

SUMMARY
Immunodeficiency is one of the most important causes of mortality associated to Wolf-Hirschhorn Syndrome (WHS), a severe rare disease originated by a deletion in chromosome 4p. The WHS candidate 1 (WHSC1) gene has been proposed as one of the main responsible for many of the alterations in WHS, but its mechanism of action is still unknown. Here, we present in vivo genetic evidence showing that Whsc1 plays an important role at several points of hematopoietic development. Particularly, our results demonstrate that both differentiation and function of Whsc1-deficient B cells are impaired at several key developmental stages due to profound molecular defects affecting B cell lineage specification, commitment, fitness and proliferation, therefore demonstrating a causal role for WHSC1 in the immunodeficiency of WHS patients.
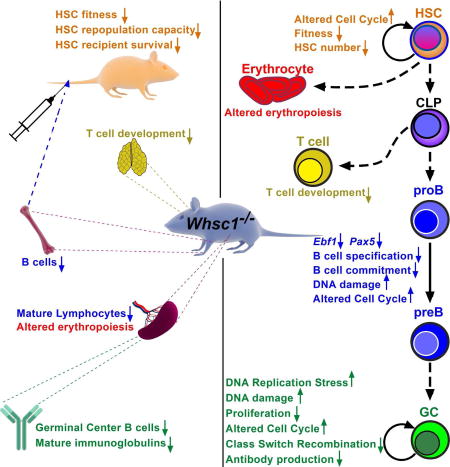
Campos-Sanchez et al. show that Whsc1 plays an important function in hematopoiesis in vivo, demonstrating a role for Whsc1 in the immunodeficiency in Wolf-Hirschhorn Syndrome. Whsc1-deficient blood cells are impaired at several developmental stages due to defects in HSC fitness and B cell lineage specification and commitment, among other problems.
INTRODUCTION
Rare diseases affecting development, beyond their medical importance, offer us a unique opportunity to identify key genes involved in development and pathology. Wolf-Hirschhorn Syndrome (WHS) is caused by the heterozygous loss of material in chromosome 4p arm (Battaglia et al., 2015; Battaglia et al., 2011). Patients suffer from serious problems including immunodeficiency, seizures, developmental delay and mental retardation. The increased susceptibility to infections is a major cause of morbidity and mortality among WHS patients (Hanley-Lopez et al., 1998). The heterozygous loss of the Wolf-Hirschhorn Syndrome Candidate-1 gene (WHSC1, also known as MMSET and NSD2) has been postulated to be the main responsible for the malformations and the immune deficiencies (Bergemann et al., 2005). WHSC1 is also involved in other pathologies affecting B lymphocytes, like multiple myeloma (Chesi et al., 1998; Stec et al., 1998) and childhood B cell acute lymphoblastic leukemias (Huether et al., 2014; Jaffe et al., 2013). Furthermore, it belongs to the protein family of Nuclear SET [Su(var)3–9, Enhancer-of-zeste, Trithorax] Domain proteins (NSD) whose other members are also involved in developmental and tumoral pathologies (Morishita and di Luccio, 2011). The WHSC1 protein contains a SET domain that confers it with histone-methyltransferase activity (Marango et al., 2008; Stec et al., 1998). Its most important in-vivo activity is to mediate H3K36 mono- and di-methylation (Kuo et al., 2011), therefore acting as an epigenetic regulator (Kuo et al., 2011). Methylation at H3K36 has been associated with regulation of transcription, splicing, DNA replication and DNA repair (Wagner and Carpenter, 2012). So far, a specific role for WHSC1 in the immune defects associated to WHS patients has not been proven and, in general, the functions of the members of the NSD family in normal hematopoiesis have not been investigated, even though they are recurrently involved in hematopoietic malignancies (Shilatifard and Hu, 2016). Here, we present in vivo genetic evidence showing that Whsc1 deficiency impairs normal hematopoietic development at several stages and lineages, and particularly affects B cell differentiation and mature B cell function. These findings reveal the role of Whsc1 as a player in hematopoietic development and also indicate that many of the immune defects associated to WHS can be directly attributed to the reduced levels of Whsc1.
RESULTS
Lymphoid differentiation is impaired in Whsc1 heterozygous mice
Since WHS patients lack only one copy of the WHSC1 gene, we first studied the hematopoietic development in heterozygous mice (Nimura et al., 2009). We could not identify any major hematopoietic change in Whsc1+/− mice at 6 months of age (Figure S1A). However, in Whsc1+/− mice above 15 months-old, a significant decrease in the percentages of B and T lymphocytes (Figure 1A) suggested a progressive defect in the long-term maintenance of the lymphoid compartment.
Figure 1.

Lymphoid and hematopoietic differentiation is impaired in Whsc1+/− and Whsc1−/− cells. (A) Representative FACS plots (out of a total of 10 experiments) of aged WT and Whsc1+/− mice, showing the reduction in the percentages of Whsc1+/− B and T cells. (B) Disadvantage of Whsc1+/− hematopoietic cells in a competitive BM reconstitution assay. The vertical axis shows the percentage of contribution of the indicated Whsc1+/− cell types to the peripheral blood of recipient mice injected with a 1:1 mix of WT:Whsc1+/− cells, as determined by flow cytometry of peripheral blood samples at the times indicated on the “x” axis. Mean ± SEM are shown (C) Percentage of contribution of the indicated Whsc1+/− cell types in recipient mice injected with a 1:1 mix of WT:Whsc1+/− cells, 3–6 months after injection. n = 5 mice for each time point. Mean ± SD are shown. (D) Kinetic delay in the reconstitution of the peripheral blood cellularity by Whsc1−/− cells (red) in comparison with Whsc1+/− (blue) or WT (black) cells, monitored by flow cytometry over 30 weeks after fetal liver transplantation. The vertical axis represents the percentage of donor cells in the peripheral blood of recipient mice at the indicated time points. n= number of mice analyzed. Mean ± SD are shown. (E) Reduced percentage of B cells of Whsc1−/− donor origin in peripheral blood, with time, as indicated in previous panel. Mean ± SD are shown. (F) Reduced absolute numbers of total blood lymphocytes in mice reconstituted with Whsc1−/− donor cells, as measured by automatic hematic biometry. Samples are from mice reconstituted with either Whsc1−/− (red dots), Whsc1+/− (blue diamonds), or WT (black filled squares) cells, 2 months after FLT, or from non-reconstituted, normal age-matched C57Bl/6 control animals (black hollow squares). Mean ± SD are shown. (G) Percentages of the different developmental stages of hematopoietic cells in the BM and spleen from mice reconstituted with either WT or Whsc1−/− cells, 3–6 months after transplant: B cell developmental stages (left graph) and other hematopoietic cell types (right graph). Mean ± SD are shown. (H) Total cellularity of BM and spleen of reconstituted mice. n = number of mice analyzed. Mean ± SEM are shown. See also Figures S1, S2 and S3.
