

Abstract
Purpose
Myopia is a common visual disorder caused by eye overgrowth, resulting in blurry vision. It affects one in four Americans, and its prevalence is increasing. The genetic mechanisms that underpin myopia are not completely understood. Here, we use genotype data and linkage analyses to identify high-risk genetic loci that are significantly linked to myopia.
Methods
Individuals from 56 Caucasian families with a history of myopia were genotyped on an exome-based array, and the single nucleotide polymorphism (SNP) data were merged with microsatellite genotype data. Refractive error measures on the samples were converted into binary phenotypes consisting of affected, unaffected, or unknown myopia status. Parametric linkage analyses assuming an autosomal dominant model with 90% penetrance and 10% phenocopy rate were performed.
Results
Single variant two-point analyses yielded three significantly linked SNPs at 11p14.1 and 11p11.2; a further 45 SNPs at 11p were found to be suggestive. No other chromosome had any significant SNPs or more than seven suggestive linkages. Two of the significant SNPs were located in BBOX1-AS1 and one in the intergenic region between ORA47 and TRIM49B. Collapsed haplotype pattern two-point analysis and multipoint analyses also yielded multiple suggestively linked genes at 11p. Multipoint analysis also identified suggestive evidence of linkage on 20q13.
Conclusions
We identified three genome-wide significant linked variants on 11p for myopia in Caucasians. Although the novel specific signals still need to be replicated, 11p is a promising region that has been identified by other linkage studies with a number of potentially interesting candidate genes. We hope that the identification of these regions on 11p as potential causal regions for myopia will lead to more focus on these regions and maybe possible replication of our specific linkage peaks in other studies. We further plan targeted sequencing on 11p for our most highly linked families to more clearly understand the source of the linkage in this region.
Keywords: myopia, linkage analysis, family studies, case-control study
Myopia, or nearsightedness, is a condition caused when light is focused in front of the retina rather directly on it, causing distant images to appear blurry. It affects approximately 25% of Americans. Prevalence rates in the United States were significantly higher in the early 2000s compared with the early 1970s (41.6% to 25.0%); this trend holds true in Caucasian individuals at the rate of 43.0% to 26.3%.1
Myopia is a complex trait and is influenced by both genetic and environmental factors.2,3 Environmental studies have identified both positive (near work level) and negative (outdoor activity) correlations with myopia.2 Multiple genetic studies have identified risk variants for myopia including population-based genome-wide association studies (GWAS).3–8 Almost all of the variants identified through GWAS are common (minor allele frequency > 0.05) and have a small effect size.
Family-based linkage studies are more effective than population-based studies at identifying rare variants with large effect sizes because family-based studies need fewer individuals to detect rare variants with sufficient power. Rare variants may be common within a given family but rare within the population as a whole. Unlike population-based association studies, family-based linkage studies are not subject to population stratification due to differing allele frequencies in different ethnic groups, because each family is analyzed as its own separate unit and not all in a group as in an association study. Each family receives its own independent logarithm of the odds (LOD) score. Being the logarithm of a ratio, LOD scores are cumulative and can be added across families (even in different populations) to get an overall LOD score. Heterogeneity LOD (HLOD) scores can also be calculated across families; HLOD scores allow for heterogeneity across families. Family-based linkage studies are susceptible to incorrect allele frequency estimates due to ethnic admixture in a set of families. Therefore, the allele frequencies used in linkage analyses should be calculated from data separated into major ethnic groups (e.g., the European-derived Caucasians in this study). Then families should be analyzed using their appropriate marker allele frequencies and LOD scores can then be summed across ethnically different groups of families. Studies have shown this maintains proper type I error rates.9–11
Another advantage of family-based linkage studies is their ability to take advantage of large linked haplotypes within family units. In population-based association studies, participants are distantly related and many meioses through the ages have broken up haplotypes. Thus, only variants that are very close together will exhibit linkage disequilibrium (LD). However, in families, the founders of each family represent a small subset of the existing haplotypes in the population at large, and these haplotypes become the founding haplotypes for every descendent in the family. Because most families in a linkage study are small (2–4 generations), only a very small number of recombinations will occur between variants that are moderately far apart on the same chromosome. The end result is a longer linked haplotype across a given genetic region. This can provide higher power to detect the existence of a causal variant along the haplotype (even when the causal variant is not genotyped). The downside is that a significant linkage region may contain a large number of candidate genes and potentially damaging variants. In general, linkage studies have good power to detect causal variants which are highly penetrant (i.e. have a large effect on disease risk). Association studies are most powerful to detect common causal genetic variants, which tend to have small effects on risk of most diseases. For a more extensive reading concerning family-based linkage studies, see Ott et al.12 and Elston.13
The family study design has been successful for linkage studies of pathogenic myopia (mean spherical equivalent [MSE] < −6.00 diopters [D]).14,15 Further studies analyzing nonpathogenic myopia (MSE < −1.00 D) in highly aggregated families identified genomic regions likely to be harboring high-risk genetic variants.16–20 Although several chromosomal regions have been identified in families with strong linkage evidence for causal variants with a large effect on myopia risk, the specific causal genes and specific risk variants have not been identified.
