

The triplet pair from singlet fission is characterized by distinct spectroscopic signature and can be difficult to break apart.
Keywords: Singlet Fission, Triplet Pair State, Solar Energy Conversion, Electron Transfer, Energy Transfer
Abstract
Singlet fission, the conversion of a singlet exciton (S1) to two triplets (2 × T1), may increase the solar energy conversion efficiency beyond the Shockley-Queisser limit. This process is believed to involve the correlated triplet pair state 1(TT). Despite extensive research, the nature of the 1(TT) state and its spectroscopic signature remain actively debated. We use an end-connected pentacene dimer (BP0) as a model system and show evidence for a tightly bound 1(TT) state. It is characterized in the near-infrared (IR) region (~1.0 eV) by a distinct excited-state absorption (ESA) spectral feature, which closely resembles that of the S1 state; both show vibronic progressions of the aromatic ring breathing mode. We assign these near-IR spectra to 1(TT)→Sn and S1→Sn′ transitions; Sn and Sn′ likely come from the antisymmetric and symmetric linear combinations, respectively, of the S2 state localized on each pentacene unit in the dimer molecule. The 1(TT)→Sn transition is an indicator of the intertriplet electronic coupling strength, because inserting a phenylene spacer or twisting the dihedral angle between the two pentacene chromophores decreases the intertriplet electronic coupling and diminishes this ESA peak. In addition to spectroscopic signature, the tightly bound 1(TT) state also shows chemical reactivity that is distinctively different from that of an individual T1 state. Using an electron-accepting iron oxide molecular cluster [Fe8O4] linked to the pentacene or pentacene dimer (BP0), we show that electron transfer to the cluster occurs efficiently from an individual T1 in pentacene but not from the tightly bound 1(TT) state. Thus, reducing intertriplet electronic coupling in 1(TT) via molecular design might be necessary for the efficient harvesting of triplets from intramolecular singlet fission.
INTRODUCTION
Singlet fission is a many-body photophysical process in molecules where the photoexcited singlet (S1) splits into two triplets (2 × T1) with spin conservation (1, 2). Since its discovery, efficient singlet fission has been reported mostly for solids and aggregates of conjugated molecules (1, 2), and a dominant mechanistic picture is the molecular dimer model (3, 4)
| (1) |
where S0 is the ground state and the intermediate 1(TT) is the correlated triplet pair with both singlet spin and double-excitation characters. Despite its prevalent use, Monahan et al. (5, 6) pointed out the inadequacy of the dimer model in describing inherently delocalized excitons in the solid state. Exciton delocalization has been cited as an important driving force for singlet fission (7–10). This problem is circumvented in recent demonstrations of efficient singlet fission in single molecules [particularly in dimers of acenes (11–16)] that allow for accurate application of the dimer model and for closely connecting experiment with theory (17). The isolation of the transient 1(TT) state in a single molecule leads to a much longer lifetime than that in the condensed phase, thus allowing spectroscopic characterization of this ambiguous and poorly understood state. This is exemplified in the detection by electron spin resonance spectroscopy in pentacene dimers of the quintet state, 5(TT), which is mixed with the 1(TT) state as predicted by the spin Hamiltonian (18).
The 1(TT) state is a singlet excited state with double-excitation characters and differs from 2 × T1 not only by the electronic and spin entangled nature of the former but also by the presence of orbital overlap, which changes its excitation energy from the sum of two triplet energies. Scholes (19) recently clarified some persistent confusion on the 1(TT) state in theoretical descriptions. The energetic difference between the correlated triplet pair state and two individual triplets, that is, the triplet pair binding energy, can be as large as 1 eV, as is known for the excited states of oligoenes (20–22), including carotenoids (23), where the tightly bound triplet pair has been called the “dark” S1 state serving as a sink for nonradiative recombination and a less tightly bound triplet pair (S*) has been associated with singlet fission (15, 24–26). In contrast, in prototypical model systems of pentacene or tetracene dimers (both covalent and van der Waals), computational analysis predicted little, if any, triplet pair binding energy (17, 27–32). However, a recent finding of similar 1(TT) lifetimes in polypentacene and bipentacene indicates that the triplet pair does not dissociate even in a long conjugated chain (33), suggesting that the correlated triplet pair state is more strongly bound than previously thought.
A major obstacle to a clear understanding of the 1(TT) state is the lack of spectroscopic signatures from experiments. Zhu and co-workers applied time-resolved two-photon photoemission spectroscopy to quantitatively determine the energetic position of the 1(TT) state from its ionization potential (IP) in crystalline pentacene (34), tetracene (10), and hexacene (6). This approach is unambiguous only for hexacene (6) where the 1(TT) state is energetically well separated from S1 but is difficult for other singlet fission systems where the 1(TT) states are in close energetic resonance with S1. The most widely used technique to probe singlet fission has been transient absorption (TA) spectroscopy, but most studies to date have only identified spectral features assigned to S1 and T1 states, and there has been little explanation as to why these TA peaks nearly always overlap (12–16, 35, 36). Exceptions to this prevalent practice can be found in the recent work of Sanders et al. (11) who found, in pentacene dimers, an excited-state absorption (ESA) peak at ~690 nm whose magnitude is strongly correlated with the strength of intertriplet electronic coupling and in the work of Pensack et al. (37) who observed near-infrared (IR) (1200 to 1400 nm) ESA in pentacene aggregates assigned to 1(TT) but not to the triplet pair labeled 1(T…T), which has lost electronic coherence but retained spin coherence. These two examples reveal the presence of spectroscopic signatures for the 1(TT) state in TA, but the origins of these transitions and their relationships to the energetics of the 1(TT) state remain unknown.
The distinct electronic structure of the 1(TT) state should be reflected not only in its spectroscopic signature but also in its chemical and physical properties. The oft-cited motivation for nearly every recent paper on this subject has been the potential “usefulness” of singlet fission to solar energy conversion. The basic argument was put forward initially by Dexter (38) for the sensitization of conventional solar cells by singlet fission chromophores in 1979, but a more recent paper by Hanna and Nozik (39) on using singlet fission to increase the solar cell efficiency above the Shockley-Queisser limit really energized the field. A number of research groups have explored the harvesting of triplet pairs from intermolecular singlet fission using solid interfaces between a singlet fission material and an electron or triplet acceptor material (34, 40–43). These efforts have also led to the successful demonstration of singlet fission–based solar cells with quantum efficiencies exceeding 100% (44). The recent demonstration of efficient intramolecular singlet fission in single molecules (11–16, 36) opens the door to new opportunities for the realization of singlet fission–sensitized solar cells (45). A more exciting opportunity is the potential for the harvesting of two electron-hole pairs for photocatalysis, for example, by coupling a singlet fission molecule to a molecular or cluster-based catalytic center (46) to enable two-electron redox reactions. Unlike intermolecular singlet fission in the solid state in which electronic delocalization (5, 47) and entropy (10) are driving forces to split the 1(TT) state to two electronically decoupled triplets (which can nonetheless retain spin coherence) on ultrafast time scales (19), the confinement in a molecular dimer or oligomer traps the two triplets in the 1(TT) state in a single molecule (18, 33). Thus, instead of individual triplets at solid-state interfaces, the harvesting of triplets in intramolecular single fission would likely come from the 1(TT) state. However, the two triplets in the 1(TT) state from intramolecular singlet fission can be tightly bound, and charge or energy transfer from each triplet may be inhibited.