Competitive bone marrow transplantation (BMT) experiments were performed injecting WT (Ly5.1+) and Whsc1+/− (Ly5.2+) cells in a 1:1 ratio into lethally irradiated Rag1−/− mice (Figure 1B). In this competitive setting, the developmental defect of Whsc1+/− B and T lymphocytes could be appreciated at an early stage (Figure 1B), since they can only contribute to about 20% of the total lymphocytes in peripheral blood (PB). By contrast, the myeloid compartment is maintained at the initial 50:50 ratio, suggesting that its development is not, or only slightly, affected. In order to determine if the lymphocyte defects could be compensated, injections were performed at a 1:4 WT:Whsc1+/− cell ratio (Figure S1B). This resulted in a compensating recovery of the lymphocyte chimerism to a 50:50 ratio. Once more, the myeloid ratios recapitulated those of the original injection ratio.
Full necropsy of 1:1-ratio injected animals (Figure 1C and Figure S1C) showed that Whsc1+/− LSK hematopoietic stem/progenitor cells (HS/PCs) and common lymphoid and myeloid progenitors (CLPs and CMPs) in the BM were present at the original 50:50 ratio of injection, like myeloid cells (Figure S1C). However, the Whsc1+/− B cell compartments beyond proB cells showed a gradual decrease, with the latest stages of B cell development being present only at a 20% ratio, like in the PB (Figure 1C). Therefore, the loss of one copy of Whsc1 leads to an impairment in lymphoid development that, under normal conditions, only manifests as the mice get older.
Whsc1 is required for normal hematopoietic development
Given that Whsc1−/− mice die at late gestational stages or in perinatal day zero (Nimura et al., 2009), in order to study the development of a full Whsc1−/− hematopoietic system, we performed embryo fetal liver (FL) transplants. Whsc1−/− cells could give rise to all the main types of hematopoietic cells (Figure S2). Therefore, Whsc1 is not strictly essential for the development of any of the hematopoietic lineages. However, there were differences in the reconstitutive capacity of Whsc1−/− cells. First, a kinetic delay could be seen in the repopulation of peripheral blood (Figure 1D), mainly due to the 3-fold reduction in the percentages of B220+ cells (Figure 1E). Automatic hematic analysis (Figure 1F and Figure S2) confirmed that this 3-fold reduction was also occurring in the absolute numbers of lymphocytes. FACS analysis showed consistent alterations in different hematopoietic compartments (Figure S3A and Figure 1G): indeed, although total bone marrow cellularity was not affected (Figure 1H), total B cell numbers were 5-fold reduced in the BM of Whsc1−/−-reconstituted animals. The myeloid compartment was again much less affected, and there was also a significant increase in BM CD71+Ter119+ erythroid progenitors (erythroblasts). Within Whsc1−/− B cells, the largest decrease took place from proB cells onwards, suggesting the existence of a non-stringent, leaky, developmental block at the proB-to-preB cell transition, allowing the differentiation to later developmental B cell types, albeit in much reduced numbers (Figure S3A and Figure 1G). This was confirmed by the more than two-fold reduction in the percentages and absolute numbers of B cells in the spleen (Figure 1G,H). T cell numbers were also decreased in the absence of Whsc1 (Figure 1G). Also in the spleen there was a strong increase in the percentages of erythroblasts (Figure S3A and Figure 1G), suggesting the presence of extramedullary erythropoiesis. Finally, these alterations also led to a reduction of total cellularity in the spleen of Whsc1−/−-reconstituted animals (Figure 1H).
Whsc1−/− hematopoietic cells are totally outcompeted in the presence of WT cells
We performed competitive fetal liver transplant (FLT) experiments in a 1:1 ratio of Whsc1−/− (Ly5.2+) cells against WT (Ly5.1+) cells. All types of Whsc1−/− blood cells were totally outcompeted, proving the existence of a developmental disadvantage in all the lineages (Figure 2A). New competitive FLTs were performed in ratios 1:2 or 1:5 (data not shown) and 1:20 (Figure S1D). At this last ratio, Whsc1−/− myeloid cells could be found in the blood at an average ratio of 40%, and B cells at around 5%. 3 months after the 1:20 transplant, 45% of the LSK population in the BM (Figure 2B) were Whsc1−/−. The myeloid compartment was present in the same LSK range (Figure S1E). However, B cells beyond the proB stage were present at only 5%, as in the blood. 7 months after transplant, Whsc1−/− cells formed only 18% of the LSK cells in the BM (Figure 2C and Figure S1F) and so did myeloid cells, while B cells were strongly decreased beyond proB cells. These results supported the previously detected developmental impairment at the proB to preB cell transition, and also revealed problems in the maintenance of the Whsc1−/− LSK compartment.
Figure 2.

Whsc1−/− cells are outcompeted in competitive transplantation. (A) Vertical axis shows the drastically reduced percentage of contribution of the indicated Whsc1−/− cell types to the peripheral blood of recipient mice injected with a 1:1mix of WT:Whsc1−/− cells, as determined by flow cytometry of peripheral blood samples at the weeks indicated on the “x” axis. (B, C) Percentage of contribution of the indicated Whsc1−/− cell types in recipient mice injected with a 1:20 mix of WT:Whsc1−/− cells, 3 months (B) or 7 months (C) after injection. The panels show the drastic reduction of B cell developmental stages beyond proB cells, and the decrease in LSK cells with time. n = 2 or 5 mice (3 and 7 months, respectively). Mean ± SD are shown. See also Figure S2.
Repopulation capacity is impaired in Whsc1−/− hematopoietic stem cells
In order to test if there was an impairment in the long-term maintenance of Whsc1−/−stem cell function, we performed serial transplantations of WT, Whsc1+/− or Whsc1−/− BM cells into secondary and tertiary recipients. The Kaplan-Meier survival plot for the tertiary transplant (Figure 3A) shows that Whsc1−/− cells have exhausted their repopulation potential, and the majority of the tertiary recipients die quickly due to bone marrow failure (see below). Whsc1−/− secondary recipients have no B cells and very few T cells (Figure 3B), confirming that lymphocytes, and especially B cells, are the most severely affected lineage a fact also supported by the clear reduction in B cell numbers seen in secondary recipients of Whsc1+/− cells (Figure 3B) (recapitulating the age-related loss of lymphocytes in Whsc1+/− mice (Figure 1A)) and by the clear impairment in B cell development beyond proB cells in secondary Whsc1−/− recipients (Figure S3C). This reduction in B cell numbers was more dramatic in tertiary recipients (Figure 3C). Additionally, the effects of the lack of Whsc1 in erythropoiesis in the long term can already be seen in secondary recipients by hematic counting, which shows reductions in red blood cells, hemoglobin, hematocrit and platelets (Figure S3B). All these effects indicate an impairment in the repopulation capacity of Whsc1−/− HSCs. Indeed, when mice with a 1:1 Whsc1−/−:WT mixture were examined at very short times after reconstitution (2d, 4d, 7d, 15d and 30d, Figure 3E), Whsc1−/− LSK cells and their descendants were quickly outcompeted. Furthermore, FACS analysis of primary recipients (Figure 3D) showed a significant, Whsc1 dose-dependent, reduction in the percentages of LSK cells in the bone marrow.
Figure 3.