One potential way to find causal variants is to focus on coding regions, currently made possible using inexpensive exome-based arrays. Here, we use dense exome single nucleotide polymorphism (SNP) and microsatellite genotype data from highly aggregated Caucasian families to attempt to find linked genes with variants that increase myopia risk.
Methods
Data Collection and Phenotype Classification
Caucasian individuals were recruited from 56 families living in Pennsylvania and New Jersey. Phenotype data were collected on 412 individuals, and genotype data were collected on 273 of these individuals. The average age was 38.58 years, with an SD of 19.67; 56% of the subjects were female. Families ranged from two to four generations. Families were identified through mailings, eye clinic interviews, and referrals from private optometrists and ophthalmologists. Eligibility was dependent on the following criteria: (1) at least three participating family members; (2) only one myopic parent, and (3) at least two myopic siblings. Children had to be at least 5 years of age to participate. Medical records were obtained for each consenting member and/or refractions were done in the absence of adequate records. Data were collected on all eligible and consenting relatives of each proband. All study participants provided informed consent. Protocols were carried out in accordance to the Declaration of Helsinki. The study was approved by the institutional review boards of the National Human Genome Research Institute and the University of Pennsylvania.
Participants underwent comprehensive eye examinations that included medical/ocular health history, visual acuity, slit-lamp biomicroscopy, dilated fundus examinations, and manifest refraction. For subjects under 41 years of age, cycloplegic refraction using 0.5% cyclopentolate or 1% tropicamide was performed; older subjects used manifest refraction. If participants were unable to be examined at a clinic, refraction data was obtained from medical records or eyeglass prescriptions. Refraction was measured using MSE in diopters, obtained by adding the spherical component of the refraction to one-half the cylindrical component and averaging for both eyes.
Individuals with an MSE of −1.00 D or lower in both eyes (based on an actual eye examination, medical records, or eyeglass prescriptions, if the person was not available for examination) were coded as affected. Individuals with an MSE of 0.00 D or greater were coded as unaffected, and any individuals with an MSE between 0.00 and −1.00 D were coded as having an unknown or missing phenotype. Any individual that had a history of systemic or ocular disease that might cause a predisposition to myopia was coded as unknown regardless of their MSE measurements.
Because of the normal developmental changes in refractive error during childhood and concomitant potential problems of misclassification, we took a more stringent approach to classification of affected versus unaffected subjects for the groups of individuals aged 5 to 21 years. Individuals with a −1.00 D or lower spherical equivalent were considered affected, as above. Individuals were considered unaffected if the MSE refraction was > +3.00 D (5–9), > +2.00 D (10–17), and > +0.50 (18–21) in both eyes because they are not likely to develop myopia. Individuals that fell between their given upper limit and −1.00 D were designated as “unknown” due to the anticipated refractive errors changes toward less hyperopia in children. This conservative approach balances the power loss that results from lack of a good segregation analysis model of age-dependent penetrance and the concomitant confusion about appropriate genotype probabilities for young unaffected subjects, with the power loss resulting from the classification of normal children as “unknown.” Classification resulted in 261 affected individuals, 68 unaffected individuals, and 83 unknown individuals. The average MSE was 2.88 D, with an SD of 3.40.
Exome Genotyping and Quality Control
The 273 individuals with DNA samples were genotyped at the Center for Inherited Disease Research at Johns Hopkins University using an Illumina ExomePlus array. Blind duplicates and HapMap controls were distributed across plates for concordance checking. Affected and unaffected individuals were evenly distributed across plates, but family members were kept on the same plate. Samples with suspected mixtures of other sample DNA or unusual X and Y patterns (e.g., XXY individuals or people with major deletions; none identified) or sex mismatch were identified and dropped before release. SNP clustering was performed on all SNPs in project, and SNP genotypes with genotype quality (GC) score less than 0.15 were recoded as missing genotypes. Autosomal SNPs with less than 85% call rate, cluster separation of less than 0.3, and heterozygote rate greater than 80% were dropped prior to laboratory release. We later further filtered markers using a 95% call rate, resulting in a mean call rate of 99%.
