

Abstract
Nuclear export of partially spliced or unspliced HIV-1 RNA transcripts requires binding of the viral protein Rev to the Rev Response Element (RRE) and subsequent oligomerization in a cooperative manner. Cellular DEAD-box helicase DDX1 plays a role in HIV replication, interacting with and affecting Rev-containing HIV transcripts in vivo, interacting directly with the RRE and Rev in vitro and promoting Rev oligomerization in vitro. Binding of DDX1 results in enhancement of Rev oligomerization on the RRE that is correlated with an RNA structural change within the RRE that persists even after dissociation of DDX1. Further, this structural transition is likely located within the three way junction of stem II of the RRE that is responsible for initial Rev binding. This discovery of the stem II structural transition leads to a model wherein DDX1 can act as an RNA chaperone, folding stem IIB into a proper Rev binding conformation.
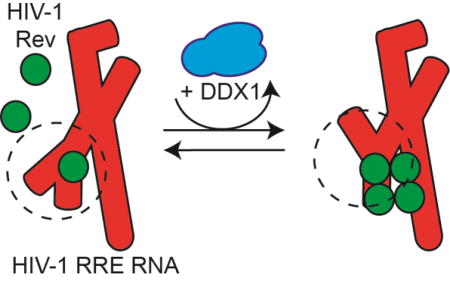
Graphical abstract
Background
In eukaryotic cells, splicing and nuclear export are functionally coupled with splicing preceding nuclear export for a majority of transcripts [1]. In the case of HIV, the export of an unspliced or partially spliced transcript circumvents this requirement by coopting the cellular export CRM1 pathway machinery [2]. This export mechanism is dependent on two key viral factors, the protein Regulator of Expression of Virion (Rev) and the RNA Rev Response Element (RRE).
Partially and fully unspliced HIV transcripts both contain the RRE RNA, located within the env coding region. Rev initially binds to the RRE at a specific high affinity binding site in stem II, followed by subsequent binding of Rev molecules to both Rev and RRE elements using lower affinity protein and RNA binding interfaces [3–8]. The resulting oligomeric Rev:RRE complex then binds to cellular protein CRM1 resulting in export via the CRM1/RanGTP-dependent pathway. While many of the fundamental cellular components necessary for CRM1-dependent export are understood, it is unclear what cellular factors play a role in Rev binding to or oligomerizing on the RRE. Cellular protein DEAD-Box Protein 1 (DDX1) has been implicated as a likely binding partner for the Rev Nuclear Diffusion Inhibitory Signal (NIS) that is responsible for nuclear retention of Rev (Figure 1d) [9, 10], although its specific role in Rev-dependent processes remains unclear [11, 12].
Figure 1. Rev/RRE/DDX1.

(a) Schematic diagram of Rev binding to RRE. RRE in red, Rev in green, and DDX1 in blue. Rev binds to the RRE and oligomerizes in a concentration dependent manner (left pathway). In the presence of DDX1, this oligomerization of Rev increases. (b) Schematic diagrams of measured binding interaction between DDX1, Rev and RRE from previously published studies [13, 14]. (1), KdRRE – dissociation constant between the first Rev molecule and the RRE. (2),KdDDX1 – dissociation constant between Rev and DDX1. (3), KdRRE – dissociation constant between the first Rev molecule and the RRE in the presence of DDX1. (4) KdRRE – dissociation constant between DDX1 and the RRE. (c) Two validated RRE234 secondary structure models. Initial Rev binding site boxed in solid black and lower affinity Rev binding site boxed in dashed black. (d) A diagram of the 117 aa Rev protein. Boxed regions represent structured alpha-helical domains, with oligomerization domains (OD) and nuclear export inhibition domain (NIS) labelled. (e) Schematic diagram of 740 aa human DDX1 protein showing SPRY domain insertion of N-term DEAD domain. Putative Rev NIS interacting region labelled below. (f) A representative electrophoretic gel mobility shift titration of Rev into radiolabeled RRE234. Putative Rev oligomerization states are labelled to the left, and a schematic of factors added to reaction on the right. Titrated species in bold.
Previous work established a role for DDX1 in the HIV lifecycle, where knockdown of DDX1 resulted in a decrease of p24 extracellularly and a decrease in RRE/Rev-dependent nuclear export intracellularly[13]. A direct binding interaction between DDX1 and Rev was established by binding assays in vitro and co-immunoprecipitation factors in vivo[13]. The association of Rev and DDX1 in vivo was abrogated in the presence of two Rev oligomerization mutants. These data taken together led to the hypothesis that one of the modes of action for DDX1 was at the level of Rev oligomerization on the RRE.
Single molecule fluorescence studies which measured directly the oligomerization state of Rev on a given RRE molecule indicated the presence of DDX1 enhances Rev oligomerization on the RRE in vitro (Figure 1a) [14]. Further, the Rev oligomerization enhancement activity of DDX1 was correlated with an increase in affinity for the first Rev/RRE binding event in the presence of DDX1 (detailed in Figure 1b). The concentration of DDX1 required for these effects correlated with the binding affinity measured between DDX1 and Rev, and not the observed binding affinity between DDX1 and the RRE [13]. These data led to a protein chaperone model of DDX1 activity, where DDX1 enhances Rev oligomerization via interacting directly with Rev. However, because of the well-known RNA chaperone activities of DEAD-Box helicases, there is an alternative possibility where DDX1 may act to remodel the RRE secondary structure between forms that have differential affinity for Rev binding[15].
The RRE is a large secondary structure element consisting of ~361 nucleotides, comprised of either 4 or 5 stem loops depending on the sequence and strain of HIV (Figure 1c, Supplemental Figure 3) [15, 16]. The length of stem I can be varied, and partial truncations of stem I retain high functionality in nuclear transport [17]. Highly conserved stem II contains a three way junction between stems IIA, IIB, and IIC, and acts as the initial high affinity Rev binding site [17–19]. Two alternative secondary structures have been identified for stems III and IV, which can be either distinct or combined RNA elements, and the formation of these two alternative structures depends on single nucleotide differences that occur naturally in viral isolates [15, 16, 20]. These two structures may both be present in a given isolate population, and the differences in the relative population of the two forms have been implicated in viral fitness, Rev binding and Rev oligomerization on the RRE.
Rev is a small 13 kD protein, with a structured helix-turn-helix motif at the N-terminus and an unstructured C-terminus (Figure 1d) [5, 6, 21]. Though small, the Rev protein contains many signal sequences located throughout its sequence, responsible for a variety of functions, including the Rev Oligomerization Domains (ODs), Nuclear Import Signal Domain (NLS), RNA Binding Domain or Arginine Rich Motif (RBD, ARM), Nuclear Export Signal (NES) and the Nuclear Diffusion Inhibitory Signal (NIS).
DDX1 is a 740 aa protein consisting of highly conserved DEAD N-terminal and C-terminal domains, with a SPRY domain that is atypically inserted within the DEAD N-terminal domain (Figure 1e). Previous studies of DDX1 showed that even with this domain insertion, DDX1 is a functional RNA-dependent ATPase, and therefore DDX1 might be expected to exhibit RNA helicase activity [13]. Because of this, we were interested in understanding the possible effects DDX1 has on the structure of the RRE, and in turn, how DDX1 could subsequently affect Rev binding and oligomerization.
Here, we demonstrate that DDX1 does indeed affect the structure of the RRE, and that the structural changes correlate strongly with enhanced Rev oligomerization. Further, the DDX1-dependent structural rearrangement occurs in both known forms of the stem III/IV region, indicating a novel RNA reorganization that is independent of the previously described stem III/IV transition. Chemical probing of the RRE structure in the presence of both Rev and DDX1 shows distinct as well as overlapping signal changes, leading to a model of DDX1 as an RNA folding chaperone that is independent of the Rev-DDX1 interaction, where DDX1 enhances the first step of Rev binding.