Impaired functionality of Whsc1−/− HSCs. (A) Kaplan-Meier survival plot of recipients of a 3ary serial BMT of cells of the indicated genotypes, showing the exhausted reconstitutive capacity of Whsc1−/− cells. n = number of transplanted animals. (B) PB from mice reconstituted with a secondary serial transplant of either WT, Whsc1+/−, or Whsc1−/− cells, 6 months after transplant. Data shown are representative FACS plots, out of at least 14 Whsc1+/− and 31 Whsc1−/−-reconstituted mice independently analyzed, from 3 different donors. (C) PB from mice reconstituted with a tertiary serial transplant of either WT, Whsc1+/−, or Whsc1−/− cells, 3.5 months after transplant. Data shown are representative FACS plots, out of at least 8 Whsc1+/− and 17 Whsc1−/−-reconstituted mice independently analyzed, from 3 different donors. (D) Percentages of LSK cells in the BM of recipients of cells of the indicated genotypes, 6–8 weeks after transplantation with fetal liver cells from littermate embryos of the indicated genotypes. FACS plots are representative of the data summarized in the graph below (mean ± SD). (E) Short-term engraftment of Whsc1−/− cells into irradiated Ly5.1/Ly5.2 heterozygous recipients. First two rows: kinetics of short-term B cell reconstitution in the BM of mice injected with either WT or Whsc1−/− cells. FACS plots are gated in total B220+ cells. Third row: competitive kinetics of short-term B cell reconstitution in the BM of mice injected with a 1:1 mix of WT:Whsc1−/− cells. FACS plots are gated in total B220+ cells. Lower row: kinetics of short-term reconstitution, at 15 and 30 days, of LSK cells in the BM of mice injected with a 1:1 mix of WT:Whsc1−/−cells. The whole experiment was repeated twice with similar results. See also Figure S3.
To discard that the developmental defects we have found might be due to transplantation-induced hematopoietic stress, fetal liver, fetal spleen and fetal thymi of Whsc1−/− E18-E21 embryos were analyzed (Figure S3D). In the fetal spleen, the percentages of CD19+ cells are reduced by half in Whsc1−/− embryos and, in the fetal thymus, a severe reduction in the percentages of CD4-SP cells can already be appreciated in heterozygous embryos, and is dramatic in Whsc1−/− embryos. Similar results were found in fetal liver (data not shown), all of them supporting the true nature of the developmental defects identified in our transplantation studies.
Class switch recombination efficiency is reduced in Whsc1−/− B cells due to a proliferative defect and increased apoptosis
The facts that WHS patients tend to present with decreased levels of switched immunoglobulins (Hanley-Lopez et al., 1998) and that it had been previously shown in CH12F3 cells in vitro that WHSC1 facilitates class switching to IgA (Pei et al., 2013), prompted us to evaluate if later stages of lymphocyte development were functionally affected. We performed ex vivo class switch recombination (CSR) experiments in competitive conditions so that equal numbers of splenic B cells of either Whsc1+/− or Whsc1−/− genotypes were plated 1:1 with WT cells (Figure 4) and stimulated with different CSR-inducing stimuli. Both Whsc1+/− and Whsc1−/− B cells were impaired in switching to all the isotypes in all the conditions tested (Figure 4A). This was confirmed by in vivo SRBC immunization of reconstituted mice (Figure 4B–C), showing that the percentage of switched cells at 13 days was greatly reduced for both Whsc1+/− and Whsc1−/− cells. Also, ELISA assays showed a reduction in the serum levels of switched IgG1 in Whsc1−/−-reconstituted, SRBC-injected mice (data not shown), although both Whsc1+/− and Whsc1−/− cells could give rise to splenic plasma cells (Figure 4D). All these results show in vivo, in lymphocytes in a genetic model, that Whsc1 is required for an efficient CSR to most of the isotypes, providing a model that really recapitulates one of the most serious complications faced by WHS patients.
Figure 4.

Impaired CSR in Whsc1−/− B cells. (A) Differences in the percentages of class-switched B cells depending on their genotype, expressed as percentages relative to WT cells (left black bar, 100%). The stimuli used and the switched immunoglobulin subtypes measured are indicated. n = number of independent experiments performed. p values refer to a two-sided Student’s t test vs. WT. Mean ± SD are shown. (B) Reduction of in vivo splenic Ig-switched GC B cells (pre-gated as B220+,PNA+,FAS+) in Whsc1+/+, Whsc1+/− or Whsc1−/− recipients. Spleens were analyzed 13 days after immunizing with SRBCs recipient mice reconstituted with cells of the indicated phenotypes. FACS plots are representative of the data summarized in the graph shown in the next panel (mean ± SD). (C) Graphical representation and statistical analysis of the percentages of in vivo IgG1+ cells for each genotype. Mean ± SD are shown. (D) Whsc1−/− cells can give rise to plasma cells; representative FACS plots are shown from three independent replicate experiments analyzing the spleen of mice reconstituted with the indicated genotypes. (E) FACS histogram showing the deconvolution, after 72 hours of stimulation with LPS, of the total cellular population in the different generations of cells, using the dilution of the CellTrace dye to separate the cells according to the number of times they have divided. The “y” axis scale in the left histogram is expressed as percentage related to the maximum, and in the right histogram is expressed as absolute numbers of cells. G0 to G4 = generations of cells. (F) Evolution of the percentage of either WT or Whsc1−/− ex vivo LPS-stimulated Ly5.2+ B cells, in competition against WT Ly5.1+ B cells, along the different generations, and relative to the initial percentage when plated. Mean ± SD are shown. (G) Reduced absolute numbers of Whsc1−/− cells obtained 72 hours after stimulation of splenic B cells in absence of competition, in the presence of a defined number of microbeads. (H) Percentages of apoptotic B cells after 48h or 72h in culture under LPS-stimulation. Left: gating strategy and representative plots of cells after 48h in culture. C57Bl6 refers to cells from WT non-reconstituted animals (n=7), while the other two lanes show plots of mice reconstituted with either WT (n=3) or Whsc1−/− (n=9) cells. Right: bar graph plot summarizing the data. See also Figure S4.
Since CSR is linked to cellular proliferation, ex vivo competitive assays were carried out after labelling the cells with CellTrace in order to follow the different cycles of division, showing that the contribution of Whsc1−/− cells is strongly reduced in all generations, and at the end it accounts for a very reduced percentage (Figure 4E–F and Figure S4), and absolute number (Figure 4G) of cells, and therefore supporting the existence of a proliferative impairment in Whsc1−/− cells. FACS analysis also showed (Figure 4H) increased levels of apoptosis in stimulated Whsc1−/− B cells.
Cell cycle alterations in Whsc1−/− B cells and HS/PCs
We performed cell cycle analysis in the presence of BrdU during the CSR reaction. After 72 hours under stimulation, in all the different cell generations there was a 3-fold increase in the number of cells in the S phase of cell cycle in Whsc1−/− B cells (Figure 5A and Figure S5A). To investigate if this alteration was also present in vivo at the other developmental points where we had found that the absence of Whsc1 led to important malfunctions, we performed in vivo BrdU labellings. The results showed that, in the BM, both B cells at all the different developmental stages (Figure 5B,F) and LSK cells (Figure 5C) also presented an increase in the number of BrdU+ S-phase cells, while Whsc1−/− myeloid cells did not present any alteration (Figure 5B).
Figure 5.