We merged the phenotype and family relationship information with the genotype data and included an additional 118 ungenotyped individuals in the data set for use in the linkage analysis. The ungenotyped individuals included some individuals who provided phenotype information but did not provide samples or failed genotyping. However, the majority of these ungenotyped individuals consisted of individuals who were known to exist (based on family history) but who did not participate in the study because they chose not to or were deceased. These individuals (who were coded as having an unknown phenotype and unknown genotypes) were added to connect disjointed pedigrees and ensure proper familial relationships. Mendelian error checking was performed on the pedigrees using the sib-pair,21 and any SNP with more than one Mendelian error was dropped. PLINK22 was used to detect sex discrepancies; samples that did not appear sufficiently matched to their recorded sex were dropped. PREST-PLUS23 was used to identify duplicate samples. SNPs with more than one error in blind duplicates or HapMap controls were dropped. Heterozygosity rates across samples were checked, and outlier samples were excluded. We also examined the samples for chromosomal abnormalities and autosomal SNPs with a sex difference in allelic frequency >0.2 or sex difference in heterozygosity >0.3 were dropped. Monomorphic variants were dropped. We merged this exome data with 367 microsatellite markers from the same genotyped individuals to increase power in the multipoint analyses; microsatellites are highly informative. All markers were mapped to the Rutgers Genetic Map version 3.24 The final set of markers for analysis contained 67,145 SNPs and 367 microsatellites. Hardy-Weinberg equilibrium was tested for all linked variants.
Allele Frequency Calculations
Prior studies9–11 have shown that separation of family data into major ethnic groups (e.g., European ancestry Caucasians) and estimation of the frequencies from the family data set properly controls the type I error rate. All the individuals in this study were self-reported European-derived Caucasian Americans, so allele frequencies were calculated for the entire data set using sib-pair.21
Parametric Linkage Analyses
Three types of parametric linkage analysis were performed: single variant two-point analysis, collapsed haplotype pattern variant two-point analysis, and multipoint analysis. All analyses assumed an autosomal dominant model with a disease allele frequency of 0.01. Penetrance was 90% for carriers and 10% for noncarriers.
Single variant two-point analyses were performed using an implementation of the Elston-Stewart algorithm by the TwoPointLods software created by Alun Thomas (http://www-genepi.med.utah.edu/~alun/software/). Each family was analyzed individually, and afterward, cumulative LOD scores and HLOD scores were calculated across all families.
Multipoint analyses were performed using SimWalk2.25–27 Previous studies have shown that multipoint linkage using a dense marker map with strong intermarker LD often lead to type I error inflation, so we pruned the data by condensing SNPs into 1-cM bins. The SNP with the highest minor allele frequency chosen to represent the bins in the subsequent multipoint analyses (because such SNPs will provide the highest information content about segregation of this region of the chromosome in the families). Further LD analysis was performed in Haploview.28 Any SNP pairs with an r2 value greater than 0.2 had one SNP of the pair removed. Microsatellites were not included in the pruning; they were retained due to their high information level and general low intermarker LD with nearby SNPs.