Results
DDX1-Enhanced Rev Oligomerization Correlates with a DDX1-Dependent RRE Species
While the full length RRE is ~361 nt. in length, a shorter ~234 nt. version is sufficient for nuclear export[17]. Using electrophoretic mobility shift assays (EMSAs), the sequential process of Rev binding and oligomerization on the RRE234 RNA can be visualized (Figure 1f). The first Rev molecule binds with nM affinity, with subsequent Rev monomers adding to the Rev:RRE complex in a stepwise manner. In previous reports, the first lower mobility species that is shifted up from the free RNA band has been ascribed to a single Rev protein bound to the RRE, and subsequent species have been assumed to represent species with 2 or more Rev proteins bound in an oligomeric complex[8, 22, 23]. Using this assay, it is possible to distinguish at least 6 distinct complexes that presumably represent various oligomeric complexes of Rev on RRE234. When the difference in gel migration distance for the sequential set of species is plotted versus the apparent oligomerization state, it is clear the migration change exhibited for the first Rev/RRE complex is distinct from those seen in subsequent oligomerization states (Supplemental Figure 1a). This basic observation is independent of the poly-acrylamide percentage and gel running conditions (data not shown). While the mobility of any complex is a function of the molecular weight, net charge, and hydrodynamic radius, there is something unique about the first Rev binding event that produces a disproportionately slower migrating species when compared to the addition of subsequent Rev molecules to the complex. One possible interpretation is that a Rev dimer is initially binding to the RRE. However, single molecule data has proven that Rev molecules overwhelmingly bind as monomers at the concentrations reported here[14, 24]. Further, the incremental change in electrostatics remains constant with each Rev binding event. Therefore, it is proposed that the change in migration rates represents a larger change in the hydrodynamic surface area upon binding of the first Rev monomer when compared to subsequent binding events.
Previous work indicated that DDX1 binds directly to the RRE in vitro, and Rev/DDX1/RRE co-binding studies hinted at a role for DDX1 in the initial Rev-binding step[13]. To further investigate DDX1 binding to the RRE, DDX1 was recombinantly fused with maltose binding protein as a solubility tag for expression and purification from E. coli cell lysates. DDX1-dependent Rev oligomerization enhancement was assessed using a previously published single molecule fluorescence technique[14, 24]. Here, fluorescent signals corresponding to fluorescently labelled Rev molecules binding to the RRE are quantified over time, and the histograms of signal occurrences in the presence and absence of DDX1 are shown in Supplemental Figure 1b. In this experiment, monomeric Rev exhibits a fluorescence intensity around 200 a.u. in TIRF (Total Internal Reflection Fluorescence) recordings, a dimer exhibits a signal around 400 a.u, and so on for each oligomeric state. At 1 nM Rev concentration, Rev binds primarily as a monomer to the full length RRE RNA, with a minor population near 400 a.u. representing dimers, and very small population at 600 a.u. (Supplemental Figure 1b, top middle panel). In the presence of 150 nM MBP-DDX1, the population of complexes with intensity at 400–800 a.u. increases notably, indicating an increase in Rev- association and oligomerization on the RRE RNA (Supplemental Figure 1b, bottom middle panel). These findings mirror previous experiments[14], indicating the presence of the MBP tag does not affect the ability of DDX1 to enhance Rev oligomerization on the RRE.
The enhanced effect of DDX1 on Rev oligomerization can also be observed using the EMSA assays described above. In the absence of DDX1, Rev oligomerizes on the RRE in a concentration dependent manner (Figure 2a, top panel). In an identical experiment where 25 nM MBP-DDX1 has been added, the amount of RNA associated with protein increases, as measured by the decrease in the RNA only species, and the number of species present at each Rev concentration also increases (Figure 2a, bottom panel; compare red, yellow and blue asterisk lanes; Supplemental Figure 2a for side by side comparison, and Supplemental Figure 2b for absolute quantification). Intriguingly, in the presence of MBP-DDX1 alone, two slower migrating species appear (Figure 2a, bottom panel, lane 2). However, the distance travelled by the slowest migrating species (corresponding to the highest oligomerization state of Rev) remains the same in both the (+) and (–) MBP-DDX1 samples (Figure 2a, Lane14 top panel, Lane 15 bottom panel). When each species is quantified and graphed on a plot of Fraction bound vs Rev concentration, it becomes apparent that each complex appears and disappears, at lower Rev concentrations for the Rev + DDX1 mixture than with Rev alone (Supplemental Figure 2b). Further, the distance migrated by the two RNA species in the DDX1-only lane have a similar migration rate to the first two Rev-dependent species (Figure 2a, lanes 9–11 top panel, lane 2 bottom panel; Figure 2d). The small change in mobility of these DDX1-dependent species is surprising since MBP-DDX1 is approximately 9 times the size of Rev. Further, previous EMSA studies conducted at lower current indicated RNA that associated with DDX1 remained in the sample well[13].
Figure 2. DDX1 Affects the Migration of the RRE in EMSA Experiments.

(a) Top panel – Electrophoretic gel mobility shift titration of Rev into radiolabeled RRE234. Lane 1, RRE234 alone; lanes 2–15, increasing concentrations of Rev as diagramed atop. Bottom panel – Identical to top panel but performed in the presence of 25 nM MBP-DDX1. Lane 1, RRE 234 alone; lane 2, RRE234 + 25 nM MBP-DDX1; lanes 3–16, increasing concentrations of Rev + 25 nM MBP-DDX1. For easier visual comparison, red, yellow and blue stars denote lanes with identical concentrations of Rev between top and bottom panels. These experiments were run on a 10% polyacrylamide gel. (b) Top panel – Electrophoretic gel mobility shift titration of MBP-DDX1 into radiolabeled RRE234. Lane 1, RRE234 alone; lanes 2–15, increasing concentrations of MBP-DDX1 as diagramed atop. Bottom panel – Identical to top panel, but performed in the presence of 7 nM Rev. Lane 1, RRE 234 alone; lane 2, RRE234 + 7 nM Rev; lanes 3–16, increasing concentrations of MBP-DDX1 + 7 nM Rev. Extra DDX1-dependent species are labelled shift 1, shift 2 and shift 3. Only species seen consistently over multiple experiments are identified and labelled. These experiments were run on a 7% polyacrylamide gel. (c) Similar to (a), but using oligomerization deficient mutant Rev V16D/I55N. See Supplemental Figure 2C for adjusted contrast. In (A–C), a schematic of factors added to the reaction is shown on the right. (d) Electrophoretic gel mobility shift assay showing side by side comparisons of RRE234 alone or in the presence of Rev or MBP-DDX1.
The mid-point of DDX1 binding to RRE234 was investigated using EMSA experiments run at higher current conditions and was determined to be ~150 nM affinity (Figure 2b, top panel), corresponding to previous data with RRE361[13]. It should be noted that excess unlabeled tRNA in these EMSA experiments acts as a competitor with RRE234 for DDX1 binding, making this assay unsuitable to determine absolute equilibrium binding affinities. In contrast to previous studies, three distinct RRE234 mobility species are observed in the presence of DDX1. One is a previously described “well-shift” species, corresponding to DDX1 binding, and two are intermediate migrating species. These intermediate species are either associated with MBP-DDX1 or have been stripped from it due to the higher current used in these experiments. Regardless, the two DDX1-dependent and Rev-dependent RRE complexes again migrate identically (Figure 2b, lane 9 top panel, lane 2 bottom panel; Figure 2d). The increased number of complexes seen in the combined presence of DDX1 and Rev has two possible explanations. First, this could represent a higher oligomerization state of Rev binding, as described in single molecule fluorescence experiments. Second, DDX1 could be responsible for some of the slower migrating complexes, and Rev responsible for subsequent shifted complexes that do not necessarily represent higher oligomerization states.
To clarify the identity of the various shifted species, the binding of a monomeric Rev mutant to the RRE was measured in the presence and absence of DDX1.The Rev V16D/I55N mutant is oligomerization-deficient, and has a monomeric binding profile in single molecule fluorescence Rev oligomerization assays[24]. In EMSA experiments, this mutant results in two protein/RNA complexes that appear simultaneously, and oligomerization is only observed at very high Rev concentrations that are not physiologically relevant (Figure 2c, Supplemental Figure 2c, top panel). The mobility shift data are consistent with either pre-dimerization of Rev in solution followed by RRE234 binding, or the existence of two alternative RNA conformations that are competent for binding. Single molecule oligomerization experiments argue against the existence of a pre-formed Rev dimer, and instead indicate that the Rev mutant is binding as a monomer and creating two structurally distinct migrating species[24].