Cell cycle alterations and DNA damage accumulation in Whsc1−/− cells. (A) FACS analysis of cell cycle in ex vivo LPS-stimulated, BrdU-labelled B cells. 72 hours after stimulation, cell culture medium was supplemented with BrdU and cells were harvested 1 hour later. Leftmost panels show the total incorporation of BrdU and the dilution of CellTrace. Central panels display the cell cycle profile (BrdU vs. 7AAD) for the generation 3 (G3) of cells, showing the 3-fold increased percentage of Whsc1−/−cells in the S phase. Bar graph shows the percentage of cells in S phase for each generation for the three indicated genotypes. Bars represent mean ± SD. n = number of independent experiments. (B) 3-fold increased percentage of Whsc1−/− total B cells in the S phase in vivo in the BM, and absence of changes in myeloid cells Mice were intraperitoneally injected with BrdU and sacrificed after two hours. Bar graphs represent the mean ± SD. (C) Increased BrdU incorporation in vivo by BM Whsc1−/−LSK cells. Bar graphs represent the mean ± SD. (D) Histograms showing the increased levels of γH2AX accumulation in ex vivo LPS-stimulated Whsc1−/− B cells, in comparison with either WT or Whsc1+/− B cells, as determined by flow cytometry. (E) Increased levels of γH2AX in vivo in total BM Whsc1−/− B cells and LSK cells, in comparison with WT counterparts. Whsc1−/− myeloid cells do not present abnormal γH2AX levels, and neither do Whsc1+/− BM cells. Representative plots are shown. (F) Cell cycle analysis through the different stages of B cell development in the BM. Upper row: gating strategy for BM B cell compartments (cells were fixed for cell cycle studies). Middle rows: representative FACS plots for WT and Whsc1−/− cells. Lower graphs: percentages of cells in S-phase for the indicated BM B cell compartments, as determined by in vivo incorporation of BrdU. (G) Impaired proliferation of Whsc1+/− and Whsc1−/− MEFs. Growth curves of freshly prepared MEFs of the indicated genotypes showing the reduced proliferative capacity of Whsc1−/− cells. Representative analyses are shown out of three independent experiments with a total of 17 MEF preparations (4Whsc1+/+, 5 Whsc1+/− and 8 Whsc1−/− embryos). (H) Impaired DNA damage repair in Whsc1−/− MEFs. WT and Whsc1−/− MEFs were irradiated with 5 Gy and γH2AX levels were measured after 1 and 5 hours. Whsc1−/− MEFs cannot efficiently remove gamma irradiation-induced γH2AX accumulation, while WT MEFS have reverted to normal levels of γH2AX within 5 hours after exposure. One representative analysis is shown out of three independent experiments with a total of 12 MEF preparations (6 Whsc1+/+and 6 Whsc1−/− embryos). See also Figure S5.
It has been reported that changes in the pattern of methylation at H3K36 can affect DNA repair (Wagner and Carpenter, 2012), and WHSC1 has been associated with DNA damage repair (Evans et al., 2016; Hajdu et al., 2011; Shah et al., 2016). We studied the levels of γH2AX, a well-known indicator of the existence of broken DNA ends. In the context of the CSR reaction (Figure 5D and Figure S5B,C), we could see progressively increased levels of γH2AX during the successive generations in Whsc1−/−cells, and this correlates with the accumulation of cells in S phase. Furthermore, also in vivo, a slight elevation in γH2AX levels can be appreciated in Whsc1−/− total B cells in the BM, especially when compared with Whsc1−/− myeloid cells that, once more, seem to be unaffected (Figure 5E). Also, a similar increase in γH2AX levels could be detected in BM Whsc1−/− LSK cells (Figure 5E). These same results could be found in a totally different cell type, Whsc1−/− mouse embryo fibroblasts (MEFs), which also presented a very compromised proliferative capacity (Figure 5G) and a severe impairment for the downregulation of γH2AX levels after DNA damage (gamma-irradiation instead of CSR, in this case) (Figure 5H).
Whsc1−/− GC B cells present severe alterations in many basic cellular processes
We next studied the variations in the transcriptome in proliferative Whsc1−/− GC cells versus WT cells. We used RNA-seq to compare Whsc1−/− and WT ex-vivo LPS-stimulated splenic B cells (Figure 6A, Figure S6 and Tables S1–3). The results demonstrate the existence of severe defects at many key levels of cellular biology. First, a basic differential gene expression analysis showed that there are several developmental genes deregulated in the absence of Whsc1, like the genes of the Hoxa cluster (Figure S6A and Tables S1–2). These developmental genes, although of great importance to the morphogenetic pathways affected in WHS patients, do not explain the B cell phenotypes that we have described. However, by using pathway analysis, we can see that many key processes like cell cycle, splicing, ribosome synthesis, DNA replication or DNA repair are very significantly altered in proliferating Whsc1−/− B cells (Fig. 6A and Table S3). Functional analysis using Gene Set Enrichment Analysis (GSEA) (Figure 6B and Figure S6B) confirmed these findings. Indeed, although B cell-specific pathways are also affected (see “BCR signaling” panels, Figure 6B and Figure S6B), and this could explain some aspects of the developmental B cell impairment, the downregulation of ribosomal proteins, the upregulation of the components of the spliceosome or of cell cycle genes (Figure 6A,B) are directly related to all the cellular phenotypes described. In concordance with the accumulation of γH2AX, a very strong deregulation of the genes involved in DNA repair could be seen (Figure 6B and Figure S6B). Among them, there was an upregulation of the Fanconi Anemia pathway, involved in the repair of DNA damage associated with DNA replication (Kais et al., 2016). In fact, one of the most significantly altered pathways was the one regulating DNA replication (Figure 6B and Figure S6B), therefore pointing towards this process as a potential trigger of the abnormal proliferative behaviour of Whsc1−/− cells.
Figure 6.

Severe alteration of biological processes in Whsc1−/− GC B cells (A) Threshold-based functional pathway analysis for the differentially expressed genes in GC B cells (Whsc1−/− vs. WT) with DAVID. Selected KEGG (green) and GO (blue) pathways are represented. Bars represent the antilogarithm of the adjusted P value (left vertical axis). Red dots represent the False Discovery Rate for the corresponding pathways (right vertical axis). (B) Gene Set Enrichment Analysis (GSEA) plots for the indicated genesets, (see their corresponding heatmaps in Figure S5B). The GSEA comparison was performed [WT minus Whsc1−/−], so the genes downregulated in the Whsc1−/− cells appear at the left of the scale on each graph, and those upregulated appear at the right side. Therefore genesets downregulated in Whsc1−/− cells have a positive Normalized Enrichment Score (NES), and genesets upregulated in Whsc1−/−cells have a negative NES. (C) Impaired DNA replication of Whsc1−/− MEFs and CSR-B cells, as determined by DNA fiber analysis. Fork rate (left panel) in Kbps/min, and Inter Origin Distance, (IOD, right panel) in Kpbs were measured for either MEFs (two leftmost columns in every graph) or CSR-stimulated B cells (two right columns) for WT (grey) or Whsc1−/− (red) cells. n= number of different clones (donors). Horizontal bar represents the median for each data column. See also Figures S5 and S6.