Collapsed haplotype pattern (CHP) variants were created using the SEQLinkage software.29 This program uses rare variants to create short regional haplotypes that serve as multiallelic pseudo-markers corresponding to specific genetic regions such as genes (determined by RefSEQ). This approach has been shown to not require pruning of markers based on intermarker linkage disequilibrium29 (required for multipoint linkage). Because SEQLinkage is known to properly control type I error rate when using rare variants, we limited this analysis to SNPs with a MAF ≤ 0.05. The SEQLinkage method can use different informative rare variants in different families to build the family-specific haplotype pseudo-markers, thus allowing retention of information that would otherwise be lost in the pruning process when all rare variants are discarded prior to multipoint linkage. MERLIN30 was used to perform two-point linkage on the pseudo-markers. Variants were annotated with ANNOVAR,31,32 and protein prediction was performed by PolyPhen-2,33 SIFT,34–39 and ClinVar.40
Results
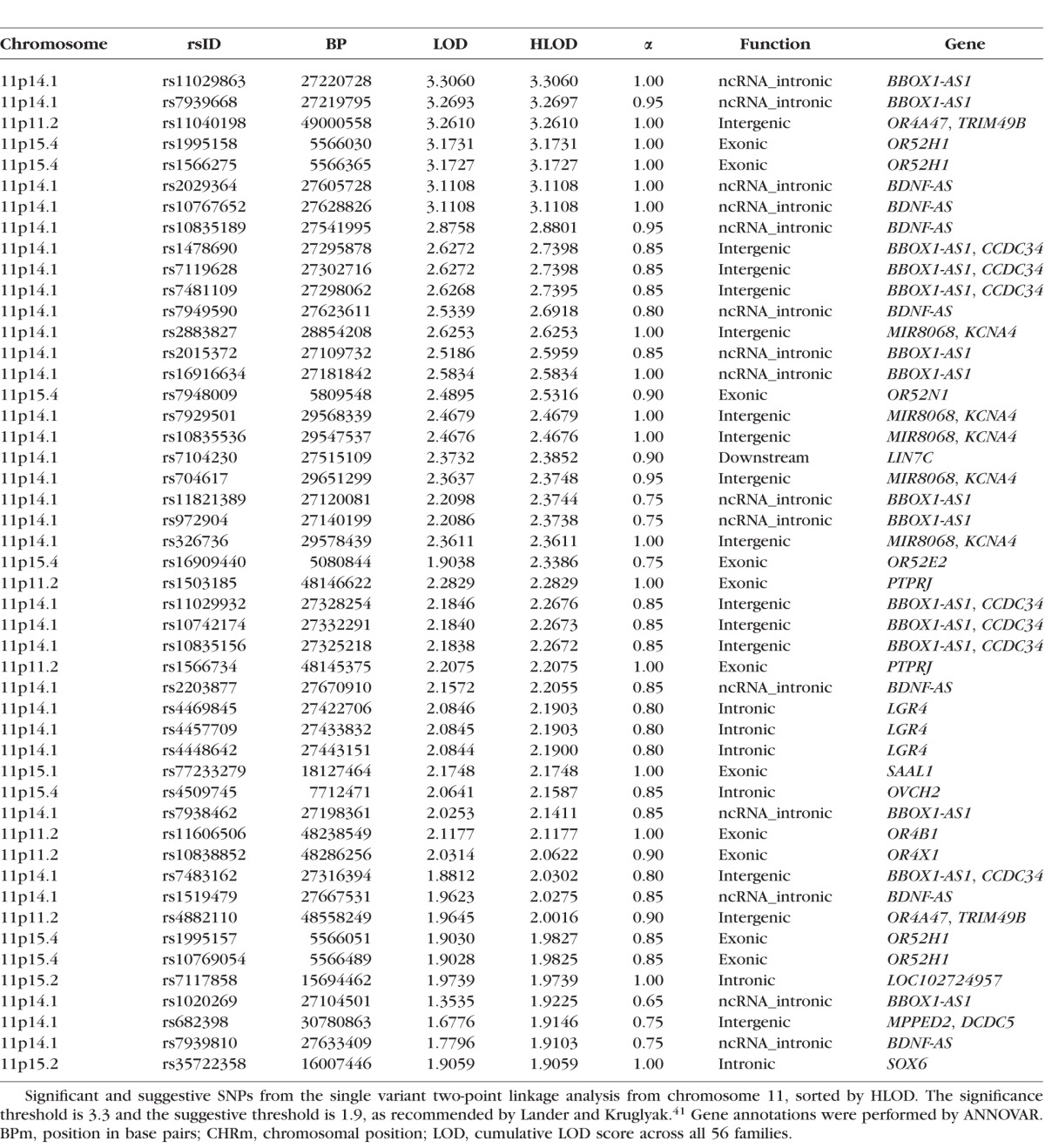
The single variant two-point analysis localized a strong linkage signal to 11p (Fig. 1). Three variants exhibited genome-wide significant HLODs, and 45 variants had suggestive HLODs in the regions around 11p15.4, 11p15.1, 11p14.1, and 11p11.2 (Fig. 2). No suggestive variants were observed on 11q. HLOD values ≥3.3, and HLODs ≥1.9 are considered genome-wide significant and suggestive as recommended by Lander and Kruglyak.41 Two significant variants (rs11029863 and rs7939668) were identified at 11p14.1 (HLOD = 3.31, 3.27) and were located in the noncoding antisense RNA BBOX1-AS1 gene. Both SNPs are common, with sample data MAFs of 0.20 (rs11029863) and 0.40 (rs7939668). Neither of the sample MAFs deviated greatly from the MAF for CEU in 1000Genomes. The final significant SNP was rs11040198 (HLOD = 3.26), located at 11p11.2 in an intergenic region between OR4A47 and TRIM49B. This SNP had a MAF of 0.1506 in the data set with no significant deviation from the 1000Genomes population frequency. Note we have considered all three of these variants to be significant because their LOD scores round to 3.3; however, only one SNP has a LOD that actually exceeds 3.3. All three of these “significant” SNPs are very close together in the 11p region, giving strong evidence of a myopia susceptibility gene in this region.
Figure 1.

Graph of the genome-wide single variant two-point HLOD scores produced by TwoPointLods. The lines at 1.9 and 3.3 represent the respective suggestive and significant thresholds recommended by Lander and Kruglyak.41
Figure 2.

Graph of the chromosome 11 single variant two-point HLOD scores produced by TwoPointLods. The lines at 1.9 and 3.3 represent the respective suggestive and significant thresholds recommended by Lander and Kruglyak.41
Other smaller and less dense suggestive signals were found throughout the genome. The top 13 HLOD scores were all located on 11p, as were 45 of the 68 overall suggestive variants. The list of suggestive SNPs on chromosome 11 can be found in Table 1, whereas the full list of all other suggestive signals (excluding chromosome 11) can be found in Supplementary Table S1.
Table 1.