If DDX1 was involved in the formation of some complexes, and Rev binding was responsible for subsequent complexes, the appearance of additional bands would be expected upon the introduction of DDX1. Instead, the only species present in MBP-DDX1/Rev V16D/I55N samples are also present in the Rev V16D/I55N samples (Figure 2c, Supplemental Figure 2c, bottom panel). Furthermore, the abundance of the RRE234 associated with the protein-RNA complexes increases at lower Rev concentrations in the presence of DDX1. These data are consistent with an enhancement of Rev association by DDX1.
Persistent Protein Binding is not Required For Most DDX1-Dependent RRE Species
Previous work has shown that the extended stem I, which is truncated in the RRE234 construct, may play a role in Rev binding at high concentrations, and may affect the global RRE conformation[25]. To understand if the DDX1-dependent RRE species formation described above can also be detected in larger RRE constructs, RRE361, or full length RRE, was used in an EMSA experiment containing MBP- DDX1 (Figure 3a, right top panel). Once again, an intermediately shifted species forms in the presence of the full length RRE, similar to that found in the shorter RRE234 species. However, it is still unclear which protein partners are contained in each shifted RRE species.
Figure 3. RRE Colocalization with DDX1/Rev and Secondary Structure Determination.

(a) Electrophoretic gel mobility shift titration of Rev (left gels) or MBP-DDX1 (right gels) into full length RRE361. RRE RNA (red, top panels) imaged using SYBR® Safe gel stain. Alexa555-labelled proteins (green, middle panels) imaged via fluorescence visualization. Bottom panels are merges of top two images. Schematic diagram of factors added to each reaction shown below. (b) Native gel electrophoresis of HIV BH10 RRE 361 imaged using Sybersafe™ stain. (c) Secondary structure model of HIV BH10 RRE361. Colored red circles represent normalized SHAPE signal intensity units.
To better understand the protein composition of each shifted RRE species, EMSA experiments were performed in the presence of fluorescently labelled MBP-DDX1 or Rev. In EMSA experiments using Rev-A555, Rev co-migrates with each RRE species (Figure 3a, left panel). However, in EMSA experiments using MBP-DDX1-A555, there is no evidence for the presence of DDX1 in the intermediate RRE species (Figure 3a, right panel). This was confirmed using western blotting techniques (data not shown).
Because the sensitivity of fluorescent or western signal may be below what is necessary to observe trace amounts of protein co-migration, an RRE361:MBP-DDX1 reaction was allowed to come to binding equilibrium, followed by incubation with proteinase K at 37C for 1 hour and visualized via EMSA (Figure 3b). There was no difference between + and − proteinase K incubated reaction mixtures. This data taken together indicate that at least one slower RNA migrating species in the presence of DDX1 is due to a change in the RNA’s hydrodynamic properties, likely due to an RNA structural change, rather a mobility shift due to continued protein binding.
While DDX1-dependent RRE structural species may persist in the absence of DDX1, previous work has demonstrated that these species are not observed under lower electrophoretic voltage or in the presence of Proteinase K [13]. Further, under the conditions reported here, the DDX1-dependent species do not accumulate over time, as one would expect (data not shown). Instead they are formed proportionally to the concentration of DDX1 used, regardless of incubation times above 10 minutes at room temperature. These data would suggest that the formation and unfolding of this structure is intimately linked to DDX1 binding and dissociation.
BH10 RRE adopts a combined stem III/IV secondary structure
Two possible secondary structure models have been presented for the RRE involving different arrangements of stems III and IV [15, 16, 25], and the RRE is capable of adopting both isoforms in equilibrium, where single nucleotide differences can shift the position of the equilibrium[15]. The EMSA experiments above indicate that the RRE from the BH10 isolate migrates as a single species (Figure 2d). However, since the RNA structure is highly sequence dependent, and folding may be dependent on ion concentrations, full length RRE361 was subjected to native gel electrophoresis in a buffer containing physiologically relevant concentrations of monovalent and divalent salts (Figure 3c). As with RRE234, this RNA migrated as a single species, indicating a preference for one secondary structure species over another.
The BH10 RRE sequences are more consistent with a combined stem III/IV secondary structure model[16]. To determine the secondary structure of the stem III/IV region, the structure of the RRE361 was probed using SHAPE chemical probing (Figure 3d, Supplemental Figure 3, Supplemental Table 1). The SHAPE-associated signal was located throughout the RRE sequence, with strong signal located in predicted loops in stems I, II, III/IV and V. Raw data were input to RNAstructure Web server model generator resulting in a model consistent with a single stem III/IV (Figure 3d, Supplemental Figure 3b). Contrary to previously published models, there is no evidence for a preformed stem II Rev binding site (nts 109–111 and 134–137) in the absence of Rev protein. We attempted to replicate previous Rev binding SHAPE studies using the BH10 sequence[16], but found that Rev used at the concentration reported resulted in a universal decrease in SHAPE modification of the RRE (Supplemental Figure 3a, Lane 4), either due to titration of the SHAPE reagent by the added protein, or large, and likely physiologically irrelevant, amounts of Rev binding throughout the RRE.
The DDX1-dependent RRE structural rearrangement is not a transition from stem III/IV to stems III and IV
Because RRE234 BH10 migrates as a single species, it is possible the structural transition seen in EMSA experiments represents stem III/IV reorganizing to separate stems III and IV. The G187A mutation of RRE234 (G250A on RRE361) has been shown to shift the secondary structure from a combined stem III/IV to a separated stems III and IV folding configuration[16]. We therefore introduced the G187A mutation into RRE234 and ran both native and denaturing poly-acrylamide gels, While WT and G187A RRE234 RNAs are similar sizes and migrate identically in a denaturing polyacrylamide gel, their gel mobility differs under native conditions (Figure 4a). The RRE234 (G187A) migrates slower than WT, indicating a tertiary structure with more solvent accessible surface area, consistent with previously published results indicating a separated stem III and IV structure[15, 16].
Figure 4. DDX1-Associated RRE Structural Rearrangement is not Due to Stem III/IV Reorganization.

(a) Polyacrylamide gel electrophoresis analysis of WT RRE (left lanes) and G187A mutant (right lanes). Predicted secondary structure models of wt RRE234 and G187A mutant RRE are in gray to the left and right of each lane respectively. Top panel - denaturing gel electrophoresis; bottom panel - native gel electrophoresis. (b) Electrophoretic gel mobility shift titration of Rev (left gels) or MBP-DDX1 (right gels) into RRE234WT (top gels) or RRE G187A (bottom gels). In each case, lane 1 is RNA alone, and subsequent lanes are increasing concentrations of coinciding protein. Schematic diagram of factors added to each binding reaction on left and right.
Previous Rev binding studies indicated a slight Rev binding and oligomerization preference for the separated stem III-stem IV configuration[15]. However, for the EMSA experiments shown in Figure 4b, left panels, there are no significant binding differences between the two. When EMSA experiments were conducted in the presence of DDX1, slower migrating RRE species were again observed for both WT and mutant RREs (Figure 4b, right panels). Further, the first species formed at similar concentrations of DDX1 for both RRE isoforms. The second species, although weaker in the G187A isoform, was also present. This data would indicate that the structure changes seen upon DDX1 binding, and strongly correlated with enhanced Rev binding and oligomerization, are not due to the structural rearrangement of stems III/IV, and indeed are present in both contexts.
ATP Does Not Affect DDX1-Dependent RRE Species Formation
A common theme of DEAD-box helicases is their dependence on ATP to catalyze RNA strand displacement and/or annealing. Indeed, single molecule fluorescence studies indicate the presence of a non-hydrolysable ATP analog, AMPPNP, and DDX1 is able to enhance Rev oligomerization even more than with no nucleotide present[14]. Further, work from our lab has shown that DDX1 acts as an RNA-stimulated ATPase in the presence of yeast tRNA or stem II of the RRE. However, no published work has demonstrated the ability of ATP to affect Rev oligomerization, or indeed DDX1 association with the RRE, though it is known that ATP does not affect the Rev:DDX1 interaction[13].