Here, we use triisopropylsilylethynyl-functionalized pentacene (TIPS-Pc) dimers, each coupled at the 2-position without or with a phenylene spacer, BP0 or BP1 (Fig. 1) (11), as well as pentacene dimers with different dihedral angles (17), as model systems to quantitatively probe the nature of the tightly bound 1(TT) state from the ESA spectra. Molecules of this type allow for the systematic tuning of electronic coupling between the two pentacene units and between the S1 and the 1(TT) states, as reflected in the singlet fission time constants of τSF = 0.76, 20, and 220 ps and triplet recombination time constants of τAN = 0.45, 16.5, and 270 ns for dimers with zero, one, and two phenylene spacers (BP0, BP1, and BP2), respectively, obtained from an analysis of TA spectra in the visible region (11). Here, we focus on the distinct ESA peak in the near-IR region (hν ~ 1 eV), which is a signature of the 1(TT) state from singlet fission in BP0. Its intensity diminishes as the intertriplet electronic coupling is lowered in BP1 or significantly decreases in bipentacene with different dihedral angles (17). The ESA peak of 1(TT) in BP0 closely resembles that of the S1 state in the near-IR region; both show vibronic progressions of the aromatic ring breathing mode and can be assigned to the 1(TT)→Sn and S1→Sn′ transitions, respectively. This finding unambiguously establishes that 1(TT) is spectroscopically distinct from 2 × T1, and such a spectroscopic signature enables one to quantitatively follow the dynamics of this critical intermediate in singlet fission.
Fig. 1. The model systems for intramolecular singlet fission and triplet harvesting.
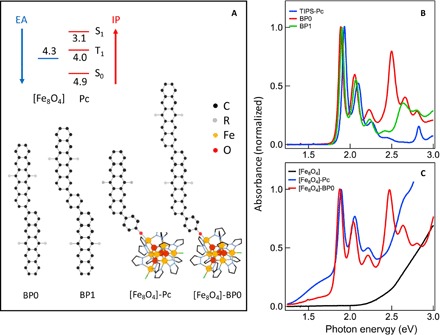
(A) Schematics of BP0, BP1, [Fe8O4]-Pc, and [Fe8O4]-BP0. R = (triisopropylsilyl)ethynyl (TIPS) for [Fe8O4]-Pc and (n-octyldiisopropyl)silylethynyl (NODIPS) for [Fe8O4]-BP0 and [Fe8O4]-BP1. The inset shows estimated IP and EA (electron affinity) from electrochemical oxidation/reduction potentials of [Fe8O4] and TIPS-pentacene. (B and C) Optical absorption spectra of (B) TIPS-Pc, BP0, and BP1 in toluene and (C) [Fe8O4], [Fe8O4]-Pc, and [Fe8O4]-BP0 in dichloromethane solutions.
To establish the distinct chemical properties of the 1(TT) state, we use the redox-active molecular cluster Fe8O4pz12Cl4 (pz, pyrazolate), which we label [Fe8O4], as an electron acceptor (48, 49) and tether BP0 to [Fe8O4] through a Fe-phenoxide bond (schematically illustrated in Fig. 1A). As a control, we replace the pentacene dimer BP0 with a pentacene monomer (Pc). Note that the formation of the Fe-phenoxide bond in both [Fe8O4]-Pc and [Fe8O4]-BP0 introduces a low-energy absorption tail (~1.3 to 1.8 eV; Fig. 1C). This spectral feature has been assigned to the phenolate-to-Fe(III) ligand-to-metal charge transfer (LMCT) transition (50) but may also have contributions from pentacene-to-[Fe8O4] charge transfer (CT) transitions. We show that electron transfer from pentacene to [Fe8O4] occurs efficiently from an individual T1 state in pentacene ([Fe8O4]-Pc), but not from the tightly bound triplet pair state in [Fe8O4]-BP0. This finding establishes that the chemical property of the 1(TT) state is distinctively different from that of an individual triplet and suggests that reducing intertriplet electronic coupling in 1(TT) might be needed for the harvesting of triplets from intramolecular singlet fission.
RESULTS AND DISCUSSION
Spectroscopic signature of the 1(TT) state
We use TA spectroscopy to probe singlet fission in BP0 and BP1 (11). We excite the S1 state of each pentacene dimer at hν1 = 2.1 eV and probe the subsequent dynamics from the TA of a white-light continuum (Fig. 2). Figure 2 (A and B) shows TA spectra at selected pump-probe delays, Δt = 0.1 ps (red), 10 ps (purple), and 100 ps (blue) for BP0. The visible parts of the TA spectra have been discussed extensively before, and the broad positive TA features at Δt < 1 ps and Δt > 2 ps are assigned to the ESA of S1 and T1, respectively (11). The latter is confirmed by the ESA spectrum of T1 obtained from sensitization (black). On the basis of the calculated triplet energies in pentacene (T3 and T4 are close in energy and are not distinguished here) (51), we assign the ESA peak at 2.42 eV to the T1→T3 transition. For the triplet pair from singlet fission, this transition corresponds to 1(T1T1)→1(T1T3). In each case, the ESA transition also shows vibronic progression (hνvib ~ 0.17 eV) similar to those in the ground-state absorption spectrum (11). The singlet decay and triplet formation can be clearly seen from kinetic profiles at probe photon energies of hν2 = 2.13 eV (gray) and 2.42 eV (green), respectively (Fig. 2C), with τSF = 0.7 ps (11); note that there is an overlapping contribution to ESA signal at hν2 = 2.42 eV from the singlet at short time scales. The two triplets confined to the pentacene dimer can be assigned to 1(TT), which decays on the time scale of τTT1 = 450 ps (see the green curve at long pump-probe delays in Fig. 2C), much shorter than the 30-μs lifetime of an individual triplet (11). Here, we focus on the near-IR region, which provides key spectroscopic insight into the triplet pair state.
Fig. 2. TA in the near-IR and visible regions reveal singlet and triplet characters of 1(TT).
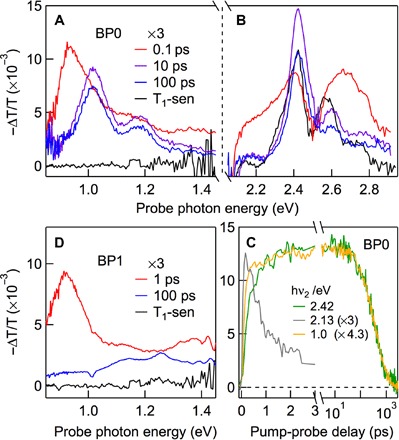
TA spectra in (A) the near-IR and (B) the visible regions for BP0 at different pump-probe delays, Δt = 0.1 ps (red), 10 ps (purple), and 100 ps (blue), following excitation at time zero by hν1 = 2.1 eV. The triplet TA spectrum from sensitization (black) is also shown in (A) and (B). (C) Kinetic profiles from TA spectra for BP0 at the indicated probe photon energies. (D) TA spectra at Δt = 1 ps (red) and 100 ps (blue) for BP1 following excitation at time zero by hν1 = 2.1 eV. The corresponding triplet spectrum (black) from sensitization is also shown.
There is a distinct ESA peak at 0.922 ± 0.005 eV when the singlet dominates at Δt = 0.1 ps (red) (Fig. 2A); this peak is also accompanied by a vibronic feature on the higher energy side, with hνvib ~ 0.17 eV, similar to the vibronic progressions of S0→S1, T1→Tn, and 1(T1T1)→1(T1Tn) discussed above. This ESA is assigned to an S1→Sn′ transition, with transition energy close to the S1→S2 transition for a single pentacene molecule. In the absorption spectrum of TIPS-Pc in Fig. 1B (blue), there is a weakly allowed S0→S2 peak at 2.82 eV, in agreement with the two-photon absorption spectrum of the same molecule (52). Given the S0→S1 peak at 1.93 eV (blue spectrum in Fig. 1B), we obtain the S1→S2 transition energy at 0.89 eV. In conjugated bipentacene dimers, the singlet states are described by linear combinations of two localized states on each pentacene chromophore (17). Although both symmetric and antisymmetric linear combinations are possible, the optically bright S1 state in BP0 is of odd parity (u). Therefore, excited state transitions must occur to Sn′ states of even parity (g). We assign the 0.92-eV peak to a transition from S1 to the symmetric linear combination of the monomer S2 states.