Significant and Suggestive SNPs From Single Variant Two-Point Analysis on Chromosome 11
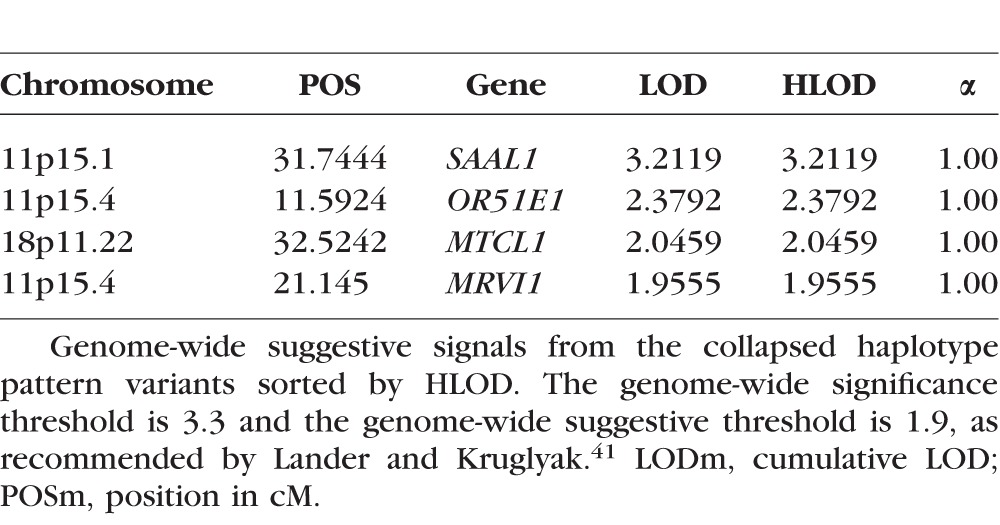
CHP variant linkage analysis did not reveal any genome-wide significant regions (Fig. 3). It did reveal four suggestive peaks, with three on 11p (Table 2). The two highest overall HLODs were centered on SAAL1 at 11p15.1 (HLOD = 3.21) and ORE15E1 at 11p15.4 (HLOD = 2.38). SAAL1 also had a suggestive signal in the two-point analysis (rs77233279). The SNP had a MAF of 0.04 and was nonsynonymous exonic, although not predicted as damaging. There was a further suggestively linked CHP variant centered in the MRVI1 gene at 11p15.4 (HLOD = 1.96).
Figure 3.

Graph of the genome-wide collapsed haplotype pattern variant HLOD scores produced by SEQLinkage and MERLIN. The lines at 1.9 and 3.3 represent the respective suggestive and significant thresholds recommended by Lander and Kruglyak.41
Table 2.
Suggestive Genome-Wide Collapsed Haplotype Pattern Variants
There were no genome-wide significant regions identified in the multipoint analysis; 16 suggestive variants were found (Fig. 4; Supplementary Table S2). The top 14 variants located on 20q13.13–20q13.31. This result is near a previous suggestive finding for myopia in Ashkenazi Jewish families at 20p12-q11.1.42 Two further suggestive variants (HLOD = 2.07) were found at 11p15.1.
Figure 4.

Graph of the genome-wide multipoint HLOD scores produced by SimWalk2. The lines at 1.9 and 3.3 represent the respective suggestive and significant thresholds recommended by Lander and Kruglyak.41
Discussion
This study has identified a cluster of significant and suggestive linkage signals along 11p15-11p11 for myopia in Caucasian families. Suggestive signals were spread throughout the region from both the single-variant and CHP two-point linkage analyses. The only significant signals were identified by the single-variant two-point analysis at 11p14.1 and 11p11.2, but the CHP analyses showed strong evidence that rare variants in 11p15.1-11p15.4 are segregating with myopia in these families. Both the two-point and CHP analyses agree that there is a strong linkage signal on 11p. The significant SNPs are novel and have not been previously reported significant in any previous association or linkage study, so replication to these specific regions are needed, and any interpretations of these specific regions should be viewed with some caution. However, the broader 11p region has a strong history of linkage with myopia and refractive error, including the Myopia-7 (MYP7) region at 11p13-11p15.1,43 11p15.1 in a subset of these Caucasian families,19 and 11p14-q14 in Ashkenazim.42
The SNPs that are individually significantly linked are common variants and possibly suggest that different rare variants within the same gene may be causal in different families or that the causal variants are located in noncoding areas in 11p15-11p11. It is possible that one of these common SNPs could be causal for a common, heterogeneous phenotype like myopia or, more likely, that these SNPs may be tagging (within each family) the segregating haplotype that may contain any rare, potentially causal, variants (probably not on this limited exome-based chip). Targeted sequencing could help to elucidate the source of the linkage signal.
Both the two-point and CHP analyses found significant or highly suggestive linkage signals on 11p. The two-point analyses did have a higher magnitude and number of signals than the CHP analyses, due mainly to the fact that the CHP analyses required SNPs with a MAF ≤0.05. Nevertheless, both analyses showed that the signals on 11p were much higher than elsewhere in the genome (Figs. 1, 3).