Rev:RRE EMSA experiments were performed in the absence and presence of 1 mM ATP (Figure 5a, top panels). The presence of excess ATP has no noticeable effect on Rev binding to or oligomerization on the RRE, as expected. However, and unexpectedly, the presence of 25 nM MBP-DDX1 in the ATP titration series had no measureable effect on the Rev-RRE interaction (Figure 5, bottom panels). Further, there seemed to be only a small decrease in alternative RRE species formation in the presence of ATP, indicating that ATP may not be required for the alternative RRE conformation’s formation or stability. However, the formal possibility exists that endogenous ATP is associated with MBP-DDX1 during protein purification and is responsible for the structural rearrangement effects observed.
Figure 5. ATP Does Not Affect RRE Structural Rearrangement Activity of DDX1 or Rev Oligomerization Enhancement.

(a) Electrophoretic gel mobility shift titration series of Rev into RRE234 in the absence (top gels) and presence (bottom gels) of DDX1. Right Gels are identical to left, but in the presence of 1 mM ATP. Red, yellow and blue asterisks denote identical concentrations of Rev and RRE234 between each series. Schematic diagram of factors added to each binding reaction on left and right.
Creating and Characterizing DDX1 Functional Mutants
DEAD-box helicases may coordinate three sometimes functionally coupled activities: ATP binding and hydrolysis, RNA binding, and protein binding. To better understand the role of each of these activities in the formation of the alternative RRE conformers, a variety of DDX1 mutations were required that could specifically disrupt each activity while leaving the others intact. DDX1, like other DEAD-Box proteins, is an RNA-dependent ATPase, comprised of two highly conserved domains and 11 essential sequence motifs (G, I-VI) (Figures 1e, 6a). DEAD-box members often have accessory domains N-term, C-term or both to facilitate broad and/or specific cellular functions. DDX1 is unique in that its accessory domain, a SPRY domain structural homolog, is inserted within the N-terminal half of the DEAD domain (Figures 1e, 6a). Using yeast two hybrid analysis, Rev was shown to interact with DDX1 in vivo [11, 13]. This interaction was shown to localize to a region comprised partially of the SPRY domain’s C-terminus and partially of the DEAD domain’s N-terminus (Figure 1e). DDX1 binds directly with Rev in vitro at low nM affinity as measured by a fluorescence polarization assay, and the Rev-binding activity of DDX1 is localized to the first 428 amino acids of DDX1, comprising the SPRY and N-terminal half of the DEAD domains [13].
Figure 6. DDX1 Mutant Biochemical Characterization.

(a) Schematic diagram of DDX1. Conserved DEAD-Domain functional motifs are boxed in black and labelled above. General positions of mutations are indicated below, while structural domains are listed above. (b) MBP-DDX1/Rev complex formation as determined by fluorescent polarization experiments. A representative binding isotherm of Rev-Alexa488 binding MBP-DDX1. (c) MBP-DDX1/nucleic acid complex formation as determined by filter binding experiments. A representative binding isotherm of MBP-DDX1 binding radiolabeled RRE361 in two different binding conditions.
To further characterize this interaction, a fluorescence polarization assay was performed with Alexa-488 or Alexa-555 labelled Rev and MBP-tagged versions of DDX1 (Figure 6b, Table 1). WT MBP-DDX1’s dissociation constant of 59 +/− 12 nM for Rev binding is consistent with DDX1 constructs from previous studies[13]. Further, when the SPRY and DEAD N-term domains were tested separately for Rev binding, only MBP-DEAD N-terminus showed a wild-type affinity at 79 +/− 7 nM, while MBP-SPRY showed no measurable binding at all (Table 1).
Table 1.
Equilibrium dissociation constants for DDX1 and mutants.
| MBPDDX1 Construct | Kd(Rev)(nM) | |
|---|---|---|
| WT | 59 +/− 11 | |
| DEAD N-term + SPRY | 43 +/− 6 | |
| SPRY | n.d. | |
| DEAD N-term | 79 +/− 7 | |
| K52N (−ATP) | 65 +/− 9 | |
| L303E (−RevA) | >700 | |
| L322E (−RevB) | >500 | |
| G325E | 29 +/− 1 | |
| RNA Bind | 38 +/− 3 | |
| Binding Condition | Kd(RRE361)(nM) | Hill |
| Buffer A | 24 +/− 1 | 1.7 |
| Buffer A + AMPPNP | 29 +/− 1 | 2.2 |
| Buffer B | 169 +/− 14 | 1.0 |
| Buffer B + AMPPNP | 429 +/− 23 | 1.7 |
| MBPDDX1 Construct | Kd(RRE234)(nM) | Hill |
| WT | 38 +/− 3 | 1.6 |
| K52N (−ATP) | 67 +/− 9 | 0.8 |
| L303E (−RevA) | 40 +/− 4 | 1.4 |
| L322E (−RevB) | 56 +/− 5 | 1.1 |
| RNA Bind | 4360 +/− 636 | 1.0 |
| Kd(γATP)(nM) | Hill | |
| WT | 221 +/− 32 | 1.2 |
| K52N (−ATP) | >10000 | |
| L303E (−RevA) | 449 +/− 52 | 1.1 |
| L322E (−RevB) | 344 +/− 25 | 1.6 |
| RNA Bind | 285 +/− 26 | 1.9 |
To generate DDX1 mutants deficient in Rev binding, a model of the conserved N-term DEAD-domain was created using the SWISS-MODEL server[26] (see materials and methods for more detail). This domain was then examined for likely Rev-binding sites given current and previous binding data[13]. Multiple mutations were created within these sites and tested for Rev binding affinity. While many mutations, such as G325E, showed no noticeable change in binding affinity, two mutations, L303E (−RevA) and L322E (−RevB), showed significant decreases in Rev binding, with Kds of >700 and >500 nM respectively (Table 1). Further, while these mutations show a defect in Rev binding, they show no defects in ATP or RNA binding, indicating that the protein fold and basal helicase functions remain intact.
DDX1 residue K52, located within motif I, has been identified as a conserved lysine involved in ATP binding to DEAD-Box helicases [27]. While MBP-DDX1 WT showed an ATP binding affinity of 220 +/− 32 nM, ATP binding to MBP-DDX1 K52N, renamed MBP-DDX1 (ATP), was almost completely undetectable at >10000 nM. Further, it has a near WT Rev binding affinity of 65 +/− 9 nM. Our data differs from previous studies in that this K52N mutation does not show an RNA binding deficiency, but rather has an RRE Kd of 67 +/− 9 nM (see below for WT comparison) (Table 1)[27].
Previous studies have indicated that DDX1 binds to different RNAs with similar affinity, including tRNA and the RRE RNA, supporting the idea of a general RNA binding motif that is not sequence specific[13]. To measure DDX1’s RRE binding affinity in the absence of competing tRNA, filter binding experiments were performed in a variety of binding buffers (Figure 6c, Table 1). As demonstrated, the measured binding affinity varies depending on the buffer composition by up to 20-fold, ranging from 24 nM in Buffer A to 169 nM in Buffer B, and 29 nM to 429 nM respectively in the presence of the non-hydrolysable ATP analog AMPPNP. However, in either buffer, the presence of AMPPNP increased the cooperativity of DDX1’s RNA binding activity. In Buffer A, the cooperativity increased from 1.7 to 2.2, while in Buffer B it increased from 1.0 to 1.7 (Table 1).
To generate an RNA binding mutant, the predicted DDX1 structure was scanned for likely nucleotide interacting amino acids within the conserved RNA binding cleft between DEAD N and C domains (see materials and methods for more detail). Four single mutations showed a slight binding defect individually and were combined to create an RNA binding defective mutant named MBP-DDX1 (−RNA Bind), with a Kd of 4.4 µM, or 100-fold lower affinity than WT DDX1, while retaining WT Rev binding at 37 nM, and ATP binding at 285 nM affinities (Table 1).