At longer pump-probe delays, for example, Δt = 10 ps (purple) or 100 ps (blue), when there is only the triplet pair state, the ESA spectrum blue-shifts to 1.012 ± 0.005 eV and the vibronic signature becomes better resolved. This ESA peak does not originate from a T1→Tn transition as it is completely absent in the triplet absorption spectrum (black) from sensitization. On the basis of the similarity of this ESA peak to that of the S1→Sn′ transition at early times, we assign the former to a 1(TT)→Sn transition. The 1(TT) state in BP0 is expected to correspond to the totally symmetric representation, as shown theoretically by Fuemmeler et al. (17); it will be of opposite parity to the S1 state and will exhibit a distinct set of excited state transitions to states of odd parity (17). Sn is expected to be close in energy to Sn′, because the difference in the S1→Sn′ and 1(TT)→Sn transition energies, ΔE = 90 meV, is close to the predicted exoergicity of ~100 to 150 meV for singlet fission in bipentacene (17, 27, 29–32). The small energy spacing implies that both Sn and Sn′ likely originate from different linear combinations of the S2 monomer state of different parity. Note that, unlike the results shown here for the pentacene dimer, the near-IR ESA assigned to 1(TT) in pentacene aggregates does not show vibronic features (37).
The ESA spectrum of the 1(TT) state reveals its delocalized singlet and localized triplet characters in the near-IR and the visible regions, respectively. We use “delocalized singlet” or “delocalization” to emphasize 1(TT) in a single electronic state, which can be approximately viewed as two T1 states (on two pentacene units) that are electronically coupled and coherent. Likewise, the term “localized triplet” or “localization” refers to a T1 state on an individual pentacene unit with physical properties that are insensitive to the presence or absence of electronic coupling and coherence with a neighboring T1 state. Spectroscopically, delocalization and localization are reflected in the transitions 1(TT)→Sn and 1(T1T1)→1(T1Tn), respectively. Note that the two notations, 1(TT) and 1(T1T1), describe the same triplet pair state. The kinetic profiles (Fig. 2C) for the 1(TT)→Sn (orange) and 1(T1T1)→1(T1Tn) (green) transitions are similar; the difference at short time scales (<1 ps) can be attributed to the different overlapping contributions from ESA of S1. Note that transitions to Sn are allowed from 1(TT) but spin-forbidden from 3(TT) or 5(TT). The perfect agreement between the decays of 1(TT)→Sn and the 1(T1T1)→1(T1Tn) signals suggests that there are negligible transitions within the triplet pair manifold, for example, 1(TT)→5(TT) or 1(TT)→3(TT), during the lifetime (450 ps) of the triplet pair state. Transitions within the triplet pair manifold are expected to occur on much longer time scales (18, 53).
Supporting the conclusion that delocalization or intertriplet electronic coupling in the 1(TT) state is reflected in the 1(TT)→Sn transition strength, we find that, in BP1, the weakening of the inter-T1 electronic coupling diminishes its delocalized character as reflected in the 1(TT)→SN transition strength (Fig. 2D) where the near-IR peak for 1(TT) at long times, for example, Δt = 100 ps (blue), becomes nonresolvable from the broad background, in distinct contrast to the S1→Sn peak at Δt = 1 ps. In contrast, the localized character represented by the 1(T1T1)→1(T1Tn) transition in the visible region remains (11).
To more quantitatively isolate the S1 spectrum from that of the 1(TT), we carry out global analysis based on a sequential kinetic model, S1→1(TT)→S0 (11). The resulting S1 (red) and 1(TT) (blue) spectra are shown in Fig. 3. The global analysis yields time constants of 0.75 ± 0.05 ps and 460 ± 10 ps for singlet fission and triplet pair annihilation, respectively, in agreement with the previous report (11). Similar to the S1-Sn′ transition, the 1(TT)-Sn transition is also characterized by vibronic peaks assigned to 0-0 and 0-1 transitions, with a vibrational energy spacing of 0.16 to 0.17 eV, which corresponds to the ring breathing mode of pentacene along the short molecular axis (54). In addition to the near-IR peak, the 1(TT) state in BP0 also features a distinct peak at 1.810 ± 0.005 eV. Similar to the transition at 1.012 ± 0.003 eV, the peak at 1.810 ± 0.005 eV diminishes as the intertriplet coupling weakens in BP1 and BP2 (11). Thus, the peak at 1.810 ± 0.005 also reflects the singlet character of the 1(TT) state and can be assigned to a 1(TT)→Sn″ transition. Because of the overlapping bleaching feature (S0→S1), we are not able to resolve vibronic progression for this transition.
Fig. 3. TA spectra of BP0 for the S1 and 1(TT) states from global analysis.
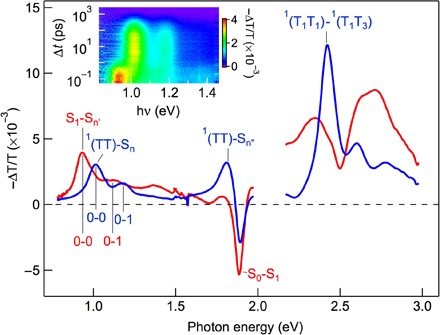
Red: Singlet state. Blue: Triplet pair state. Inset: 2D pseudocolor (intensity) plot of TA spectra following excitation at time zero by hν1 = 2.1 eV. The transitions, along with vibronic progressions, are shown on each spectrum.
Further supporting the conclusion that the near-IR 1(TT)→Sn″ transition is a spectroscopic signature of the tightly bound triplet pair state, we turn to modified BP0 molecules with different dihedral angles. In this approach, we control the dihedral angle twist by steric hindrance from the phenyl group attached to the 1-position of one or both pentacene units in the bipentacene molecule, as shown schematically in the insets in Fig. 4 (17). Computational analysis gives dihedral angles between the two pentacene molecules of 42° and 57°, and these two molecules are therefore labeled as BP-42 and BP-57, respectively (17). For comparison, the dihedral angle in BP0 is 37°; thus, BP0 ≡ BP-37. Theoretical analysis showed that the intertriplet electronic coupling decreases with increasing dihedral angle (17). The singlet fission time constants are τSF = 0.76, 1.69, and 3.38 ps, and the corresponding triplet-triplet annihilation time constants are τSF = 0.45, 1.6, and 5.2 ns for BP-37, BP-42, and BP-57, respectively (17). Figure 4 shows the near-IR region of the S1 (red) and 1(TT) (blue) ESA spectra for BP-42 (top) and BP-57 (bottom). We multiply the 1(TT) spectra by factors of 2.5 and 4.6 for BP-42 and BP-57, respectively, to normalize the 1(TT)-Sn peak intensity to the S1-Sn′ intensity in each case. For comparison, the normalization factor would be 1.25 for BP-37 in Fig. 3. Thus, relative to the S1-Sn′ transition, the 1(TT)-Sn transition strength is 80, 40, and 22% for BP-37, BP-42, and BP-57, respectively. This confirms the correlation between the 1(TT)-Sn ESA transition strength and the intertriplet electronic coupling in the 1(TT) state.