The multipoint analysis identified a suggestive signal on 11p and a suggestive signal of higher magnitude on 20q (Fig. 4). This discrepancy was likely caused by pruning; only SNPs with the highest MAF (i.e., most informative) were retained, and large amounts of information were lost, resulting in the decreased signal on 11p. The increased evidence for linkage on 20q in the multipoint analysis suggests that the common markers were jointly tagging segregating haplotypes in these families. It is possible that these haplotypes may harbor potentially causal variants, but replication will be needed to rule out the possibility that the linkage on 20q is a false positive.
Given that multiple studies, including this one, have reported significant evidence of linkage to the broader 11p region, we briefly examine a few potential candidate genes implicated in our study. We note that we do not infer causality of any of these genes or variants; we are simply reporting some potential candidates for future work and replication.
Two of the significant SNPs and 13 suggestive SNPs from the two-point analysis were located at 11p14.1 in or near the noncoding RNA BBOX1-AS1 gene, which has no known associations to myopia. Another noncoding RNA gene at 11p14.1, BDNF-AS, contained seven suggestively linked SNPs. The target gene, BDNF (at 11p14.1) has well-known protective and healing properties in the retina.44–46
A significantly linked SNP at 11p11.2 was located in the intergenic region between the olfactory receptor OR4A47 and TRIM49B. The protein tyrosine phosphatase receptor PTPRJ (11p11.2) contained two suggestive linkages at exonic SNPs (HLODs = 2.28, 2.21). One suggestive SNP322580 (rs1566734) was nonsynonymous and predicted damaging by ClinVar and PolyPhen2. The mitochondrial protein tyrosine phosphatase PTPMT1 (11p11.2) was slightly below the suggestive threshold (HLOD = 1.63) in the CHP analysis. Other protein tyrosine phosphatases such as PTPRR and PPFIA2 (PTPRF) have been associated with myopia and average dioptric sphere measurements.47
11p15 was the site of two highly suggestive linkage signals, both located in the exons of the olfactory receptor OR52H1 at 11p15.4. MRVI1 (11p15.4) was suggestive for linkage in the CHP analyses; one study found it to be overexpressed in keratoconic corneas.48
SAAL1 (11p15.1) was suggestive for linkage in both the single-variant and CHP analyses. Its function is not well known, although it has been reported to accelerate synoviocyte proliferation in joints.49 Multipoint analysis also found a suggestive linkage signal to 11p15.1. Both findings recapitulate a previous linkage signal found on 11p15.1 in a subset of these Caucasian families.19
We did not see any suggestive evidence of linkage at 11p13, the site of the maximum LOD score of 6.1 for the MYP7 locus from the original study of Hammond et al.43 of dizygotic twins that identified this locus. However, in this study, they actually showed genome-wide significant linkage to the entire 11p13-11p15.1 region and simply suggested PAX6 as a candidate gene. Later studies have both supported50,51 and disputed52,53 the causality of PAX6. Simpson et al.52 suggests that no priority be given to PAX6 over other genes in the region. The significant region of the study of Hammond et al.43 overlaps the location of the genome-wide significant and suggestive linkages on 11p found in this study, and thus our study replicates their linkage. Because it is known that linkage peaks are broad and that the causal genes for a given peak in a particular study may not be located at the position with the highest LOD score, it is possible that the same gene could be responsible for all published linkages in 11p. However, this is speculative, and much additional work is still needed to confirm the existence and identity of such a causal locus in 11p.
We presented genome-wide significant evidence for linkage of a susceptibility gene for myopia on 11p in Caucasian families. Novel genome-wide significant two-point linkage signals were identified at 11p14.1 and at 11p11.2. Highly suggestive signals were identified at 11p15.4 and 11p15.1. Further suggestive signals were observed in all these regions. This replicates evidence of linkage to this region from several previous studies.19,42,43,52 The 11p region contains a large number of good potential candidate genes for future study. Targeted sequencing of the 11p region is planned to search for any possible causal variants.
Supplementary Material
Acknowledgments
The authors thank all study participants and their families.
Supported in part by National Eye Institute Grant R01 EY020483 (DS and EBC) and the Intramural Program of the National Human Genome Research Institute, National Institutes of Health (AMM, CLS, BAM, KAL, LP, FM, JEBW).