Absence of Whsc1 leads to increased DNA replicative stress in GC B cells
In cell lines, Whsc1 has been associated with DNA damage repair (Evans et al., 2016; Hajdu et al., 2011; Shah et al., 2016). Also, cell lines derived from WHS patients present delayed cell-cycle progression and impaired DNA replication (Kerzendorfer et al., 2012), but which one among the many genes lost in WHS is responsible for this phenotype is not yet clear. It has been shown that correct H3K36me3 patterns are required to prevent replicative stress in cancer cells lines (Kanu et al., 2015), and H3K36 de-methylation has been shown to control proliferation, cell cycle and hematopoietic development (Andricovich et al., 2016; He et al., 2008). Therefore, as suggested by the RNA-seq results, we decided to test in our in vivo model whether the proliferative impairment could be due to increased levels of DNA replicative stress. We performed DNA fiber analysis (Flach et al., 2014) in Whsc1−/− MEFs and in stimulated B cells. There was a reduction both in the fork rate and in the inter-origin distances in the absence of Whsc1 (Figure 6C), confirming an impairment in the advancement of the replication fork, coupled with the activation of new dormant origins. We also cultured the cells in the presence of increasing concentrations of the DNA replication inhibitor aphidicolin (Figure S5D,E). Whsc1−/− cells were more affected than WT cells by aphidicolin (Figure S5D) but, more significantly, cell cycle analysis showed that, in the presence of aphidicolin, Whsc1−/− B cells could not correctly exit S phase (Figure S5E), presenting a prolonged S phase without properly entering G2/M. These results confirm replication stress as the main molecular mechanism behind the proliferative impairment in the absence of Whsc1.
Specification and commitment to the B cell lineage are strongly impaired in Whsc1−/− B cell progenitors
In order to identify if the proB to preB cell transition impairment was due to the same molecular mechanism, we sorted BM proB cells and performed RNA-seq followed by differential gene expression and GSEA. We found that Whsc1−/− proB cells share some of the defects found in GC B cells but with different severity and, more significantly, they also present proB cell stage-specific transcriptional defects (Tables S4–6, Figure 7 and Figure S7). Indeed, Whsc1−/− proB cells also present significant alterations in DNA repair, DNA replication and cell cycle gene sets (Figure 7A and Figure S7A), and downregulation of genes involved in nucleosome and chromatin organization (Figure 7A and Figure S7A). Since there are not statistically significant differences in the percentages of cells in the S phase between WT and Whsc1−/− BM proB cells (Figure 5F) these RNA-seq differences most probably reflect intrinsic, cell-specific differences between the two genotypes; there was a significant downregulation of key early B cell developmental genes (Table S5) like Ikzf3, Tcf3/E2a, Ebf1, Pax5, Rag1, Rag2, Il7r or Foxo1, being particularly important in the case of Ebf1 (16-fold downregulated in Whsc1−/− cells) and Pax5 (9.5-fold downregulated). Since these genes are key regulators of the specification and commitment to the B cell lineage (Cobaleda et al., 2007), we generated specific genes sets with the most relevant of their published targets (Pongubala et al., 2008; Revilla et al., 2012; Treiber et al., 2010) and evaluated them by GSEA (Figure 7B,C and Figure S7B). The results showed a strong downregulation of a significant majority of the Ebf1- and Pax5-upregulated target genes in Whsc1−/− proB cells, affecting most of the biological processes necessary for the specification and commitment of progenitors to the B cell lineage (Cobaleda et al., 2007). These data therefore correlate with the developmental block at this early phase of B cell differentiation and show that Whsc1 participates in different mechanisms that control differentiation and function at several stages of B cell life.
Figure 7.

Downregulation of essential B cell genes in developmentally impaired Whsc1−/− BM proB cells (A) Gene Set Enrichment Analysis (GSEA) plots for the indicated genesets for the differentially expressed genes in Whsc1−/− vs. WT BM proB cells, (see their corresponding heatmaps in Figure S7A). The GSEA comparison was performed [WT minus Whsc1−/−], so the genes downregulated in the Whsc1−/− cells appear at the left of the scale on each graph, and those upregulated appear at the right side. Therefore genesets downregulated in Whsc1−/− cells have a positive Normalized Enrichment Score (NES), and genesets upregulated in Whsc1−/− cells have a negative NES. (B) GSEA plots for the genesets corresponding to genes upregulated by either Ebf1 or Pax5 during normal early B cell development (see main text for the generation of the lists of genes). The GSEA analysis shows the global decrease in the levels of expression of these genes in Whsc1−/− proB cells. (C) Partial heatmaps corresponding to the GSEA analyses shown in panel B. For clarity, only the part corresponding to the genes that become downregulated in Whsc1−/− proB cells is shown. The full heatmaps can be seen in Figure S7B. (D) The introduction of a rearranged VHDHJH B1–8 allele in the Whsc1−/− proB cells does not rescue the leaky developmental block at the proB-to-preB cell transition. The bar graph represents the percentages of the indicated B cell developmental stages in the BM of recipient mice transplanted with cells of the indicated genotypes. n: number of mice analyzed. See also Figure S7.
DISCUSSION
An increased susceptibility to infections is one of the main pathological characteristics affecting WHS patients, and the molecular basis for this immunodeficiency is so far unknown. Here we present in vivo genetic evidence of the involvement of the Whsc1 gene in hematopoietic development and in B cell differentiation and function at several stages of differentiation. Importantly, we show the existence of an impairment in the development of Whsc1+/− lymphocytes that manifests subtly, but progressively, with age. This finding could have serious implications for the long-term prognosis of immunodeficiency in WHS patients (hemizygous for WHSC1), because it implies that their immune response at late stages of life might be seriously impaired.
The total loss of Whsc1 affects the development and function of different blood cell types, especially HS/PCs and B cells at several developmental stages. There is a strong block at the proB-to-preB cell transition. Blocked Whsc1−/− proB cells present severely reduced levels of the key B cell transcription factors Ebf1 and Pax5. This correlates to global downregulation of Ebf1 and Pax5 positively regulated target genes, necessary for controlling essential molecular aspects of early B cell development: immunoglobulin genes (Figure 7B,C and Figure S7B,C), receptors, signal transducers and kinases (Blnk, Bcar3, Cd19, etc.), or transcription factors and nuclear proteins (Bach2, E2f2, Rb1, Lef1, etc.). The combined decreased expression of early B cell transcriptional regulators has been previously shown to interfere with the normal development of the B cell lineage (Ungerback et al., 2015). We have tried to rescue this proB-to-preB cell block by breeding Whsc1−/− mice with animals carrying a rearranged VHDHJH gene (B1–8) inserted in the JH locus (B1–8i) (Sonoda et al., 1997). However, this, by itself, is not enough to overcome the block (Figure 7D), therefore confirming its dependence from many deficient signaling molecules and B cell specific factors beyond immunoglobulin rearrangements. In summary, Whsc1−/− cells can generate CLPs, pro-B cells, and later B cell stages too, but they don’t do it efficiently; in the absence of competition, the impairment exists, but it is leaky and allows for the generation of mature B cells, although with a much reduced efficiency. Therefore, B cell specification and commitment can take place in the absence of Whsc1, but they are seriously compromised and become insufficient when challenged in any way.