Fig. 4. TA of the 1(TT) state in the near-IR region depends on electronic coupling.
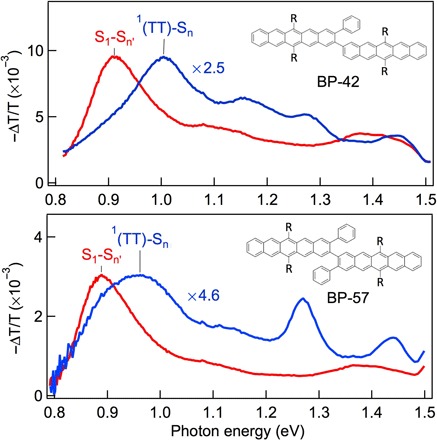
Near-IR TA spectra of BP-42 (top) and BP-57 (bottom). The 1(TT) spectra (blue) have been multiplied by factors of 2.5 and 4.6 for BP-42 and BP-57, respectively, to normalize the peak intensities of 1(TT) to those of S1 (red).
In all the pentacene dimers investigated here, the decay rate of the triplet pair state is also found to be strongly correlated with the extent of delocalization in the 1(TT) state, which is reflected in the 1(TT)-Sn transition strength. This is understood as the rate of T1-T1 annihilation is determined by the inter-pentacene electronic coupling strength, as addressed in detail elsewhere (11, 17).
The relative amplitudes of the 0-0 and 0-1 transitions allow us to estimate the Huang-Rhys factor (S) in each case and, thus, the relative shifts in the potential energy surfaces (PESs) involved. The Huang-Rhys factor is related to the offset (ΔQe) in the equilibrium positions of the two PESs in an optical transition: S = 0.5α (ΔQe)2, where α = μω/ℏ; μ is the reduced mass, and ω is the angular frequency of the vibration (55). In the harmonic oscillator and low-temperature approximation appropriate for the pentacene ring breathing mode at room temperature, the ratio in the Franck-Condon factors (and the ratio in peak intensities) between the 0-1 and 0-0 transition is equal to the Huang-Rhys factor (55). Thus, we obtain S = 0.36 ± 0.05 and 0.45 ± 0.05 for the S1→S3 and 1(TT)→S3 transitions, respectively, from the near-IR ESA spectra for BP0 in Fig. 3. For comparison, we obtain from the optical absorption spectrum a value of S = 0.55 ± 0.05 for the S0-S1 transition (11). For the pentacene ring breathing mode, we neglect the difference in equilibrium geometries between Sn′ and Sn, because they both likely come from the linear combination of S2 in each pentacene chromophore. The spectroscopic results obtained above allow us to construct PESs for singlet fission in BP0. Although there are four possible arrangements of the PES from experimental ΔQ values, Fig. 5 shows the scenario that is more consistent with the expectation of increasing nuclear displacement with excitation energy. The offset in equilibrium positions of the S1 PES and the 1(TT) PES (ΔQe ~ 0.081 Å) is also consistent with theoretical results on the covalent dimer (17). The barrierless nature of the crossing point between S1 and 1(TT) explains the fast singlet fission rate for BP0. Furthermore, the PES of 1(TT) crosses that of S0 with only two vibrational quanta on the former; this opens up an efficient nonradiative decay pathway. The nonradiative lifetime of 1(TT) (450 ps in BP0) is shorter than that of the radiative lifetime (~13 ns) of S1 in TIPS-pentacene (11, 56). Although Fig. 5 is only an approximation given the uncertainties in the spectroscopic determination of Huang-Rhys factors, it represents the first estimation of PES for singlet fission from experimental data.
Fig. 5. Estimated PES for BP0 molecule.
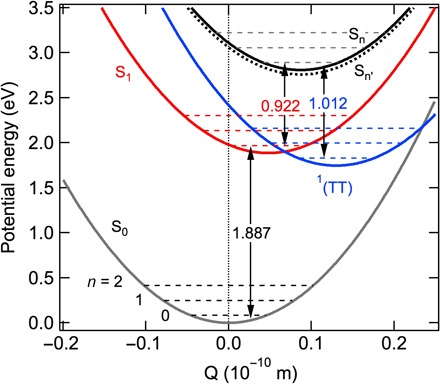
The barrier-less nature for the crossing from S1 (red) to 1(TT) (blue) facilitates the fast singlet fission for BP0. The near-IR transition for BP0 can be explained by the transition from 1(TT) to Sn, which is similar to the transition from S1 to Sn′.
Distinct chemical property of the 1(TT) state
The above spectroscopic analysis of singlet fission in BP0 provides evidence for a strong coupled triplet pair state, 1(TT), whose delocalized and localized characters are revealed in ESA in the near-IR and visible regions, respectively. Here, we show that the tightly bound triplet pair state exhibits chemical properties that are different from those of an individual triplet.
The inset in Fig. 1A shows the estimated values for the IP and EA for TIPS-Pc and [Fe8O4], respectively. These values are obtained from the electrochemical oxidation potentials for TIPS-Pc (57) and [Fe8O4] (58), respectively, based on the reference value of the Ag/AgCl electrochemical potential at 4.4 eV below vacuum energy (EV) (59). Also shown are the estimated IPs of S1 and T1 states from the excitation energies of TIPS-Pc. The use of IPs and EAs of both ground and excited states allows us to accurately put all relevant energy levels on the same single-particle diagram, as discussed in detail by Zhu (60). Note that the energy levels obtained from electrochemistry are adiabatic single-particle energies and can be used to approximate the vertical single-particle energies, that is, highest occupied molecular orbital and lowest unoccupied molecular orbital, when the reorganization energies are negligible (60). Given this approximate energy level diagram, we expect efficient electron transfer from either the T1 or the S1 in pentacene to the [Fe8O4] cluster. Figure 1C compares the optical absorption spectra of the [Fe8O4] cluster (black) and those of compounds [Fe8O4]-Pc (red) and [Fe8O4]-BP0 (blue). The absorption spectra of both [Fe8O4]-Pc and [Fe8O4]-BP0 primarily arise from the sum of the absorption spectra of [Fe8O4] and that of pentacene or bipentacene. An additional broad feature below 1.75 could contain a CT state from the Pc-PhO-ligand or the BP0-PhO-ligand to the [Fe8O4] cluster, in addition to the more local LMCT transition (50).
The energy level alignment in Fig. 1A suggests that, in addition to direct photoexcitation of the CT state, electron transfer can occur from T1 in pentacene to [Fe8O4] to indirectly form the CT state. We find that CT and T1 are strongly coupled resonantly. When we directly excite the CT state in [Fe8O4]-Pc or [Fe8O4]-BP0 at hν = 1.65 eV (Fig. 6A), we observe in each case a TA spectrum characteristic of the T1 state in pentacene, including an ESA peak at ~2.4 eV and a ground-state bleaching at 1.88 and 2.05 eV (see fig. S1 for complete TA data for [Fe8O4]-Pc). Although we observe small differences in the TA spectra for [Fe8O4]-Pc (green) and [Fe8O4]-BP0 (red), they all match their T1 spectra obtained by sensitization very well. For clarity, here, we only present the T1 spectrum of [Fe8O4]-BP0 (gray); see the Supplementary Materials for T1 spectra of the other molecules. Note that neither the isolated [Fe8O4] nor the uncoupled pentacene molecules absorb light below ~1.75 eV. Excitation of isolated [Fe8O4] at higher photon energies results in completely different TA spectra (fig. S2). The ultrafast formation of T1 within experimental time resolution (~100 fs) from the selective excitation of CT indicates that the cluster and pentacene ligands are strongly electronically coupled. Supporting this conclusion, we found in a triplet sensitization experiment that the observable T1 signal from [Fe8O4]-Pc is an order of magnitude lower than that from TIPS-Pc (fig. S3).