DDX1-Associated RRE Structure Change is Dependent on DDX1 RNA Binding
EMSA assays were performed to test the effects that each of these DDX1 functional mutants has on the structure of the RRE. Slower migrating RRE species form at lower DDX1 concentrations than the protein/RRE complex found in the well (Figure 7a, Top panel). Similar results were observed for DDX1 K52N, DDX1 (−RevA) and DDX1 (−RevB) (Figure 7a, 2nd, 4th and 5th panels). Since ATP is less likely to copurify with DDX1 during protein production, it seems very unlikely that the DDX1-induced RRE structural shift is dependent on the presence of ATP (though it may be enhanced by it in vivo). Further, mutations which affect Rev binding do not affect the formation of this alternative RRE-structured species. As expected, neither the intermediate migrating RRE species nor protein-associated species was observed with the DDX1 (−RNA) mutant, indicating that the proposed DEAD-Box domain:RRE interaction is likely necessary for the RNA chaperone activity (Figure 7a, 3rd panel).
Figure 7. DDX1 and Rev Have Overlapping Areas of Activity on the RRE.

(a) Electrophoretic gel mobility shift titrations of MBP-DDX1 WT (top panel) (as seen in figure 2b), MBP-DDX1 K52N (second panel), MBP-DDX1 (−RNA Bind) (third panel), MBP-DDX1 (−Rev A) (fourth panel) and MBP-DDX1 (−Rev B) (fifth panel) into radiolabeled RRE234. (b) Secondary structure model of HIV BH10 RRE361 stem II using 1M7 SHAPE probing data (see Supplemental Figure 4). Colored circles denote normalized SHAPE signal intensity (c,d) SHAPE difference map and predicted secondary structure models of RRE361 stem II when Rev (c) or MBP-DDX1 (d) is present. Blue color denotes a decrease in SHAPE reactivity, while yellow indicates an increase. Dotted lines indicate predicted purine-purine interaction based on previously published NMR and X-ray crystallography data5,29.
DDX1 and Rev Have Partially Overlapping Regions of Chemical Protection on the RRE
Previous SHAPE chemical probing studies of the RRE have used both NMIA, a reagent with a reaction time in minutes, as well as 1M7, a reagent with a reaction time in seconds[16, 25, 28]. Because RNA structures can be transient, the secondary structure of the RRE was probed on both the long and shorter time scales using each reagent. RRE361 probed using the 1M7 reagent (Figure 7b, Supplemental Figure 4, Supplemental Table 1) showed some minor differences in reactivity compared to NMIA-treated RRE361. Primarily, lower overall reactivity was observed for the linker region between stems III/IV and V, as well as the apical loop of stem V. However, the majority of reactive nucleotides remained consistent between reagents, leading to identical secondary structure models.
To better understand the change of chemical probing signal in the presence of Rev, as well as to probe possible secondary structure changes, SHAPE analysis was performed on RRE361 using reagent 1M7 in the presence of Rev. Rev was added at a concentration consistent with > 90% Rev-bound, but below full Rev binding (Figure 1f, Lane 5), corresponding to 160 nM Rev (Figure 7c, Supplemental Figure 4c, left panel, Supplemental Table 1). While no statistically significant and reproducible signal enhancements were observed, there was distinct decreased signal located within the three-way junction of stem II, as well as within an internal bulge in stem I at G90. These sites both correlate with previously described Rev binding sites[18, 22, 29].
SHAPE probing was again performed on a sample containing RRE361, but in the presence of MBP-DDX1. The chosen DDX1 concentration ensured a greater than 50% RRE bound species (Figure 4b), corresponding to 360 nM MBP-DDX1 (Figure 7d, Supplemental Figure 4c, right panel, Supplemental Table 1). Unlike Rev, the DDX1-associated SHAPE signal showed distinct enhancement in three areas. Namely, in stem I (nts. 261 and 262), stem III/IV (nts. 193–195) and stem IIA (nts. 106, 107, 162 and 163). The enhancements for nucleotides located in stem I and stem III/IV are increases to enhancements already observed in the free RNA, either indicating a stabilization of the single stranded nature in these positions, or an increase of signal due to helicase binding. However, the nucleotides in stem IIA represent a large, statistically significant and reproducible increase from no or weak to high 1M7 reactivity, corresponding to a shift from double stranded to single stranded character. Further 1M7 reactivity decreased, usually to undetectable levels, within the stem II three way junction, at nucleotides 108–111, 133, 134, 136 and 138 (Figure 6d, Supplemental Table 1). Though these signal decreases are not as dramatic as those seen for the DDX1 dependent signal enhancements, they are present over multiple experiments and are statistically significant. Because both of these experiments were done in the presence of DDX1 or Rev proteins, it is difficult to differentiate between changes in RNA structure and protein footprint signal. However, it is clear that the changes observed for both protein sets overlap specifically in the stem II binding site.
Discussion
Rev binding and oligomerization on the RRE is a key step in the HIV RNA nuclear translocation pathway as demonstrated by Rev oligomerization defective mutants that negatively affect Rev-dependent nuclear export[3]. However, the nuclear Rev concentrations required for export are in the low nM range, corresponding to 1–2 Rev molecules bound for every RRE-containing RNA in in vitro experiments[30–32]. In vivo evidence indicates that both oligomerization domains are required for efficient export, implying a complex including not less than 3 Rev monomers[3, 33].
The introduction of DDX1 into the Rev:RRE assembly, as demonstrated by single molecule experiments, enhances the oligomerization of Rev on the RRE, though the mechanism by which these effects are produced is not well understood. Single molecule studies, in combination with our previous work, suggested a DDX1 protein chaperone activity explanation for the observed enhancement of Rev oligomerizing on the RRE, as the functional DDX1 concentration matched that for Rev binding, but was below that observed for DDX1-RNA interaction (Figure 1b)[13, 14]. Our previous binding data indicated a DDX1/RRE interaction in the ~250 nM affinity range, likely due to the presence of competing tRNA species in the EMSA experiments themselves[13]. However, using a direct binding assay we can now see that DDX1 interacts with the RRE RNA at mid-nM concentrations, with this binding being highly sensitive to the buffer conditions. This implies that DDX1 could be affecting Rev binding at either/both the RNA and protein binding levels.
Here, we have demonstrated an RNA chaperone activity for DDX1, resulting in an RRE species which migrates slower in polyacrylamide gels than the unchanged RNA (Figure 2a). Further, this species is structurally stable even after dissociating from DDX1, although it is possible that DDX1 is stripped from complexes during the EMSA experiments (Figure 3a and 3b). Intriguingly, these species migrate identically to the first two Rev/RRE species (Figure 2d), and correlate strongly with increased Rev oligomerization (Figures 2a–b). These data taken together are consistent with a model wherein an RRE structural change coincides with Rev binding, and DDX1 is able to induce this structural change in the absence of Rev. This does not, however, preclude the idea that the protein:protein and protein:RNA binding processes may, or do, happen concurrently within the cell.
To understand the DDX1-dependent RRE conformational change requires a thorough understanding of the RRE structure. Previous experiments using SHAPE have focused on two HIV strains, NL4-3 and ARV-2/SF2, and have demonstrated some sequence dependent structural plasticity within the RRE itself, particularly within stems III-V. Stem II, the location of the high affinity binding site, is highly conserved amongst HIV strains. However, even with this conservation, models based on SHAPE data disagree on the level of secondary structure formation occurring in the three way junction between stems IIA, IIB and IIC. In our studies, the RRE stem II junction shows a large amount of SHAPE-dependent signal, in both fast and slow reacting reagent regimes, consistent with either a highly unstructured or conformationally dynamic RNA region (Figure 3d, 7b). Analysis of this region is likely impaired partially by the presence of a large RT stop located within this junction, making SHAPE quantification difficult. This stop may be portrayed as half of a nucleotide base pair because the coinciding SHAPE data is discounted. Based on the SHAPE data, we propose that the Rev binding site is either unstructured, or highly flexible in the absence of a protein binding partner.