Disclosure: A.M. Musolf, None; C.L. Simpson, None; B.A. Moiz, None; K. A. Long, None; L. Portas, None; F. Murgia, None; E.B. Ciner, None; D. Stambolian, None; J.E. Bailey-Wilson, None
References
- 1. Vitale S,, Sperduto RD,, Ferris FL., III. Increased prevalence of myopia in the United States between 1971-1972 and 1999-2004. Arch Ophthalmol. 2009; 127: 1632–1639. [DOI] [PubMed] [Google Scholar]
- 2. Verhoeven VJ,, Hysi PG,, Wojciechowski R,, et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet. 2013; 45: 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kiefer AK,, Tung JY,, Do CB,, et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet. 2013; 9: e1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simpson CL,, Wojciechowski R,, Oexle K,, et al. Genome-wide meta-analysis of myopia and hyperopia provides evidence for replication of 11 loci. PLoS One. 2014; 9: e107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klein AP,, Duggal P,, Lee KE,, et al. Linkage analysis of quantitative refraction and refractive errors in the Beaver Dam Eye Study. Invest Ophthalmol Vis Sci. 2011; 52: 5220–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simpson CL,, Wojciechowski R,, Yee SS,, Soni P,, Bailey-Wilson JE,, Stambolian D. Regional replication of association with refractive error on 15q14 and 15q25 in the Age-Related Eye Disease Study cohort. Molec Vis. 2013; 19: 2173–2186. [PMC free article] [PubMed] [Google Scholar]
- 7. Stambolian D,, Wojciechowski R,, Oexle K,, et al. Meta-analysis of genome-wide association studies in five cohorts reveals common variants in RBFOX1, a regulator of tissue-specific splicing, associated with refractive error. Hum Molec Genet. 2013; 22: 2754–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verhoeven VJ,, Hysi PG,, Saw SM,, et al. Large scale international replication and meta-analysis study confirms association of the 15q14 locus with myopia. The CREAM consortium. Hum Genet. 2012; 131: 1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandal DM,, Sorant AJ,, Atwood LD,, Wilson AF,, Bailey-Wilson JE. Allele frequency misspecification: effect on power and Type I error of model-dependent linkage analysis of quantitative traits under random ascertainment. BMC Genet. 2006; 7: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mandal DM,, Wilson AF,, Bailey-Wilson JE. Effects of misspecification of allele frequencies on the power of Haseman-Elston sib-pair linkage method for quantitative traits. Am J Med Genet. 2001; 103: 308–313. [PubMed] [Google Scholar]
- 11. Mandal DM,, Wilson AF,, Elston RC,, Weissbecker K,, Keats BJ,, Bailey-Wilson JE. Effects of misspecification of allele frequencies on the type I error rate of model-free linkage analysis. Hum Heredity. 2000; 50: 126–132. [DOI] [PubMed] [Google Scholar]
- 12. Ott J,, Wang J,, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet. 2015; 16: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elston RC. Linkage and association. Genet Epidemiol. 1998; 15: 565–576. [DOI] [PubMed] [Google Scholar]
- 14. Guo H,, Tong P,, Liu Y,, et al. Mutations of P4HA2 encoding prolyl 4-hydroxylase 2 are associated with nonsyndromic high myopia. Genet Med. 2015; 17: 300–306. [DOI] [PubMed] [Google Scholar]
- 15. Zhou L,, Li T,, Song X,, Li Y,, Li H,, Dan H. NYX mutations in four families with high myopia with or without CSNB1. Molec Vis. 2015; 21: 213–223. [PMC free article] [PubMed] [Google Scholar]
- 16. Ibay G,, Doan B,, Reider L,, et al. Candidate high myopia loci on chromosomes 18p and 12q do not play a major role in susceptibility to common myopia. BMC Med. Genet. 2004; 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stambolian D,, Ibay G,, Reider L,, et al. Genomewide linkage scan for myopia susceptibility loci among Ashkenazi Jewish families shows evidence of linkage on chromosome 22q12. Am J Hum Genet. 2004; 75: 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wojciechowski R,, Moy C,, Ciner E,, et al. Genomewide scan in Ashkenazi Jewish families demonstrates evidence of linkage of ocular refraction to a QTL on chromosome 1p36. Hum Genet. 2006; 119: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciner E,, Ibay G,, Wojciechowski R,, et al. Genome-wide scan of African-American and white families for linkage to myopia. Am J Ophthalmol. 2009; 147: 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrew T,, Maniatis N,, Carbonaro F,, et al. Identification and replication of three novel myopia common susceptibility gene loci on chromosome 3q26 using linkage and linkage disequilibrium mapping. PLoS Genet. 2008; 4: e1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duffy D. SIB-PAIR: A program for simple genetic analysis v1.00.beta. Queensland Institute of Medical Research. Brisbane, Australia. Available at: https://genepi.qimr.edu.au/staff/davidD/#sib-pair. [Google Scholar]
- 22. Purcell S,, Neale B,, Todd-Brown K,, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun L. Detecting pedigree relationship errors. In: Elston RC, Satagopan JM, Sun S, eds. Statistical Human Genetics: Methods and Protocols. New York: Humana Press; 2012: 25–46. [DOI] [PubMed] [Google Scholar]