Fig. 6. TA reveals the strong coupling of CT state to T1.
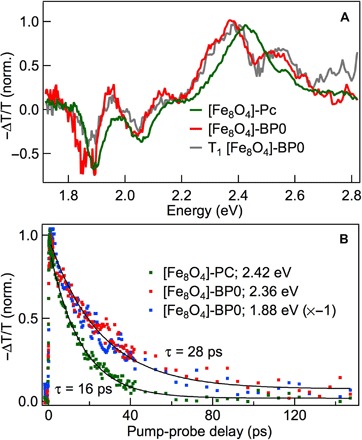
(A) TA spectra at 1 ps for [Fe8O4]-Pc (green) and [Fe8O4]-BP0 (red) upon CT excitation of 1.65 eV. The gray curve is the triplet spectrum of [Fe8O4]-BP0 from triplet sensitization. (B) Triplet decay dynamics for [Fe8O4]-Pc (green) and [Fe8O4]-BP0 (red and blue for ESA and ground-state bleaching, respectively). The solid curves are single-exponential fits with the indicated lifetimes (τ = 16 ± 2 ps for [Fe8O4]-Pc and 28 ± 3 ps for [Fe8O4]-BP0).
The coupled T1-CT state features first-order decay kinetics well described by single-exponential decays (solid curves in Fig. 6B), with time constants of τCT-T1 = 28 ± 3 ps and 16 ± 2 ps for [Fe8O4]-BP0 and [Fe8O4]-Pc, respectively. The simple first-order kinetics is reflected in both the decay in T1-like ESA signal (red dots) and the recovery in ground-state bleaching (blue dots) for [Fe8O4]-BP0 in Fig. 6B. The T1-CT decay constant is five orders of magnitude shorter than that of an individual T1 state in pentacene or bipentacene molecules (11). Because no fluorescence emission is observed for any of the cluster-pentacene complexes, we assign the fast decay in the T1-CT state to nonradiative recombination. Both CT across the pentacene-cluster interface and the presence of paramagnetic Fe atoms can couple to electron spins, thus facilitating recombination (61).
Unlike the strong coupling of individual T1 in pentacene or bipentacene to the CT state at their interfaces to [Fe8O4], we find that the triplet state in the tightly bound 1(TT) in BP0 does not undergo CT to the electron accepting cluster. Figure 7A shows TA spectra for [Fe8O4]-BP0 as a function of pump-probe delay, following initial photoexcitation at hν1 = 2.1 eV. Figure 7B shows horizontal cuts at selected pump-probe delays (Δt = 0, 10, and 100 ps), along with a T1 spectrum obtained from sensitization of [Fe8O4]-BP0. At this excitation photon energy, BP0 is known to undergo efficient singlet fission (11), and the results for [Fe8O4]-BP0 are nearly identical to those in BP0. Initially (Δt = 0 ps; red spectrum in Fig. 7B), the TA spectrum is that of S1 characterized by the broad ESA in the visible region and a vibronically resolved ESA in the near-IR region. The singlet exciton decay and triplet rise in [Fe8O4]-BP0 are both characterized by a single-exponential lifetime of τSF = 0.55 ± 0.02 ps, which is slightly shorter than the corresponding process in BP0 (τSF = 0.76 ps) (11). Figure 7D compares the 1(TT) decay dynamics in [Fe8O4]-BP0, as monitored by the decays of ESA signals attributed to both triplet (2.36 eV, red) and singlet (0.97 eV, green) characters. For comparison, we also show in Fig. 7D the 1(TT) decay dynamics in BP0 (2.36 eV, blue). The three decay traces are superimposable. The data for [Fe8O4]-BP0 are well described by a single-exponential decay with a time constant of τTT = 0.42 ± 0.03 ns, which is, within experimental uncertainty, identical to that of BP0. In stark contrast to the efficient CT from an individual T1 state in [Fe8O4]-Pc, there is no measurable CT from the tightly bound 1(TT) state in [Fe8O4]-BP0.
Fig. 7. TA spectra and dynamics of [Fe8O4]-BP0 under 2.1 eV excitation.
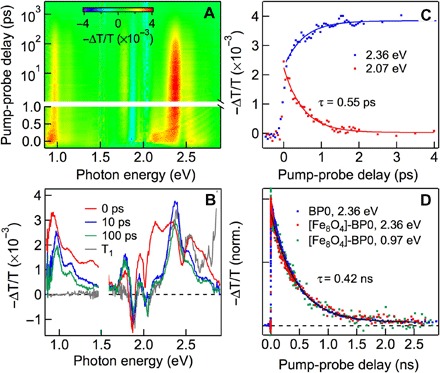
(A) 2D pseudocolor plot of TA (= −ΔT/T; T, transmission) as a function of pump-probe delay (Δt) and probe photon energy. (B) TA spectra at Δt = 0 ps (red), 10 ps (blue), and 100 ps (green), along with T1 spectrum from sensitization (gray). (C) Singlet fission dynamics, as represented by S1 decay at 2.07 eV (red) or 1(TT) buildup at 2.36 eV (blue). (D) Comparison of 1(TT) decay dynamics for [Fe8O4]-BP0 and BP0.
In summary, using covalently linked pentacene dimers as model systems, we show evidence for a tightly bound triplet pair state, which reveals its delocalized 1(TT) and localized T1 characters in the near-IR and visible ESA spectra, respectively. The near-IR ESA spectra can be assigned the 1(TT)→Sn transition, which is similar to the S1→Sn′ transition, with vibrational progression corresponding to the well-known aromatic ring breathing mode. The 1(TT)→Sn transition is an indicator of the intertriplet coupling strength; when a phenylene spacer is inserted between the pentacene moieties (BP1) or varies the angle between the pentacene moieties (BP45, BP90, and 1,2-BP) to decrease this coupling, we find that the 1(TT)→Sn ESA peak decreases. This is in contrast to the spectrum in the visible region, assigned to the 1(T1T1)→1(T1T3) transition present with similar intensities for all bipentacene molecules. Using an electron-accepting iron oxide molecular cluster [Fe8O4] linked to pentacene and bipentacene (BP0), we find that electron transfer to the cluster occurs efficiently from an individual T1 but not from the 1(TT) state. Thus, the tightly bound 1(TT) state exhibits a distinctively different chemical reactivity from that of an individual T1 state. A viable strategy to efficiently harvest triplets from intramolecular singlet fission is to control the intertriplet electronic coupling via molecular design.
MATERIALS AND METHODS
Synthesis
The synthesis of TIPS-Pc, BP0, and BP1 molecules (11); BP0 with different dihedral angles (17); and the [Fe8O4] cluster (49) has been previously described. To install the pentacene-based ligands on [Fe8O4], we first deprotonated the pendent phenol group with an excess of sodium hydride in tetrahydrofuran (THF). The reaction mixture was filtered through a 0.2-μm syringe filter and added dropwise to a solution of [Fe8O4] in THF. We used a 1:1 stoichiometric ratio of the ligand to [Fe8O4] to prepare the monosubstituted clusters, which were purified by reversed-phase chromatography. Additional synthetic details and characterization data can be found in the Supplementary Materials.