Upon addition of low concentrations of Rev, the SHAPE signal decreases in this region, specifically in nucleotides associated with the Rev/RRE initial interaction. These data are consistent with both NMR and X-ray crystallography data showing a highly organized stem II upon Rev binding[5, 34]. Intriguingly, a similar pattern of protections is observed in the presence of DDX1. Here, nucleotides found in stem IIA show an increase in SHAPE signal, while those in the three way junction show a decrease comparable to that observed in the presence of Rev (Figure 7b–d).
SAXS data has led to an A-form model of the RRE, wherein the entire stem II is localized opposite stem IA [35]. We propose a model consistent with the data here which suggest an additional detail to the SAXS-based RRE structural model. Here stem IIB becomes highly structured upon Rev binding, leading to a change in migration in EMSA assays. DDX1 would either facilitate pre-formation of this RNA binding platform or stabilize the formed structure, leading to an increased affinity of Rev for the RRE stem II (Figure 8). However, it is not clear what the relationship of the local conformational changes seen here is to the A-form model based on SAXS.
Figure 8. A Mechanistic Model of DDX1/RRE/Rev Interaction and Effects.

(a) A Schematic model for Rev (green) binding to RRE (red). RRE stems are labelled I-V. In the presence of DDX1 (blue), stem IIB is able to preform the extended helical structure required for Rev binding, and stem IIA is partially unwound. This preformed site is now able to bind Rev with higher affinity than the unstructured junction.
It is interesting that, in the experiments presented here, ATP had little or no effect in the formation of RRE intermediates or on apparent DDX1-dependent Rev oligomerization enhancement on the RRE. While many studies have indicated an ATP-dependent nature of RNA chaperone activity for DEAD-box helicases (for a review see [36]), others have described ATP-independent RNA unfolding and annealing activities for some DEAD-box helicases[37]. However, though not required for the RNA folding activity described here in vitro, it is clear that within a cell ATP is present, and therefore may play a role which is not apparent using the biophysical techniques described here. An obviously interesting experiment would be probing the structure of the RRE over time in the presence of DDX1 and ATP. However, current SHAPE reagents also react with the large excess of ATP required, making an alternative method for study of the RRE conformation necessary.
It is difficult to directly assess the impact of each of the DDX1 functional mutations created for this study on the binding and subsequent oligomerization of Rev using mobility shift assays, since it is now apparent that the effects of protein binding and RNA conformation on mobilities are convolved. Therefore, additional experiments are needed using single molecule methods to better understand the contribution of each of these activities in Rev/RRE complex assembly, and these experiments are currently underway.
Further, while the goal of this work was to understand the mechanism of DDX1-dependent Rev oligomerization enhancement described in in vitro experiments, it is still unclear what the implications this may have in vivo. Because of the sequence-independent nature of DDX1:RNA binding as well as the high degree of conservation between many DEAD-box helicases and DDX1 RNA binding cleft, it seems possible that the RNA chaperone activity described in this study could easily be embodied by a number of other DEAD-box helicases. Further, the binding interface between Rev and DDX1 also resides on a conserved DEAD-domain, indicating a general mechanism of interaction between DEAD-box proteins and Rev.
While an RNA chaperone activity may be involved, previous studies have implied that another role for DDX1 in RRE-dependent export may be in the localization of Rev to the RRE within the nucleus[11, 12, 38]. Indeed, when DDX1 nuclear localization is disrupted, as is naturally the case in astrocytes or artificially mislocalized by expressing a tat-mutant, Rev also mislocalizes followed by a decrease in Rev-dependent export. It has been difficult to study this effect because mutations in Rev which disrupt direct DDX1 interaction are also known to disrupt Rev oligomerization on the RRE[3, 8, 13]. However, here we have described DDX1 mutations which are able to disrupt DDX1:Rev complex formation while leaving Rev:RRE interactions intact. Further work detailing the role DDX1 plays in vivo during Rev-dependent export is required.
In conclusion, all available data support a model where DDX1 can remodel the RRE into a form that has improved Rev binding and oligomerization, but that the DDX1-Rev interaction is not required for this effect in vitro.
Materials and Methods
HIV strains
All HIV proteins and RNAs were derived from viral isolate BH10.
Rev proteins
S10C Rev is described previously[14]. Rev V16D and V16DI55N were created using standard overlapping-primer point mutagenesis (Agilent Technologies). Expression and purification were performed as described[14, 23, 24].
Fluorescently labeled proteins
The native and mutant forms of Rev and DDX1 were fluorescently labelled as previously described with a few modifications[14, 24]. Purified proteins were placed in reducing buffer (20 mM HEPES pH 7.8, 800 mM NaCl, 10 mM DTT) at 4°C overnight. Ammonium sulfate was then added to a final concentration of 70% saturation, and resulting precipitate was centrifuged and separated from supernatant. Protein precipitate was washed twice with 70% ammonium sulfate, and allowed to air dry. The pellet was resuspended in Alexa labelling buffer (50 mM Sodium Phospate Buffer pH 7.2, 500 mM NaCl and 5 Molar excess Alexa-maleimide fluorophore (Thermo-Fisher)) and subsequent labelling and purification proceeded as previously described[14, 24]. Rev-Alexa labelling efficiency was >95% for MBP-DDX1, WT Rev, and Rev mutants per manufacturer quantification methods.
MBP-DDX1 expression
The insert from the previously described DDX1 expression construct [13] was PCR amplified and inserted into Gateway cloning vector pDonr221 using BP Clonase (Invitrogen). The sequence was verified, and the resulting gene was recombined into Gateway cloning vector pDonr-566 using LR Clonase (Invitrogen). The resulting gene encodes from the N-terminus a 6X-His tag, Maltose Binding Protein (MBP) tag, flexible linker, TEV protease cleavage site and wt DDX1 gene. Expression and purification were performed as described previously[13]. Subsequent domain and point mutations were made using overlapping primer mutagenesis. The Gateway parent vectors were a generous gift of the Dominic Esposito Lab at the National Cancer Institute.
Fluorescence polarization experiments
Fluorescence polarization titration experiments and analysis followed protocols established previously, with a few modifications[13]. 15 nM Alexa488-labeled Rev and varying amounts of WT MBP-DDX1, or DDX1 mutants, were suspended in FP buffer (10 mM Hepes (pH 7.5), 150 mM KCl, 2 mM MgCl2, 5 mM β-mercaptoethanol, and 10% glycerol) at room temperature, with a final sample volume of 200 µl per condition. Samples were equilibrated in 96-well opaque low-binding microtiter plates (Grenier), and the fluorescence polarization was determined using an Envision 2104 Multilabel Plate Reader (Perkin Elmer). The reported error is 1 SD calculated from the averaged titrations of 3 or more technical replicates. Because the constant concentration of Rev, R0, used was close to the measured 50% bound concentrations of DDX1 constructs, the dissociation constant, Kd, of each titration was determined by fitting the FP data using the following quadratic binding isotherm for a bimolecular complex:
Here, FB is the bound Rev-Alexa488 fraction, mP = polarization value in millipolarization units, mPmax = maximum mP value within a titration, mPmin = minimum mP value within a titration, P0 = MBP-DDX1 concentration, R0 = Rev-Alexa488 concentration and Kd = equilibrium dissociation constant.
RRE234 and RRE361 cloning/expression/purification
The initial RRE-containing plasmid is previously described[23]. RRE234 and RRE361 constructs were created using overlapping primer PCR and contained (from the 5’ end) an EcoRI restriction site, T7 RNA polymerase promoter, hammerhead ribozyme, RRE234 or RRE361, hepatitis delta ribozyme and a BamHI restriction site. Restriction fragments were created using BamHI and EcoRI restriction enzymes (NEB) and purified using phenol chloroform extraction and ethanol precipitation. The resulting product was then ligated into pUC19 plasmid, and sequence verified by DNA sequencing. RNA transcription, purification, and concentration all followed previously described methods[39, 40].