Regarding later stages of B cell development, we could also show that there is an impairment in the CSR reaction that is dose-dependent in relation to the absence of the Whsc1 alleles. This is related to an aberrant proliferation of Whsc1-deficient cells, which showed a 3-fold increase in the percentage of cells in S phase, coupled with an accumulation of DNA damage. Whsc1-deficient cells have large deregulations of gene sets involved in key biological processes, among them the genes involved in DNA repair and DNA replication. They also present a significantly decreased fork rate counteracted by local increases in origin density suggesting that a higher frequency of stalled forks induces compensatory activation of dormant origins and triggers a DNA damage response (Maya-Mendoza et al., 2009). The fact that the Fanconi Anemia (FA) pathway (responsible for repairing DNA as a response of replication stress (Kais et al., 2016)) is significantly altered further supports this idea. This replicative stress leads to the accumulation of DNA damage, as indicated by increased γH2AX levels; however, this does not result in increased numbers of chromosomal aberrations as measured by DNA FISH on the immunoglobulin locus (Cobaleda and Skok, unpublished observations). There is also an accumulation of Whsc1-deficient apoptotic cells (Figure 4G), therefore suggesting that those cells that cannot cope with the replication stress die and, in the surviving cells, the increased DNA repair mechanisms, together with the increased numbers of replication origins and an elongated S phase, allow them to correctly finish the cell cycle, although the global fitness of the population is severely affected, as shown by all the different competition experiments. Additionally, one of the cellular components most affected by replicative stress is the ribosome, both at the rRNA and protein levels (Golomb et al., 2014), and stimulated Whsc1-deficient cells show a massive downregulation of the expression of integral ribosomal proteins, most likely as a reflection of this fact.
According to the methyltransferase activity of Whsc1, it has been demonstrated in different tumor-related experimental settings that the alteration of WHSC1 activity levels leads to parallel global changes in the patterns of H3K36 methylation (Garcia-Carpizo et al., 2016; Kuo et al., 2011; Oyer et al., 2014; Popovic et al., 2014). In our Whsc1−/− mouse model, it has already been shown by western blot and by immunohistochemistry that the levels of H3K36 methylation are reduced in Whsc1-KO cells (Nimura et al., 2009; Sarai et al., 2013). Therefore, the changes in gene expression revealed by RNA-seq experiments are most likely downstream of changes in H3K36 methylation patterns and include indirect effects of an altered epigenetic landscape. In the context of B cell differentiation, the proB to preB transition and the germinal center reaction are the two stages corresponding to the maximum peaks of expression of Whsc1 in hematopoiesis [http://www.immgen.org (Heng and Painter, 2008)] therefore suggesting that the high levels of Whsc1 expression makes these cells particularly vulnerable to the loss of this gene. Conversely, the comparative RNA-seq analysis of gene expression in non-proliferative, ex vivo sorted, resting lymph node B cells (data not shown, 2 WT vs. 4 KO samples) did not show any significant changes in the pathways that were greatly altered in proliferative CSR B cells or proB cells, only in some of the morphogenetic developmental genes.
We also found the existence of a stem cell functional defect in the absence of Whsc1. First, there is a dose-dependent decrease in the percentages of LSK cells in the absence of Whsc1 (Figure 3D). Since the total BM cellularity is relatively constant between WT and Whsc1−/− animals (Figure 1H), it can therefore be concluded that the absolute numbers of LSK cells are significantly decreased in Whsc1−/− mice. Second, in competitive FL transplantations mixing WT and Whsc1−/− cells at a 1:20 ratio (Figure 2 and Figure S1), the percentage of Whsc1−/− LSK cells steadily decreases with time (45% at 3 months, 18% at 7 months). Third, the serial transplantation experiments lead to BM failure and death in tertiary recipients (Figure 3A), a paradigmatic demonstration of stem cell failure. Fourth, the presence in Whsc1−/− BM LSKs of a 3-fold increased percentage of BrdU+ (S-phase) cells (Figure 5C), is indicative of a cell cycle problem similar to the one observed in stimulated (CSR) B cells or BM B cells. Fifth is the presence in Whsc1−/− BM LSKs of an unresolved accumulation of γH2AX, similarly to stimulated (CSR) B cells, BM B cells (Figure 5E) or irradiated Whsc1−/− MEFs (Figure 5H). The data from CSR B cells show that this increased γH2AX correlates with increased replicative stress and increased percentage of cells in the S-phase. Correspondingly, the data from MEFs show that Whsc1−/− MEFs are almost non-proliferative in culture (Figure 5G) and have increased DNA replicative stress (Figure 6C) and impaired DNA repair (Figure 5H). Finally, the results from short-term analysis of mice transplanted in a 1:1 WT:Whsc1−/− competition (Figure 3E) show that Whsc1−/−LSK cells are quickly outcompeted after transplant. All these functional and cellular evidences, strongly suggest that Whsc1−/− HS/PCs have a cell cycle disadvantage that leads to a progressive reduction in their numbers with time. This reduced fitness becomes quickly evident when Whsc1−/− cells are challenged in a serial transplantation or in a competitive setting. However, any number of events, such as the mentioned reduced number, the impaired homing, the altered self-renewal potential, or abnormal differentiation, could account for the impaired reconstituting potential of Whsc1-deficient HS/PCs, and this should be the focus of future work.
Altogether, our results show that Whsc1 is involved in hematopoietic development at several stages and implicating different cellular lineages. Furthermore, it participates in the regulation of different molecular mechanisms throughout these different stages and cell types, from hematopoietic stem cell function to B cell lineage specification and commitment, fitness or cellular proliferation. Our results highlight Whsc1 as a player in the control of hematopoietic and, especially, B cell development, and also indicate that the immune defects associated to WHS can be directly attributed to the reduced levels of Whsc1. These findings provide a framework for the understanding, prognosis and potential future treatment of immunodeficiency in Wolf-Hirschhorn Syndrome.
EXPERIMENTAL PROCEDURES
Mice
The following mice were maintained in the C57BL/6 background and, when required, genotyped as described: Whsc1−/− (Nimura et al., 2009), Rag1−/− (Mombaerts et al., 1992) and B6.SJL-PtprcaPep3b/BoyJ. All mice were housed in a special pathogen-free (SPF) barrier facility. All experimental procedures conducted on mice were approved by the CBMSO’s Institutional and Spanish Regional Committees on Ethics of animal research. Mice with the indicated genotypes were included in the study without any further preselection or formal randomization and comprised balanced numbers from both genders; we used age-matched mice. Investigators were not blinded to genotype group allocations. All mice were maintained in C57BL/6J background.
FACS Analysis and Sorting Purification
Contaminating red blood cells were lysed using ACK Lysing Buffer. The remaining cells were washed in FACS buffer (1X PBS with 2% heat-inactivated fetal bovine serum (HI-FBS)). After staining, all cells were washed once in FACS buffer and resuspended in the same buffer containing 2 mg/mL propidium iodide (PI) to allow dead cells to be excluded. The samples and the data were acquired in a FACSCantoII Flow Cytometer, sorted in a FACSAria Fusion and analyzed using FlowJo software (TreeStar). Preincubation of cells with CD16/CD32 (2.4G2) Fc-block solution (BD Biosciences) was used to avoid nonspecific antibody binding. BM proB cells were purified in a FACSAria Fusion (BSII, Becton-Dickinson) as B220+ CD19+ c-Kit+ CD25− PI−. The different antibodies used and the surface markers defining the different populations are indicated in Supplementary Experimental Procedures.
