-1. [DOI] [PubMed] [Google Scholar]
- 18.Wieprecht T., Apostolov O., Seelig J. Membrane binding and pore formation of the antibacterial peptide PGLa: thermodynamic and mechanistic aspects. Biochemistry. 2000;39:442–452. doi: 10.1021/bi992146k. [DOI] [PubMed] [Google Scholar]
- 19.Strandberg E., Wadhwani P., Ulrich A.S. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys. J. 2006;90:1676–1686. doi: 10.1529/biophysj.105.073858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bechinger B., Kim Y., Opella S.J. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR. 1991;1:167–173. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 21.Bechinger B., Zasloff M., Opella S.J. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 1998;74:981–987. doi: 10.1016/S0006-3495(98)74021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strandberg E., Tremouilhac P., Ulrich A.S. Synergistic transmembrane insertion of the heterodimeric PGLa/magainin 2 complex studied by solid-state NMR. Biochim. Biophys. Acta. 2009;1788:1667–1679. doi: 10.1016/j.bbamem.2008.12.018. (BBA) [DOI] [PubMed] [Google Scholar]
- 23.Bechinger B., Gierasch L.M., Opella S.J. Orientations of helical peptides in membrane bilayers by solid state NMR spectroscopy. Solid State Nucl. Magn. Reson. 1996;7:185–191. doi: 10.1016/0926-2040(95)01224-9. [DOI] [PubMed] [Google Scholar]
- 24.Glaser R.W., Sachse C., Ulrich A.S. Orientation of the antimicrobial peptide PGLa in lipid membranes determined from 19F-NMR dipolar couplings of 4-CF3-phenylglycine labels. J. Magn. Reson. 2004;168:153–163. doi: 10.1016/j.jmr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Glaser R.W., Sachse C., Ulrich A.S. Concentration-dependent realignment of the antimicrobial peptide PGLa in lipid membranes observed by solid-state 19F-NMR. Biophys. J. 2005;88:3392–3397. doi: 10.1529/biophysj.104.056424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afonin S., Mikhailiuk P.K., Ulrich A.S. Evaluating the amino acid CF3-bicyclopentylglycine as a new label for solid-state 19F-NMR structure analysis of membrane-bound peptides. J. Pept. Sci. 2007;13:614–623. doi: 10.1002/psc.854. [DOI] [PubMed] [Google Scholar]
- 27.Tremouilhac P., Strandberg E., Ulrich A.S. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid state 2H-NMR. Biochim. Biophys. Acta. 2006;1758:1330–1342. doi: 10.1016/j.bbamem.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 28.Tremouilhac P., Strandberg E., Ulrich A.S. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 2006;281:32089–32094. doi: 10.1074/jbc.M604759200. [DOI] [PubMed] [Google Scholar]
- 29.Salnikov E.S., Bechinger B. Lipid-controlled peptide topology and interactions in bilayers: structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys. J. 2011;100:1473–1480. doi: 10.1016/j.bpj.2011.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuzaki K., Mitani Y., Miyajima K. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry. 1998;37:15144–15153. doi: 10.1021/bi9811617. [DOI] [PubMed] [Google Scholar]
- 31.Afonin S., Grage S.L., Ulrich A.S. Temperature-dependent transmembrane insertion of the amphiphilic peptide PGLa in lipid bilayers observed by solid state 19F NMR spectroscopy. J. Am. Chem. Soc. 2008;130:16512–16514. doi: 10.1021/ja803156d. [DOI] [PubMed] [Google Scholar]
- 32.Strandberg E., Horn D., Ulrich A.S. 2H-NMR and MD simulations reveal membrane-bound conformation of magainin 2 and its synergy with PGLa. Biophys. J. 2016;111:2149–2161. doi: 10.1016/j.bpj.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salnikov E.S., Aisenbrey C., Bechinger B. Membrane topologies of the PGLa antimicrobial peptide and a transmembrane anchor sequence by dynamic nuclear polarization/solid-state NMR spectroscopy. Sci. Rep. 2016;6:20895. doi: 10.1038/srep20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marquette A., Salnikov E.S., Bechinger B. Magainin 2-PGLa interactions in membranes - two peptides that exhibit synergistic enhancement of antimicrobial activity. Curr. Top. Med. Chem. 2016;16:65–75. doi: 10.2174/1568026615666150703115701. [DOI] [PubMed] [Google Scholar]
- 35.Glattard E., Salnikov E.S., Bechinger B. Investigations of the synergistic enhancement of antimicrobial activity in mixtures of magainin 2 and PGLa. Biophys. Chem. 2016;210:35–44. doi: 10.1016/j.bpc.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Strandberg E., Zerweck J., Ulrich A.S. Influence of hydrophobic residues on the activity of the antimicrobial peptide magainin 2 and its synergy with PGLa. J. Peptide Sci. 2015;21:436–445. doi: 10.1002/psc.2780. [DOI] [PubMed] [Google Scholar]
- 37.Leontiadou H., Mark A.E., Marrink S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006;128:12156–12161. doi: 10.1021/ja062927q. [DOI] [PubMed] [Google Scholar]
- 38.Sengupta D., Leontiadou H., Marrink S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta. 2008;1778:2308–2317. doi: 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Bennett W.F., Hong C.K., Tieleman D.P. Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers. J. Chem. Theory Comput. 2016;12:4524–4533. doi: 10.1021/acs.jctc.6b00265. [DOI] [PubMed] [Google Scholar]