Optical absorption
The TIPS-Pc, BP0, and BP1 samples were dissolved in dry toluene (with a concentration of <100 μM) and kept free from oxygen and moisture for optical measurements on a Shimadzu UV 1800 UV-Vis Spectrophotometer. Ultaviolet-visible (UV-Vis) absorption spectra of BP0 and BP1 (Fig. 1B) showed a slight red shift from that of TIPS-Pc but contained otherwise nearly identical vibronic features near the absorption threshold (S0→S1) (11). Solutions of [Fe8O4], [Fe8O4]-Pc, or [Fe8O4]-BP0 in chloroform were used for absorption measurements. Optical absorption spectra of [Fe8O4], [Fe8O4]-Pc, and [Fe8O4]-BP0 in Fig. 1C will be discussed later.
Transient absorption
To investigate singlet fission and triplet transfer, we used femtosecond TA (fs-TA) spectroscopy. The samples were dissolved in dry toluene and kept free from oxygen and moisture. The pump pulse came from an optical parametric amplifier (tunable from UV to the near-IR, 100-fs pulse width, 1 kHz rep rate). The probe pulse was a white-light supercontinuum (from 450 to 850 nm and from 850 to 1600 nm for the visible and near-IR range, respectively). The delay between pump and probe pulses was controlled by a translational stage with a delay time up to 3 ns. The detection consisted of a pair of multichannel detector arrays coupled to a high-speed data acquisition system (HELIOS, Ultrafast System Inc.). The sample solution was at room temperature during measurement. The nanosecond-microsecond TA measurements were carried out on the same setup as fs-TA with the same pump pulse. The probe pulse was a white-light supercontinuum (from 400 to 1600 nm) generated by a supercontinuum laser (Leukos). The laser pulse width was ≤1 ns at 2 kHz. The pump-probe delay was controlled electrically.
The triplet-sensitizing experiment was carried out on the same setup except for the fact that the white-light probe beams were generated by a picosecond laser and the pump-probe delay was controlled electrically. A mixture of a (bi)pentacene compound and an excess of anthracene was dissolved in toluene with the concentration of anthracene ~100× that of (bi)pentacene. Photoexcitation at 3.35 eV created singlets in anthracene, which underwent intersystem crossing to form triplets. The triplets in anthracene subsequently transferred to (bi)pentacene molecules via diffusional collisions on a time scale of 1 to 2 μs (see the Supplementary Materials).
Supplementary Material
Acknowledgments
We thank E. Fuemmeler, T. J. H. Hele, and N. Ananth for discussions about the irreducible representation of the triplet pair state reported. Funding: The work on spectroscopic signatures (Figs. 2 to 5) of the triplet pair state was supported by the U.S. Department of Energy (DOE) (grant DE-SC0014563 to X.-Y.Z.). The work on [Fe8O4] clusters (Figs. 6 and 7) was supported by the U.S. Air Force Office of Scientific Research (grant FA9550-14-1-0381 to X.-Y.Z. and X.R.). Research was carried out in part at the Center for Functional Nanomaterials, which is a DOE Office of Science Facility, at Brookhaven National Laboratory under contract no. DE-SC0012704. A.B.P. and S.N.S. thank the NSF Graduate Research Fellowship Program for support (DGE 11-44155). Author contributions: M.T.T. and M.Y.S. carried out all TA experiments. A.P., A.B.P., and E.K., supervised by L.M.C. and X.R., carried out all synthesis and characterization. M.T.T., S.N.S., M.Y.S., and X.-Y.Z. analyzed the data. M.T.T. and X.-Y.Z. wrote the manuscript, with inputs from all authors. Competing interests: L.M.C., M.Y.S., S.N.S., E.K., and A.B.P. are inventors on a patent related to this work, filed through Columbia University (Publication No. WO2016/100754; 23 June, 2016). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700241/DC1
Transient absorption
Triplet-sensitizing experiments
Compound synthesis
fig. S1. Transient absorption (TA) spectra and dynamics of [Fe8O4]-Pc.
fig S2. Transient absorption for Fe8O4pz12Cl4 cluster (pumped at 2.58 and 2.07 eV) and [Fe8O4]-Pc (pumped at 1.65 eV).
fig. S3. Triplet-sensitizing experiments.
fig. S4. Synthetic route for compound 2.
fig. S5. Synthetic route for compound 3.
fig. S6. Synthetic route for compound Pc-Phenol.
fig. S7. Synthetic route for compound BP0-Phenol.
fig. S8. Synthetic route for compound BP1-Phenol.
fig. S9. Synthetic route for compound [Fe8O4]-Pc, [Fe8O4]-BP0, and [Fe8O4]-BP1.
fig. S10. Infrared spectra.
fig. S11. Absorption spectra.
fig. S12. Normalized absorption spectra.
fig. S13. Normalized absorption spectra.
fig. S14. NMR spectrum (0–9.5 ppm), compound 2.
fig. S15. NMR spectrum (0–145 ppm), compound 2.
fig. S16. NMR spectrum (0–9.5 ppm), compound 3.
fig. S17. NMR spectrum (0–145 ppm), compound 3.
fig. S18. NMR spectrum (0–9.5 ppm), Pc-Phenol.
fig. S19. NMR spectrum (0–155 ppm), Pc-Phenol.
fig. S20. NMR spectrum (0–9.5 ppm), BP0-Phenol.
fig. S21. NMR spectrum (0–155 ppm), BP0-Phenol.
fig. S22. NMR spectrum (0–9.5 ppm), BP1-Phenol.
fig. S23. NMR spectrum (0–160 ppm), BP1-Phenol.
fig. S24. NMR spectrum (−40 to 55 ppm), [Fe8O4]-Pc, before solvent addition.
fig. S25. NMR spectrum (−40 to 55 ppm), [Fe8O4]-Pc, after solvent addition.
fig. S26. Negative and positive mode NMR spectra (1000 to 5000 m/z), [Fe8O4]-BP0.
fig. S27. Negative and positive mode NMR spectra (1000 to 5000 m/z), [Fe8O4]-BP1.