- 24. Matise TC,, Chen F,, Chen W,, et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007; 17: 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sobel E,, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet. 1996; 58: 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 26. Sobel E,, Papp JC,, Lange K. Detection and integration of genotyping errors in statistical genetics. Am J Hum Genet. 2002; 70: 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sobel E,, Sengul H,, Weeks DE. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Heredity. 2001; 52: 121–131. [DOI] [PubMed] [Google Scholar]
- 28. Barrett JC,, Fry B,, Maller J,, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005; 21: 263–265. [DOI] [PubMed] [Google Scholar]
- 29. Wang GT,, Zhang D,, Li B,, Dai H,, Leal SM. Collapsed haplotype pattern method for linkage analysis of next-generation sequence data. Eur J Hum Genet. 2015; 23: 1739–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abecasis GR,, Cherny SS,, Cookson WO,, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002; 30: 97–101. [DOI] [PubMed] [Google Scholar]
- 31. Chang X,, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012; 49: 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang K,, Li M,, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adzhubei I,, Jordan DM,, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protocols Hum Genet. 2013: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J,, Ng PC. SIFT. Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS One. 2013; 8: e77940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar P,, Henikoff S,, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protocols. 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 36. Ng PC,, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sim NL,, Kumar P,, Hu J,, Henikoff S,, Schneider G,, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012; 40: W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vaser R,, Adusumalli S,, Leng SN,, Sikic M,, Ng PC. SIFT missense predictions for genomes. Nat Protocols. 2016; 11: 1–9. [DOI] [PubMed] [Google Scholar]
- 39. Zhao Y,, Zhao F,, Zong L,, et al. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss. PLoS One. 2013; 8: e69549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Landrum MJ,, Lee JM,, Benson M,, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44: D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lander E,, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995; 11: 241–247. [DOI] [PubMed] [Google Scholar]
- 42. Simpson CL,, Wojciechowski R,, Ibay G,, Stambolian D,, Bailey-Wilson JE. Dissecting the genetic heterogeneity of myopia susceptibility in an Ashkenazi Jewish population using ordered subset analysis. Molec. Vis. 2011; 17: 1641–1651. [PMC free article] [PubMed] [Google Scholar]
- 43. Hammond CJ,, Andrew T,, Mak YT,, Spector TD. A susceptibility locus for myopia in the normal population is linked to the PAX6 gene region on chromosome 11: a genomewide scan of dizygotic twins. Am J Hum Genet. 2004; 75: 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Afarid M,, Torabi-Nami M,, Zare B. Neuroprotective and restorative effects of the brain-derived neurotrophic factor in retinal diseases. J Neurol Sci. 2016; 363: 43–50. [DOI] [PubMed] [Google Scholar]
- 45. Binley KE,, Ng WS,, Barde YA,, Song B,, Morgan JE. Brain-derived neurotrophic factor prevents dendritic retraction of adult mouse retinal ganglion cells. Eur J Neurosci. 2016; 44: 2028–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cerri E,, Origlia N,, Falsini B,, et al. Conjunctivally applied BDNF protects photoreceptors from light-induced damage. Trans Vis Sci Tech. 2015; 4 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hawthorne F,, Feng S,, Metlapally R,, et al. Association mapping of the high-grade myopia MYP3 locus reveals novel candidates UHRF1BP1L, PTPRR, and PPFIA2. Invest Ophthalmol Vis Sci. 2013; 54: 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee JE,, Oum BS,, Choi HY,, Lee SU,, Lee JS. Evaluation of differentially expressed genes identified in keratoconus. Molec Vis. 2009; 15: 2480–2487. [PMC free article] [PubMed] [Google Scholar]
- 49. Sato T,, Fujii R,, Konomi K,, et al. Overexpression of SPACIA1/SAAL1, a newly identified gene that is involved in synoviocyte proliferation, accelerates the progression of synovitis in mice and humans. Arthrit Rheum. 2011; 63: 3833–3842. [DOI] [PubMed] [Google Scholar]
- 50. Jiang B,, Yap MK,, Leung KH,, et al. PAX6 haplotypes are associated with high myopia in Han chinese. PLoS One. 2011; 6: e19587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miyake M,, Yamashiro K,, Nakanishi H,, et al. Association of paired box 6 with high myopia in Japanese. Molec Vis. 2012; 18: 2726–2735. [PMC free article] [PubMed] [Google Scholar]
- 52. Simpson CL,, Hysi P,, Bhattacharya SS,, et al. The roles of PAX6 and SOX2 in myopia: lessons from the 1958 British Birth Cohort. Invest Ophthalmol Vis Sci. 2007; 48: 4421–4425. [DOI] [PubMed] [Google Scholar]
- 53. Dai L,, Li Y,, Du CY,, et al. Ten SNPs of PAX6, Lumican, and MYOC genes are not associated with high myopia in Han Chinese. Ophthalmic Genet. 2012; 33: 171–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.