Fetal Liver or Bone Marrow Transplantation Experiments
Total BM cells (flushing from the long bones) or Fetal Liver (FL) cells (taken from E21 embryos or P0 newborns) were injected intravenously into lethally irradiated recipients, either Rag1−/−(Ly5.2+), or B6.SJL-PtprcaPep3b/BoyJ Ly5.1+ or Ly5.1+/Ly5.2+ WT, depending on the experiment. For all transplantations, total BM or FL donor cells were counted using a handheld automated cell counter, ScepterTM [Millipore®, 40µm tips] and the injection ratio was checked by flow-cytometry.
Proliferation, Damage and Cell Cycle analysis
The Apoptosis, DNA Damage and Cell proliferation Kit [Cat No 562253, BD PharmigenTM] was use to assess the proliferative capacity of the cells, as well as cell cycle dynamics and DNA damage accumulation. Anti-BrdU-FITC or Anti-BrdU-APC vs. 7AAD were used to study cell cycle, and CellTraceTM Violet Cell Proliferation Kit [Cat No C34557, InvitrogenTM] to monitor cell proliferation. CountBrightTM Absolute Counting Beads for flow cytometry [MP 36950, Molecular ProbesTM, Invitrogen] were used to measure absolute cell yield in culture.
Class Switch Recombination assays
Mouse primary B cells were purified from the spleens of the indicated strains of mice by immunomagnetic depletion with anti-CD43 beads [Miltenyi Biotec] and were cultured in 50 µM 2-β-Mercaptoethanol (Invitrogen), 10 mM Hepes [Invitrogen], 1 mM Glutamine, antibiotics, Non-Essential Aminoacids and 10% HI-FBS RPMI medium, supplemented with either: i) 25 µg/ml LPS [Sigma-Aldrich], ii) 25 µg/ml LPS and 20 ng/ml IL4 [PeproTech], iii) 25 µg/ml LPS and TGF-Beta1 2 ng/ml [R&D Systems] iv) 20 ng/ml IL4 [PeproTech] and 1 µg/ml anti-CD40 [eBioscience] or non-supplemented (only growth medium). Cells were counted using a handheld automated cell counter, ScepterTM [Millipore®, 40 µm tips] and CellTraceTM Violet Cell Proliferation Kit [Cat No C34557, InvitrogenTM] was used as proliferation tracer.
RNA extraction
Proliferating B cells were obtained by in vitro LPS stimulation during 72 hours as previously described. BM proB cells were sorted as previously described. Sorting strategy and purity are shown in Figure S7D. RNA was extracted following commercial Trizol® Reagent [Cat No 15596-026, Life TecnologiesTM, GibcoBRL] recommendations. It chemical quality was measure by Nanodrop 1000 Spectrophotometer [Thermo ScientificTM] and its integrity was confirmed using the Agilent 2100 Bioanalyzer. Libraries for RNA-seq were prepared as indicated in Supplementary Experimental Procedures.
DNA fiber spread
Replication track analyses were performed as described in (Flach et al., 2014) with only minor modifications. MEFs were traced at exponential growth during their third passage, and proliferating B cells were obtained after 48 hours of in vitro culture under LPS stimulation. Both cell types were incubated during 20 minutes with each thymidine analogue (first CIdU and then IdU) and washing them three times with warm 1X PBS (37°C). Spreading buffer incubation was for 6 min, primary antibody incubation timing was extended to overnight and secondary antibody for 2 hours. Tracks were imaged on a Fluorescence Resonance Energy Transfer (F.R.E.T) Zeiss microscope coupled to a Hamamatsu Camera. All pictures were taken at 40x magnification. Fork Rate (FR) and Inter Origin Distance (IOD) were calculated based on the length of the IdU tracks measured using ImageJ software and the already published formulas: FR (Kb min−1) = (2.59 (Kb µm−1) × length (µm)/ pulse time (min)) and IOD (Kb) = length between two contiguous origins (µm). When aphidicolin was used, it was added at 1 µg/ml with the second pulse of thymidine analogue, IdU.
Statistical analysis
Exact sample sizes and statistical tests used for comparisons are indicated in each figure. Kaplan–Meier survival curves were created and analyzed using Prism (GraphPad Software), which used the log-rank test to calculate significance. Samples were allocated to their experimental groups according to their predetermined type (i.e., mouse genotype) and, therefore, there was no randomization. Sample sizes chosen are indicated in the individual figure legends and were not based on formal power calculations to detect pre-specified effect sizes. Data analysis was not blinded.
Supplementary Material
Table S1. Results of the Differential Gene Expression Analysis for GC B cells. Related to Figure 6.
Table S2. RNA-seq counts, as normalized with RSEM for GC B cells. Related to Figure 6.
Table S3. Functional enrichment analysis from DAVID for KEGG pathways and Gene Ontology biological processes for GC B cells. Related to Figure 6.
Table S4. Results of the Differential Gene Expression Analysis for BM proB cells. Related to Figure 7.
Table S5. RNA-seq counts, as normalized with RSEM f for BM proB cells. Related to Figure 7.
Table S6. Functional enrichment analysis from DAVID for KEGG pathways and Gene Ontology biological processes for BM proB cells. Related to Figure 7.
Highlights.
Hemizygous loss of Whsc1 causes a progressive decline of lymphocyte numbers with age
Whsc1 deficiency reduces HSC fitness and repopulation capacity
Whsc1−/− B cell precursors have defective B cell lineage specification and commitment
Whsc1−/− GC cells present impaired CSR, altered cell cycle and DNA replicative stress
Acknowledgments
We are indebted to Meinrad Busslinger and Isidro Sanchez-Garcia for useful discussions and for their critical reading of the manuscript. We thank J.Mendez and S.Rodriguez for helpful discussions and help with the DNA fiber analysis, H.Tagoh and M.Fischer for RNA-seq analysis experiments of mature LN B cells, T.Graf, M.Busslinger, A.Rodriguez-Ramiro and I.Sanchez-Garcia for providing transgenic mouse strains, B.Pintado and the Transgenesis Service of CNB-CBMSO UAM/CSIC for help with the mice, B.Raposo and S.Andrade for FACS sorting, and the “Servicio de Microscopía Óptica y Confocal (SMOC)” from CBMSO for their help. Research at C.C.’s lab was partially supported by FEDER, “Fondo de Investigaciones Sanitarias/Instituto de Salud Carlos III” (PI13/00160 and PI14/00025), “Fundación Inocente Inocente”, the ARIMMORA EU/FP7 project, donations from the “Asociación Española del Síndrome de Wolf Hirschhorn” (AESWH), the “Fundación Síndrome de Wolf-Hirschhorn or 4p-” (FSWH4p), from FECYT “Precipita”, and from “Fundación Ramón Areces”. E.C.-S. was partially supported by a JAE-PreDoc fellowship from the Spanish National Research Council (CSIC, FEDER), and was a “Residencia de Estudiantes” Fellow. N.D-S. was partially supported by a JAE-intro studentship from the Spanish National Research Council (CSIC) (JAEINT_15_01981). Research at K.Ura´s lab was partially supported by JSTP PRESTO program (4201) and Grant-in Aids for Scientific Research (22131003, 15H01345) from MEXT of Japan. P.P.R. is a National Cancer Center and American Society of Hematology Fellow. J.A.S. is a Leukemia & Lymphoma Society (LLS) scholar, NCC and ASH fellow, and is supported by NIH grants R01 GM086852 and R01GM112192. Research at M.L.M.F.’s lab was partially supported by “Instituto de Salud Carlos III” and “Fundación 1.000 sobre Defectos Congénitos”. CIBERER is an initiative of ISCIII, Ministry of Economy and Competitiveness of Spain. V.D. was partially supported by Ministry of Science and Innovation through the “Programa tecnicos de apoyo” (PTA) from 2012–2014. A.E.-C. is funded by the RED-BIO project of the Spanish National Bioinformatics Institute (INB) under grant number PT13/0001/0044. The INB is funded by the Spanish National Health Institute Carlos III (ISCIII) and the Spanish Ministry of Economy and Competitiveness (MINECO). M.D. was funded by the Ministry of Economy and Competitiveness of Spain (PTA2014-09515-I). HH is a Miguel Servet (CP14/00229) researcher funded by the Spanish Institute of Health Carlos III (ISCIII). P.P.R. was financed by NIH grant K99GM117302.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The accession numbers for the RNA-seq data reported in this paper are GEO GSE84878 and GSE88970.