REFERENCES AND NOTES
- 1.Smith M. B., Michl J., Singlet fission. Chem. Rev. 110, 6891–6936 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Smith M. B., Michl J., Recent advances in singlet fission. Annu. Rev. Phys. Chem. 64, 361–86 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Johnson R. C., Merrifield R. E., Effects of magnetic fields on the mutual annihilation of triplet excitons in anthracene crystals. Phys. Rev. B 1, 896–902 (1970). [Google Scholar]
- 4.Suna A., Kinematics of exciton-exciton annihilation in molecular crystals. Phys. Rev. B 1, 1716–1739 (1970). [Google Scholar]
- 5.Monahan N., Zhu X.-Y., Charge transfer–mediated singlet fission. Annu. Rev. Phys. Chem. 66, 601–618 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Monahan N. R., Sun D., Tamura H., Williams K. W., Xu B., Zhong Y., Kumar B., Nuckolls C., Harutyunyan A. R., Chen G., Dai H.-L., Beljonne D., Rao Y., Zhu X.-Y., Dynamics of the triplet pair state reveals the likely co-existence of coherent and incoherent singlet fission in crystalline hexacene. Nat. Chem. 9, 341–346 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Pensack R. D., Tilley A. J., Parkin S. R., Lee T. S., Payne M. M., Gao D., Jahnke A. A., Oblinsky D. G., Li P.-F., Anthony J. E., Seferos D. S., Scholes G. D., Exciton delocalization drives rapid singlet fission in nanoparticles of acene derivatives. J. Am. Chem. Soc. 137, 6790–6803 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Wang R., Zhang C., Zhang B., Liu Y., Wang X., Xiao M., Magnetic dipolar interaction between correlated triplets created by singlet fission in tetracene crystals. Nat. Commun. 6, 8602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teichen P. E., Eaves J. D., Collective aspects of singlet fission in molecular crystals. J. Chem. Phys. 143, 044118 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Chan W.-L., Ligges M., Zhu X.-Y., The energy barrier in singlet fission can be overcome through coherent coupling and entropic gain. Nat. Chem. 4, 840–845 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Sanders S. N., Kumarasamy E., Pun A. B., Trinh M. T., Choi B., Xia J., Taffet E. J., Low J. Z., Miller J. R., Roy X., Zhu X.-Y., Steigerwald M. L., Sfeir M. Y., Campos L. M., Quantitative intramolecular singlet fission in bipentacenes. J. Am. Chem. Soc. 137, 8965–8972 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Busby E., Xia J., Wu Q., Low J. Z., Song R., Miller J. R., Zhu X.-Y., Campos L. M., Sfeir M. Y., A design strategy for intramolecular singlet fission mediated by charge-transfer states in donor–acceptor organic materials. Nat. Mater. 14, 426–33 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Margulies E. A., Miller C. E., Wu Y., Ma L., Schatz G. C., Young R. M., Wasielewski M. R., Enabling singlet fission by controlling intramolecular charge transfer in π-stacked covalent terrylenediimide dimers. Nat. Chem. 8, 1120–1125 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zirzlmeier J., Lehnherr D., Coto P. B., Chernick E. T., Casillas R., Basel B. S., Thoss M., Tykwinski R. R., Guldi D. M., Singlet fission in pentacene dimers. Proc. Natl. Acad. Sci. U.S.A. 112, 5325–5330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trinh M. T., Zhong Y., Chen Q., Schiros T., Jockusch S., Sfeir M. Y., Steigerwald M., Nuckolls C., Zhu X., Intra- to intermolecular singlet fission. J. Phys. Chem. C 119, 1312–1319 (2015). [Google Scholar]
- 16.Korovina N. V., Das S., Nett Z., Feng X., Joy J., Haiges R., Krylov A. I., Bradforth S. E., Thompson M. E., Singlet fission in a covalently linked cofacial alkynyltetracene dimer. J. Am. Chem. Soc. 136, 617–627 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Fuemmeler E. G., Sanders S. N., Pun A. B., Kumarasamy E., Zeng T., Miyata K., Steigerwald M. L., Zhu X.-Y., Sfeir M. Y., Campos L. M., Ananth N., A direct mechanism of ultrafast intramolecular singlet fission in pentacene dimers. ACS Cent. Sci. 2, 316–324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tayebjee M. J. Y., Sanders S. N., Kumarasamy E., Campos L. M., Sfeir M. Y., McCamey D. R., Quintet multiexciton dynamics in singlet fission. Nat. Phys. 13, 182–188 (2017). [Google Scholar]
- 19.Scholes G. D., Correlated pair states formed by singlet fission and exciton–exciton annihilation. J. Phys. Chem. A 119, 12699–12705 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Koutecký J., Paldus J., Quantum chemical study of transannular interaction. I. Model of (n, n) paracyclophanes not considering the benzene rings distortion. Collect. Czech. Chem. Commun. 27, 599–618 (1962). [Google Scholar]
- 21.Hosteny R. P., Dunning T. H. Jr., Gilman R. R., Pipano A., Shavitt I., Ab initio study of the π-electron states of trans-butadiene. J. Chem. Phys. 62, 4764–4779 (1975). [Google Scholar]
- 22.Tavan P., Schulten K., Electronic excitations in finite and infinite polyenes. Phys. Rev. B. 36, 4337–4358 (1987). [DOI] [PubMed] [Google Scholar]
- 23.Polívka T., Sundström V., Dark excited states of carotenoids: Consensus and controversy. Chem. Phys. Lett. 477, 1–11 (2009). [Google Scholar]
- 24.Gradinaru C. C., Kennis J. T. M., Papagiannakis E., van Stokkum I. H. M., Cogdell R. J., Fleming G. R., Niederman R. A., van Grondelle R., An unusual pathway of excitation energy deactivation in carotenoids: Singlet-to-triplet conversion on an ultrafast timescale in a photosynthetic antenna. Proc. Natl. Acad. Sci. U.S.A. 98, 2364–2369 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papagiannakis E., Kennis J. T. M., van Stokkum I. H. M., Cogdell R. J., van Grondelle R., An alternative carotenoid-to-bacteriochlorophyll energy transfer pathway in photosynthetic light harvesting. Proc. Natl. Acad. Sci. U.S.A. 99, 6017–6022 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antognazza M. R., Lüer L., Polli D., Christensen R. L., Schrock R. R., Lanzani G., Cerullo G., Ultrafast excited state relaxation in long-chain polyenes. Chem. Phys. 373, 115–121 (2010). [Google Scholar]
- 27.Berkelbach T. C., Hybertsen M. S., Reichman D. R., Microscopic theory of singlet exciton fission. II. Application to pentacene dimers and the role of superexchange. J. Chem. Phys. 138, 114103 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Feng X., Casanova D., Krylov A. I., Intra-and intermolecular singlet fission in covalently linked dimers. J. Phys. Chem. C 120, 19070–19077 (2016). [Google Scholar]
- 29.Coto P. B., Sharifzadeh S., Neaton J. B., Thoss M., Low-lying electronic excited states of pentacene oligomers: A comparative electronic structure study in the context of singlet fission. J. Chem. Theory Comput. 11, 147–156 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Zeng T., Hoffmann R., Ananth N., The low-lying electronic states of pentacene and their roles in singlet fission. J. Am. Chem. Soc. 136, 5755–5764 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Aryanpour K., Shukla A., Mazumdar S., Theory of singlet fission in polyenes, acene crystals, and covalently linked acene dimers. J. Phys. Chem. C 119, 6966–6979 (2015). [Google Scholar]
- 32.Feng X., Luzanov A. V., Krylov A. I., Fission of entangled spins: An electronic structure perspective. J. Phys. Chem. Lett. 4, 3845–3852 (2013). [Google Scholar]
- 33.Sanders S. N., Kumarasamy E., Pun A. B., Steigerwald M. L., Sfeir M. Y., Campos L. M., Singlet fission in polypentacene. Chem. 1, 505–511 (2016). [Google Scholar]