RNA 5’ phosphorylation
RNA was 5’ radiolabeled with γP32-ATP, and subsequently purified, as previously described[39, 40].
EMSA assays
Electrophoretic mobility shift assays were performed similar to previously described protocols with a few modifications[13, 23]. Radiolabeled RRE234 radioactive signal was quantified using a Bioscan QC-2000. 50 cpm of RRE234 was diluted in QT buffer (10 mM HEPES pH 7.5, 150 mM KCl, 5 mM DTT, 10% glycerol, 50 µg/mL yeast tRNA, 100 µg/mL bovine serum albumin) to a final volume of 10 µL, corresponding to low-mid pM concentration of RNA. Rev stocks were separately diluted in 10µL QT buffer samples. 10 µL of RRE234 sample was added to 10 µL of each Rev dilution series and allowed to incubate at room temperature for 20 minutes to come to binding equilibrium. Samples were then loaded on either a .5X TBE 7% or 10% polyacrylamide gel that had been prerun for 1 hr at 600 V at 4°C. Gels were then run for 1–2 hours at 600 V, transferred to filter paper and dried. Gels were exposed overnight to a Phosphorscreen and imaged using a STORM® Phosphorimager. Each experiment was replicated two or more times, with one representative of each experiment being presented in this work.
RNA gel electrophoresis
RNA native and denaturing gel electrophoresis procedures were described previously[40]. Each experiment was run two or more times, with one representative gel presented in this work.
TIRF microscopy
Analysis for TIRF microscopy data was performed as previously described[14]. The full-length 361 nt RRE with extensions at both the 3’ and 5’ ends was transcribed in vitro using T7 RNA polymerase and was purified using 6% denaturing gel electrophoresis. The 3’ end of RRE transcript was hybridized with a 28 nt biotinylated oligonucleotide. A 500 pM solution containing an RRE-DNA hybrid was immobilized on a streptavidin-coated quartz surface, as described previously[24]. The solution was incubated on the slide for 5 minutes, followed by rinsing with buffer only to elute unbound RNA/DNA complexes. The quartz surface was coated with polyethylene glycol to inhibit nonspecific adsorption of Rev or DDX1 proteins, as described[41]. 1 nm Alexa 555 (A555) labeled Rev, with or without 150 nM DDX1, in 50 mM Hepes buffer (pH7.5) containing 2 mM Trolox, 150 mm KCl, 10 mM K2SO4, 2 mM MgCl2, 2 mM DTT, and oxygen scavenging system (50 µg/ml glucose oxidase, 10 µg/ml catalase, and 2–5% wt/vol glucose) was introduced into the solution. A 532 nm laser was used to excite Alexa-555 (A555) labeled Rev using a custom-built prism-based TIRF microscope. Emission from A555 was collected through a water immersion objective and recorded on an intensified CCD camera with 100 ms integration time[42, 43]. A single-molecule data acquisition package (https://physics.illinois.edu/cplc/software/) was used to record data. Fluorescence intensity traces from individual Rev complexes were extracted from the CCD camera movie files and processed using a custom program written in Matlab. Binned fluorescence intensity histograms were compiled from more than 100 individual traces using IGOR Pro. software (WaveMetrics). One technical replicate was performed for every reported reaction.
Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE)
SHAPE analysis was performed as previously described with a few modifications[40]. NMIA and 1M7 concentrations were added to a folded RRE361 or RRE361/protein mixture to a final concentration of 25 mM, and allowed to react at 37°C for 60 min or 5 min respectively. RNA was purified three times using PCIA extraction and ethanol precipitation. Resulting RNA was then combined with primers denoted in Supplemental Figure 3b and Supplemental Figure 4b, and reverse transcription performed as described. Resulting samples were run on a 10% polyacrylamide gel (1XTBE, 7M Urea) for 2, 4, 6 and 8 hours, dried on filter paper, and exposed to phosphorscreens and imaged on a phosphorimager. Data was analyzed using SAFA_WINv11b and resulting data normalized so >98% of reproducible SHAPE-dependent stops would fall between .3 and 2. Signal enhancements and protections were defined as greater than .3 enhancement or reduction in absolute signal in all replicates. Secondary structure models based on SHAPE data were created using RNAstructure Web Servers (http://rna.urmc.rochester.edu/RNAstructureWeb/index.html). At least three experiments were performed for every SHAPE reaction, with one representative analysis of each being presented in this work. All data represent highly reproducible results.
Filter binding assay for RNA binding and nucleotide binding
Filter binding assays and analysis were performed as previously described, with γ-labeled ATP, RRE234 and RRE361 acting as the limiting substrates [44]. Buffer A (10 mM HEPES, pH 7.5, 150 mM KCl, 2 mM MgCl2, 5 mM βME, 10%Glycerol) and Buffer B(10 mM Tris, pH 8.0, 100 mM NaAcetate, 200 mM KCl, 2.5 mM MgCl2, 2 mM DTT) were used to assess buffer effects on DDX1/RRE binding. Buffer A was used in all subsequent nucleotide and RNA binding assays. RNA and nucleotide concentrations ranged from 1–500 pM (5–100 cpm radioactive signal at time of use). Filters included HT Tuffryn® filter (Pall laboratory), Hybond™-N+ Nylon filter (GE Healthcare), Supported Nitrocellulose Membrane (Bio-Rad) and Fisherbrand™ filter paper sheets. Reaction mixtures were placed through filter sandwich using a Schleicher & Schuell microsample filtration manifold and vacuum pump. Because the constant concentration of RNA or nucleotides used was well below the measured 50% bound concentrations of DDX1 constructs, the dissociation constant, Kd, of each titration was determined by fitting the binding data using the following binding isotherm:
Here, FB is the bound RRE or nucleotide fraction, P0 = MBP-DDX1(or mutant) concentration, n = Hill coefficient, Kd = dissociation constant, FBmax = maximum fraction of RNA or nucleotide bound within a titration series and FBmin = minimum fraction of RNA or nucleotide bound within a titration series. Each binding series was performed with three technical replicates.
DDX1 model production
Full length and DEAD-domain models of DDX1 were generated using the SWISS-MODEL server hosted by Biozentrum. Full length DDX1 (GeneID NP_004930) acted as the model input, and generated 3D PDB-format models allowed to form given no constraints. Likely models were those based on other DEAD-box protein structures (including DBP5, and DDX16), and resulting model was scanned for likely mutational analysis using Pymol™ Molecular Vision Software. For likely RNA binding mutants, the solved structural model for Drosophila Vasa bound to RNA (pdb ID 2DB3) was scanned for likely RNA-interacting residues, and corresponding residues located on SWISS-MODEL generated DDX1 models.
Supplementary Material
Highlights.
DDX1 has been shown to increase oligomerization of HIV-1 Rev on the cognate RNA Rev Response Element, though the mechanism is unclear.
DDX1-associated Rev oligomerization enhancement correlates strongly with an RNA structural transition.
This RNA structural transition is DDX1-mediated, and persists even after DDX1 dissociation.
Chemical probing experiments indicate that Rev and DDX1 binding effects overlap in the RRE RNA within the high affinity binding site stem II.