AUTHOR CONTRIBUTIONS
E.C.-S. performed the majority of the experiments. M.G and H.H. generated RNA-seq libraries. E.C-S., N.D.-S, E.C.-S.-P., A.E-C., M.D. and C.C. analyzed bioinformatic data, K.U., Y.K. and K.N. provided the Whsc1-KO mouse. P.P.R., JH.K., A.A. and J.A.S. designed and performed IgH DNA FISH experiments (data not shown). V.D. collaborated in the generation of mouse models (data not shown). C.C. conceived and supervised the project and secured funding. M.L.M.F. provided conceptual advice. E.C.S. and C.C. designed the experiments and analyzed and interpreted the data. E.C.S., M.L.M.F. and C.C. wrote and edited the manuscript. All authors reviewed the manuscript and provided final approval for submission.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- Andricovich J, Kai Y, Peng W, Foudi A, Tzatsos A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J Clin Invest. 2016;126:905–920. doi: 10.1172/JCI84014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: A review and update. Am J Med Genet C Semin Med Genet. 2015;169:216–223. doi: 10.1002/ajmg.c.31449. [DOI] [PubMed] [Google Scholar]
- Battaglia A, South S, Carey JC. Clinical utility gene card for: Wolf-Hirschhorn (4p-) syndrome. Eur J Hum Genet. 2011;19 doi: 10.1038/ejhg.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8:463–470. doi: 10.1038/ni1454. [DOI] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- Evans DL, Zhang H, Ham H, Pei H, Lee S, Kim J, Billadeau DD, Lou Z. MMSET is dynamically regulated during cell-cycle progression and promotes normal DNA replication. Cell Cycle. 2016;15:95–105. doi: 10.1080/15384101.2015.1121323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Carpizo V, Sarmentero J, Han B, Grana O, Ruiz-Llorente S, Pisano DG, Serrano M, Brooks HB, Campbell RM, Barrero MJ. NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci Rep. 2016;6:32952. doi: 10.1038/srep32952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014;588:2571–2579. doi: 10.1016/j.febslet.2014.04.014. [DOI] [PubMed] [Google Scholar]
- Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc Natl Acad Sci U S A. 2011;108:13130–13134. doi: 10.1073/pnas.1110081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley-Lopez J, Estabrooks LL, Stiehm R. Antibody deficiency in Wolf-Hirschhorn syndrome. J Pediatr. 1998;133:141–143. doi: 10.1016/s0022-3476(98)70194-5. [DOI] [PubMed] [Google Scholar]
- He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b) Nat Struct Mol Biol. 2008;15:1169–1175. doi: 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, Painter MW. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45:1386–1391. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kais Z, Rondinelli B, Holmes A, O’Leary C, Kozono D, D’Andrea AD, Ceccaldi R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016;15:2488–2499. doi: 10.1016/j.celrep.2016.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanu N, Gronroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, Maya-Mendoza A, Mistrik M, Rowan AJ, Patel H, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. 2015;34:5699–5708. doi: 10.1038/onc.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerzendorfer C, Hannes F, Colnaghi R, Abramowicz I, Carpenter G, Vermeesch JR, O’Driscoll M. Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum Mol Genet. 2012;21:2181–2193. doi: 10.1093/hmg/dds033. [DOI] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Tang CW, Pombo A, Jackson DA. Mechanisms regulating S phase progression in mammalian cells. Front Biosci (Landmark Ed) 2009;14:4199–4213. doi: 10.2741/3523. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Morishita M, di Luccio E. Cancers and the NSD family of histone lysine methyltransferases. Biochim Biophys Acta. 2011;1816:158–163. doi: 10.1016/j.bbcan.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28:198–201. doi: 10.1038/leu.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H, Wu X, Liu T, Yu K, Jelinek DF, Lou Z. The histone methyltransferase MMSET regulates class switch recombination. J Immunol. 2013;190:756–763. doi: 10.4049/jimmunol.1201811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongubala JM, Northrup DL, Lancki DW, Medina KL, Treiber T, Bertolino E, Thomas M, Grosschedl R, Allman D, Singh H. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol. 2008;9:203–215. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, Hua Y, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10:e1004566. doi: 10.1371/journal.pgen.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revilla IDR, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, Smeenk L, Trupke J, Sommer A, Jaritz M, et al. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J. 2012;31:3130–3146. doi: 10.1038/emboj.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarai N, Nimura K, Tamura T, Kanno T, Patel MC, Heightman TD, Ura K, Ozato K. WHSC1 links transcription elongation to HIRA-mediated histone H3.3 deposition. EMBO J. 2013;32:2392–2406. doi: 10.1038/emboj.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MY, Martinez-Garcia E, Phillip JM, Chambliss AB, Popovic R, Ezponda T, Small EC, Will C, Phillip MP, Neri P, et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene. 2016 doi: 10.1038/onc.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A, Hu D. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016;30:2021–2041. doi: 10.1101/gad.284109.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, Rajewsky K. B cell development under the condition of allelic inclusion. Immunity. 1997;6:225–233. doi: 10.1016/s1074-7613(00)80325-8. [DOI] [PubMed] [Google Scholar]
- Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, Altherr MR, den Dunnen JT. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet. 1998;7:1071–1082. doi: 10.1093/hmg/7.7.1071. [DOI] [PubMed] [Google Scholar]
- Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, Grosschedl R. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity. 2010;32:714–725. doi: 10.1016/j.immuni.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Ungerback J, Ahsberg J, Strid T, Somasundaram R, Sigvardsson M. Combined heterozygous loss of Ebf1 and Pax5 allows for T-lineage conversion of B cell progenitors. J Exp Med. 2015;212:1109–1123. doi: 10.1084/jem.20132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of the Differential Gene Expression Analysis for GC B cells. Related to Figure 6.
Table S2. RNA-seq counts, as normalized with RSEM for GC B cells. Related to Figure 6.
Table S3. Functional enrichment analysis from DAVID for KEGG pathways and Gene Ontology biological processes for GC B cells. Related to Figure 6.
Table S4. Results of the Differential Gene Expression Analysis for BM proB cells. Related to Figure 7.
Table S5. RNA-seq counts, as normalized with RSEM f for BM proB cells. Related to Figure 7.
Table S6. Functional enrichment analysis from DAVID for KEGG pathways and Gene Ontology biological processes for BM proB cells. Related to Figure 7.