- 34.Chan W.-L., Ligges M., Jailaubekov A., Kaake L., Miaja-Avila L., Zhu X.-Y., Observing the multiexciton state in singlet fission and ensuing ultrafast multielectron transfer. Science 334, 1541–1545 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Yost S. R., Lee J., Wilson M. W. B., Wu T., McMahon D. P., Parkhurst R. R., Thompson N. J., Congreve D. N., Rao A., Johnson K., Sfeir M. Y., Bawendi M. G., Swager T. M., Friend R. H., Baldo M. A., Van Voorhis T., A transferable model for singlet-fission kinetics. Nat. Chem. 6, 492–497 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Lukman S., Musser A. J., Chen K., Athanasopoulos S., Yong C. K., Zeng Z., Ye Q., Chi C., Hodgkiss J. M., Wu J., Friend R. H., Greenham N. C., Tuneable singlet exciton fission and triplet – triplet annihilation in an orthogonal pentacene dimer. Adv. Funct. Mater. 25, 5452–5461 (2015). [Google Scholar]
- 37.Pensack R. D., Ostroumov E. E., Tilley A. J., Mazza S., Grieco C., Thorley K. J., Asbury J. B., Seferos D. S., Anthony J. E., Scholes G. D., Observation of two triplet-pair intermediates in singlet exciton fission. J. Phys. Chem. Lett. 7, 2370–2375 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Dexter D. L., Two ideas on energy transfer phenomena: Ion-pair effects involving the OH stretching mode, and sensitization of photovoltaic cells. J. Lumin. 18, 779–784 (1979). [Google Scholar]
- 39.Hanna M. C., Nozik A. J., Solar conversion efficiency of photovoltaic and photoelectrolysis cells with carrier multiplication absorbers. J. Appl. Phys. 100, 1–8 (2006). [Google Scholar]
- 40.Chan W.-L., Tritsch J. R., Zhu X.-Y., Harvesting singlet fission for solar energy conversion: One-versus two-electron transfer from the quantum mechanical superposition. J. Am. Chem. Soc. 134, 18295–18302 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Tritsch J. R., Chan W.-L., Wu X., Monahan N. R., Zhu X.-Y., Harvesting singlet fission for solar energy conversion via triplet energy transfer. Nat. Commun. 4, 2679 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Thompson N. J., Wilson M. W. B., Congreve D. N., Brown P. R., Scherer J. M., Bischof T. S., Wu M., Geva N., Welborn M., Van Voorhis T., Bulović V., Bawendi M. G., Baldo M. A., Energy harvesting of non-emissive triplet excitons in tetracene by emissive PbS nanocrystals. Nat. Mater. 13, 1039–1043 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Tabachnyk M., Ehrler B., Gélinas S., Böhm M. L., Walker B. J., Musselman K. P., Greenham N. C., Friend R. H., Rao A., Resonant energy transfer of triplet excitons from pentacene to PbSe nanocrystals. Nat. Mater. 13, 1033–1038 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Congreve D. N., Lee J., Thompson N. J., Hontz E., Yost S. R., Reusswig P. D., Bahlke M. E., Reineke S., Van Voorhis T., Baldo M. A., External quantum efficiency above 100% in a singlet-exciton-fission–based organic photovoltaic cell. Science 340, 334–337 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Paci I., Johnson J. C., Chen X., Rana G., Popović D., David D. E., Nozik A. J., Ratner M. A., Michl J., Singlet fission for dye-sensitized solar cells: Can a suitable sensitizer be found? J. Am. Chem. Soc. 128, 16546–16553 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Esswein A. J., Nocera D. G., Hydrogen production by molecular photocatalysis. Chem. Rev. 107, 4022–4047 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Bardeen C. J., The structure and dynamics of molecular excitons. Annu. Rev. Phys. Chem. 65, 127–148 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Turkiewicz A., Paley D. W., Besara T., Elbaz G., Pinkard A., Siegrist T., Roy X., Assembling hierarchical cluster solids with atomic precision. J. Am. Chem. Soc. 136, 15873–15876 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Baran P., Boča R., Chakraborty I., Giapintzakis J., Herchel R., Huang Q., McGrady J. E., Raptis R. G., Sanakis Y., Simopoulos A., Synthesis, characterization, and study of octanuclear iron-oxo clusters containing a redox-active Fe4O4-cubane core. Inorg. Chem. 47, 645–655 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alhashmialameer D., Collins J., Hattenhauer K., Kerton F. M., Iron amino-bis (phenolate) complexes for the formation of organic carbonates from CO2 and oxiranes. Catal. Sci. Technol. 6, 5364–5373 (2016). [Google Scholar]
- 51.Chakraborty H., Shukla A., Theory of triplet optical absorption in oligoacenes: From naphthalene to heptacene. J. Chem. Phys. 141, 164301 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Kamada K., Ohta K., Kubo T., Shimizu A., Morita Y., Nakasuji K., Kishi R., Ohta S., Furukawa S.-i., Takahashi H., Nakano M., Strong two-photon absorption of singlet diradical hydrocarbons. Angew. Chem. Int. Ed. 46, 3544–3546 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Piland G. B., Burdett J. J., Dillon R. J., Bardeen C. J., Singlet fission: From coherences to kinetics. J. Phys. Chem. Lett. 5, 2312–2319 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Bakulin A. A., Lovrincic R., Yu X., Selig O., Bakker H. J., Rezus Y. L. A., Nayak P. K., Fonari A., Coropceanu V., Brédas J.-L., Cahen D., Mode-selective vibrational modulation of charge transport in organic electronic devices. Nat. Commun. 6, 7880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Jong M., Seijo L., Meijerink A., Rabouw F. T., Resolving the ambiguity in the relation between Stokes shift and Huang–Rhys parameter. Phys. Chem. Chem. Phys. 17, 16959–16969 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Walker B. J., Musser A. J., Beljonne D., Friend R. H., Singlet exciton fission in solution. Nat. Chem. 5, 1019–24 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Payne M. M., Delcamp J. H., Parkin S. R., Anthony J. E., Robust, soluble pentacene ethers. Org. Lett. 6, 1609–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Raptis R. G., Georgakaki I. P., Hockless D. C. R., A FeIII/oxo cubane contained in an octanuclear complex of T symmetry that is stable over five oxidation states. Angew. Chem. Int. Ed. 38, 1632–1634 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Janietz S., Bradley D. D. C., Grell M., Giebeler C., Inbasekaran M., Woo E. P., Electrochemical determination of the ionization potential and electron affinity of poly (9, 9-dioctylfluorene). Appl. Phys. Lett. 73, 2453–2455 (1998). [Google Scholar]
- 60.Zhu X.-Y., How to draw energy level diagrams in excitonic solar cells. J. Phys. Chem. Lett. 5, 2283–2288 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Lower S. K., El-Sayed M. A., The triplet state and molecular electronic processes in organic molecules. Chem. Rev. 66, 199–241 (1966). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700241/DC1
Transient absorption
Triplet-sensitizing experiments
Compound synthesis
fig. S1. Transient absorption (TA) spectra and dynamics of [Fe8O4]-Pc.
fig S2. Transient absorption for Fe8O4pz12Cl4 cluster (pumped at 2.58 and 2.07 eV) and [Fe8O4]-Pc (pumped at 1.65 eV).
fig. S3. Triplet-sensitizing experiments.
fig. S4. Synthetic route for compound 2.
fig. S5. Synthetic route for compound 3.
fig. S6. Synthetic route for compound Pc-Phenol.
fig. S7. Synthetic route for compound BP0-Phenol.
fig. S8. Synthetic route for compound BP1-Phenol.
fig. S9. Synthetic route for compound [Fe8O4]-Pc, [Fe8O4]-BP0, and [Fe8O4]-BP1.
fig. S10. Infrared spectra.
fig. S11. Absorption spectra.
fig. S12. Normalized absorption spectra.
fig. S13. Normalized absorption spectra.
fig. S14. NMR spectrum (0–9.5 ppm), compound 2.
fig. S15. NMR spectrum (0–145 ppm), compound 2.
fig. S16. NMR spectrum (0–9.5 ppm), compound 3.
fig. S17. NMR spectrum (0–145 ppm), compound 3.
fig. S18. NMR spectrum (0–9.5 ppm), Pc-Phenol.
fig. S19. NMR spectrum (0–155 ppm), Pc-Phenol.
fig. S20. NMR spectrum (0–9.5 ppm), BP0-Phenol.
fig. S21. NMR spectrum (0–155 ppm), BP0-Phenol.
fig. S22. NMR spectrum (0–9.5 ppm), BP1-Phenol.
fig. S23. NMR spectrum (0–160 ppm), BP1-Phenol.
fig. S24. NMR spectrum (−40 to 55 ppm), [Fe8O4]-Pc, before solvent addition.
fig. S25. NMR spectrum (−40 to 55 ppm), [Fe8O4]-Pc, after solvent addition.
fig. S26. Negative and positive mode NMR spectra (1000 to 5000 m/z), [Fe8O4]-BP0.
fig. S27. Negative and positive mode NMR spectra (1000 to 5000 m/z), [Fe8O4]-BP1.