- 40.White S.H., von Heijne G. How translocons select transmembrane helices. Annu. Rev. Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- 41.Bond P.J., Parton D.L., Sansom M.S. Coarse-grained simulations of the membrane-active antimicrobial peptide maculatin 1.1. Biophys. J. 2008;95:3802–3815. doi: 10.1529/biophysj.108.128686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ulmschneider J.P., Smith J.C., Strandberg E. Reorientation and dimerization of the membrane-bound antimicrobial peptide PGLa from microsecond all-atom MD simulations. Biophys. J. 2012;103:472–482. doi: 10.1016/j.bpj.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y., Zhao T., Ulmschneider J.P. How reliable are molecular dynamics simulations of membrane active antimicrobial peptides? Biochim. Biophys. Acta. 2014;1838:2280–2288. doi: 10.1016/j.bbamem.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Pino-Angeles A., Leveritt J.M., 3rd, Lazaridis T. Pore structure and synergy in antimicrobial peptides of the magainin family. PLOS Comput. Biol. 2016;12:e1004570. doi: 10.1371/journal.pcbi.1004570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Chen C.H., Ulmschneider J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016;7:13535. doi: 10.1038/ncomms13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yandek L.E., Pokorny A., Almeida P.F.F. Wasp mastoparans follow the same mechanism as the cell-penetrating peptide transportan 10. Biochemistry. 2009;48:7342–7351. doi: 10.1021/bi9008243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pokorny A., Birkbeck T.H., Almeida P.F.F. Mechanism and kinetics of δ-lysin interaction with phospholipid vesicles. Biochemistry. 2002;41:11044–11056. doi: 10.1021/bi020244r. [DOI] [PubMed] [Google Scholar]
- 48.Pokorny A., Almeida P.F.F. Kinetics of dye efflux and lipid flip-flop induced by δ-lysin in phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, α-helical peptides. Biochemistry. 2004;43:8846–8857. doi: 10.1021/bi0497087. [DOI] [PubMed] [Google Scholar]
- 49.Yandek L.E., Pokorny A., Almeida P.F. Mechanism of the cell-penetrating peptide transportan 10 permeation of lipid bilayers. Biophys. J. 2007;92:2434–2444. doi: 10.1529/biophysj.106.100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marks J.R., Placone J., Wimley W.C. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc. 2011;133:8995–9004. doi: 10.1021/ja2017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He J., Hristova K., Wimley W.C. A highly charged voltage-sensor helix spontaneously translocates across membranes. Angew. Chem. Int. Ed. Engl. 2012;51:7150–7153. doi: 10.1002/anie.201202741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cruz J., Mihailescu M., Hristova K. A membrane-translocating peptide penetrates into bilayers without significant bilayer perturbations. Biophys. J. 2013;104:2419–2428. doi: 10.1016/j.bpj.2013.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheaten S.A., Ablan F.D.O., Almeida P.F. Translocation of cationic amphipathic peptides across the membranes of pure phospholipid giant vesicles. J. Am. Chem. Soc. 2013;135:16517–16525. doi: 10.1021/ja407451c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ablan F.D., Spaller B.L., Almeida P.F. Charge distribution fine-tunes the translocation of α-helical amphipathic peptides across membranes. Biophys. J. 2016;111:1738–1749. doi: 10.1016/j.bpj.2016.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 56.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 57.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 58.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 60.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 61.Huang H.W. Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim. Biophys. Acta. 2006;1758:1292–1302. doi: 10.1016/j.bbamem.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Ulmschneider J.P., Smith J.C., Ulmschneider M.B. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 2011;133:15487–15495. doi: 10.1021/ja204042f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulmschneider M.B., Doux J.P.F., Ulmschneider J.P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 2010;132:3452–3460. doi: 10.1021/ja909347x. [DOI] [PubMed] [Google Scholar]
- 64.Ulmschneider M.B., Smith J.C., Ulmschneider J.P. Peptide partitioning properties from direct insertion studies. Biophys. J. 2010;98:L60–L62. doi: 10.1016/j.bpj.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ulmschneider M.B., Ulmschneider J.P., White S.H. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat. Commun. 2014;5:4863. doi: 10.1038/ncomms5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacCallum J.L., Bennett W.F.D., Tieleman D.P. Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J. Gen. Physiol. 2007;129:371–377. doi: 10.1085/jgp.200709745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song C., Weichbrodt C., Zeth K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. USA. 2013;110:4586–4591. doi: 10.1073/pnas.1214739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perrin B.S., Jr., Fu R., Pastor R.W. Simulations of membrane-disrupting peptides II: AMP piscidin 1 favors surface defects over pores. Biophys. J. 2016;111:1258–1266. doi: 10.1016/j.bpj.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi H., Rangarajan N., Weisshaar J.C. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microbiol. 2016;24:111–122. doi: 10.1016/j.tim.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sochacki K.A., Barns K.J., Weisshaar J.C. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc. Natl. Acad. Sci. USA. 2011;108:E77–E81. doi: 10.1073/pnas.1101130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.