Acknowledgments
We thank Stephen Edgcomb, Larry Gerace and Hui-Yi Chu for helpful discussions on this project. Funding for this work was provided by the National Institute of General Medical Sciences (grant P50 GM082545; W. Sundquist PI, to J.R.W and D.P.M.), the American Cancer Society (fellowship award 121633-PF-11-222-01-RMC to J.A.H) and California HIV/AIDS Research Program (fellowship award F12-SRI-210 to R.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Custodio N, Carmo-Fonseca M, Geraghty F, Pereira HS, Grosveld F, Antoniou M. Inefficient processing impairs release of RNA from the site of transcription. EMBO J. 1999;18:2855–66. doi: 10.1093/emboj/18.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–60. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 3.Jain C, Belasco JG. A structural model for the HIV-1 Rev-RRE complex deduced from altered-specificity rev variants isolated by a rapid genetic strategy. Cell. 1996;87:115–25. doi: 10.1016/s0092-8674(00)81328-8. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci U S A. 2010;107:12481–6. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayaraman B, Crosby DC, Homer C, Ribeiro I, Mavor D, Frankel AD. RNA-directed remodeling of the HIV-1 protein Rev orchestrates assembly of the Rev-Rev response element complex. Elife. 2014;3:e04120. doi: 10.7554/eLife.04120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiMattia MA, Watts NR, Stahl SJ, Rader C, Wingfield PT, Stuart DI, et al. Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc Natl Acad Sci U S A. 2010;107:5810–4. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–7. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- 8.Jain C, Belasco JG. Structural model for the cooperative assembly of HIV-1 Rev multimers on the RRE as deduced from analysis of assembly-defective mutants. Mol Cell. 2001;7:603–14. doi: 10.1016/s1097-2765(01)00207-6. [DOI] [PubMed] [Google Scholar]
- 9.Kubota S, Pomerantz RJ. The nuclear function of the nuclear diffusion inhibitory signal of human immunodeficiency virus type 1: critical roles in dominant nuclear localization and intracellular stability. J Hum Virol. 2000;3:173–81. [PubMed] [Google Scholar]
- 10.Kubota S, Pomerantz RJ. A cis-acting peptide signal in human immunodeficiency virus type I Rev which inhibits nuclear entry of small proteins. Oncogene. 1998;16:1851–61. doi: 10.1038/sj.onc.1201738. [DOI] [PubMed] [Google Scholar]
- 11.Fang J, Kubota S, Yang B, Zhou N, Zhang H, Godbout R, et al. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology. 2004;330:471–80. doi: 10.1016/j.virol.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, Acheampong E, Dave R, Wang F, Mukhtar M, Pomerantz RJ. The RNA helicase DDX1 is involved in restricted HIV-1 Rev function in human astrocytes. Virology. 2005;336:299–307. doi: 10.1016/j.virol.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Edgcomb SP, Carmel AB, Naji S, Ambrus-Aikelin G, Reyes JR, Saphire AC, et al. DDX1 is an RNA-dependent ATPase involved in HIV-1 Rev function and virus replication. J Mol Biol. 2012;415:61–74. doi: 10.1016/j.jmb.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson-Anderson RM, Wang J, Edgcomb SP, Carmel AB, Williamson JR, Millar DP. Single-molecule studies reveal that DEAD box protein DDX1 promotes oligomerization of HIV-1 Rev on the Rev response element. J Mol Biol. 2011;410:959–71. doi: 10.1016/j.jmb.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherpa C, Rausch JW, Le Grice SF, Hammarskjold ML, Rekosh D. The HIV-1 Rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucleic Acids Res. 2015;43:4676–86. doi: 10.1093/nar/gkv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legiewicz M, Badorrek CS, Turner KB, Fabris D, Hamm TE, Rekosh D, et al. Resistance to RevM10 inhibition reflects a conformational switch in the HIV-1 Rev response element. Proc Natl Acad Sci U S A. 2008;105:14365–70. doi: 10.1073/pnas.0804461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mann DA, Mikaelian I, Zemmel RW, Green SM, Lowe AD, Kimura T, et al. A molecular rheostat. Cooperative rev binding to stem I of the rev-response element modulates human immunodeficiency virus type-1 late gene expression. J Mol Biol. 1994;241:193–207. doi: 10.1006/jmbi.1994.1488. [DOI] [PubMed] [Google Scholar]
- 18.Tiley LS, Malim MH, Tewary HK, Stockley PG, Cullen BR. Identification of a high-affinity RNA-binding site for the human immunodeficiency virus type 1 Rev protein. Proc Natl Acad Sci U S A. 1992;89:758–62. doi: 10.1073/pnas.89.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–8. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- 20.Fernandes J, Jayaraman B, Frankel A. The HIV-1 Rev response element: an RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol. 2012;9:6–11. doi: 10.4161/rna.9.1.18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010;17:1337–42. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31:824–34. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgcomb SP, Aschrafi A, Kompfner E, Williamson JR, Gerace L, Hennig M. Protein structure and oligomerization are important for the formation of export-competent HIV-1 Rev-RRE complexes. Protein Sci. 2008;17:420–30. doi: 10.1110/ps.073246608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pond SJ, Ridgeway WK, Robertson R, Wang J, Millar DP. HIV-1 Rev protein assembles on viral RNA one molecule at a time. Proc Natl Acad Sci U S A. 2009;106:1404–8. doi: 10.1073/pnas.0807388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai Y, Tambe A, Zhou K, Doudna JA. RNA-guided assembly of Rev-RRE nuclear export complexes. Elife. 2014;3:e03656. doi: 10.7554/eLife.03656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 27.Popow J, Jurkin J, Schleiffer A, Martinez J. Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature. 2014;511:104–7. doi: 10.1038/nature13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mortimer SA, Weeks KM. Time-resolved RNA SHAPE chemistry. J Am Chem Soc. 2008;130:16178–80. doi: 10.1021/ja8061216. [DOI] [PubMed] [Google Scholar]
- 29.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, et al. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–51. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 30.Heaphy S, Dingwall C, Ernberg I, Gait MJ, Green SM, Karn J, et al. HIV-1 regulator of virion expression (Rev) protein binds to an RNA stem-loop structure located within the Rev response element region. Cell. 1990;60:685–93. doi: 10.1016/0092-8674(90)90671-z. [DOI] [PubMed] [Google Scholar]
- 31.Reddy B, Yin J. Quantitative intracellular kinetics of HIV type 1. AIDS Res Hum Retroviruses. 1999;15:273–83. doi: 10.1089/088922299311457. [DOI] [PubMed] [Google Scholar]
- 32.Pomerantz RJ, Seshamma T, Trono D. Efficient replication of human immunodeficiency virus type 1 requires a threshold level of Rev: potential implications for latency. J Virol. 1992;66:1809–13. doi: 10.1128/jvi.66.3.1809-1813.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas SL, Oft M, Jaksche H, Casari G, Heger P, Dobrovnik M, et al. Functional analysis of the human immunodeficiency virus type 1 Rev protein oligomerization interface. J Virol. 1998;72:2935–44. doi: 10.1128/jvi.72.4.2935-2944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battiste JL, Tan R, Frankel AD, Williamson JR. Binding of an HIV Rev peptide to Rev responsive element RNA induces formation of purine-purine base pairs. Biochemistry. 1994;33:2741–7. doi: 10.1021/bi00176a001. [DOI] [PubMed] [Google Scholar]
- 35.Fang X, Wang J, O'Carroll IP, Mitchell M, Zuo X, Wang Y, et al. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155:594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip Rev RNA. 2011;2:135–52. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, Jain C. DEAD-box proteins from Escherichia coli exhibit multiple ATP-independent activities. J Bacteriol. 2011;193:2236–41. doi: 10.1128/JB.01488-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin MH, Sivakumaran H, Jones A, Li D, Harper C, Wei T, et al. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology. 2014;11:121. doi: 10.1186/s12977-014-0121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammond JA, Rambo RP, Filbin ME, Kieft JS. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA. 2009;15:294–307. doi: 10.1261/rna.1360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammond JA, Rambo RP, Kieft JS. Multi-domain packing in the aminoacylatable 3’ end of a plant viral RNA. J Mol Biol. 2010;399:450–63. doi: 10.1016/j.jmb.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamichhane R, Daubner GM, Thomas-Crusells J, Auweter SD, Manatschal C, Austin KS, et al. RNA looping by PTB: Evidence using FRET and NMR spectroscopy for a role in splicing repression. Proc Natl Acad Sci U S A. 2010;107:4105–10. doi: 10.1073/pnas.0907072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamichhane R, Berezhna SY, Gill JP, Van der Schans E, Millar DP. Dynamics of site switching in DNA polymerase. J Am Chem Soc. 2013;135:4735–42. doi: 10.1021/ja311641b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berezhna SY, Gill JP, Lamichhane R, Millar DP. Single-molecule Forster resonance energy transfer reveals an innate fidelity checkpoint in DNA polymerase I. J Am Chem Soc. 2012;134:11261–8. doi: 10.1021/ja3038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7:194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.