

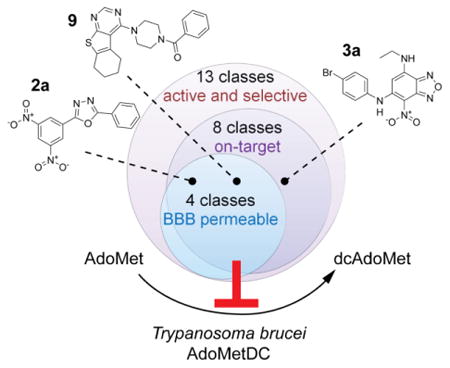
Abstract
Human African trypanosomiasis (HAT) is a fatal infectious disease caused by the eukaryotic pathogen Trypanosoma brucei. Available treatments are difficult to administer and have significant safety issues. S-Adenosylmethionine decarboxylase (AdoMetDC) is an essential enzyme in the parasite polyamine biosynthetic pathway. Previous attempts to develop TbAdoMetDC inhibitors into anti-HAT therapies failed due to poor brain exposure. Here, we describe a large screening campaign of two small-molecule libraries (∼400,000 compounds) employing a new high-throughput (∼7 s per sample) mass spectrometry-based assay for AdoMetDC activity. As a result of primary screening, followed by hit confirmation and validation, we identified 13 new classes of reversible TbAdoMetDC inhibitors with low-micromolar potency (IC50) against both TbAdoMetDC and T. brucei parasite cells. The majority of these compounds were >10-fold selective against the human enzyme. Importantly, compounds from four classes demonstrated high propensity to cross the blood-brain barrier in a cell monolayer assay. Biochemical analysis demonstrated that compounds from eight classes inhibited intracellular TbAdoMetDC in the parasite, though evidence for a secondary off-target component was also present. The discovery of several new TbAdoMetDC inhibitor chemotypes provides new hits for lead optimization programs aimed to deliver a novel treatment for HAT.
Keywords: Human African trypanosomiasis, Trypanosoma brucei, AdoMetDC, high-throughput screening, mass spectrometry
Graphical abstract
Introduction
Human African trypanosomiasis (HAT) is a debilitating disease, usually fatal without treatment. It is caused by two geographically separated subspecies of the protozoan parasite Trypanosoma brucei (T.b. gambiense and T.b. rhodesiense)1-3. T. brucei is transmitted by the Glossina fly and proliferates extracellularly in blood and lymph, eliciting nonspecific influenza-like symptoms in the early stage of the disease. The advanced stage, which occurs months to years after infection, ensues from the parasite crossing the blood-brain barrier (BBB) and is characterized by severe neurological symptoms4. The World Health Organization (WHO) estimated that there were fewer than 3000 cases of T.b. gambiense in 20155, highlighting the success of current disease control strategies6. However, if historic patterns are any indication, recent gains may yet reverse in response to environmental or socioeconomic factors7. The recent discovery of asymptomatic human T.b. gambiense infections complicates efforts to fully eliminate the disease8, 9. Moreover, the zoonotic nature of T.b. rhodesiense makes its elimination even more challenging6. Finally, animal infections with related non-human parasites such as T.b. brucei and T. vivax lead to difficulty growing livestock in endemic areas contributing to malnutrition and adverse socioeconomic conditions, necessitating development of new veterinary trypanocides10.
Effective chemotherapies are key to both treatment and elimination efforts, but none of the current drugs can be used to treat all of the clinical manifestations as treatment options are both species- and stage-dependent11. Treatment of late-stage disease is particularly problematic. Nifurtimox-eflornithine combination therapy (NECT) is the frontline treatment for late-stage T.b. gambiense infection, and while effective and relatively safe, its use is hindered by the need for IV dosing of large quantities of eflornithine to overcome rapid elimination. For T.b. rhodesiense, the efficacy of eflornithine has been questioned leaving the highly toxic organoarsenic compound melarsoprol as the only existing option11. Two new entities with broad-spectrum activity are currently in clinical development12, 13 but in the absence of an additional approved therapy there remains a clear need for new HAT treatments to support both clinical and elimination efforts.
Eflornithine is a time-dependent inhibitor of the polyamine biosynthetic enzyme ornithine decarboxylase (ODC), validating the pathway (Figure 1) as a target for treatment of HAT11, 14. The success of eflornithine suggests that other enzymes in the pathway may also be useful drug targets, and may provide a better opportunity to develop drug-like reversible inhibitors than ODC, which has a small highly charged substrate binding pocket. S-Adenosylmethionine decarboxylase (AdoMetDC) catalyzes formation of decarboxylated AdoMet, which serves as the aminopropyl donor for formation of spermidine, an essential metabolite in all eukaryotes15, 16. In T. brucei, genetic knockdown showed that AdoMetDC is required for parasite survival17. Additionally, the time-dependent AdoMetDC inhibitor MDL 73811 and a related analog Genz-644131 (1) were shown to have efficacy against early stage disease in rodent models18, 19. These studies validated AdoMetDC as an essential and druggable target. However, MDL 73811 was not useful against late stage infection due to poor CNS exposure, and efforts to improve brain penetration of the series were unsuccessful19-23. Thus, if AdoMetDC is to be advanced as a drug target for the treatment of HAT, a new chemotype needs to be identified with the physiochemical properties that support CNS activity.
Figure 1. The polyamine biosynthetic pathway in kinetoplastids.
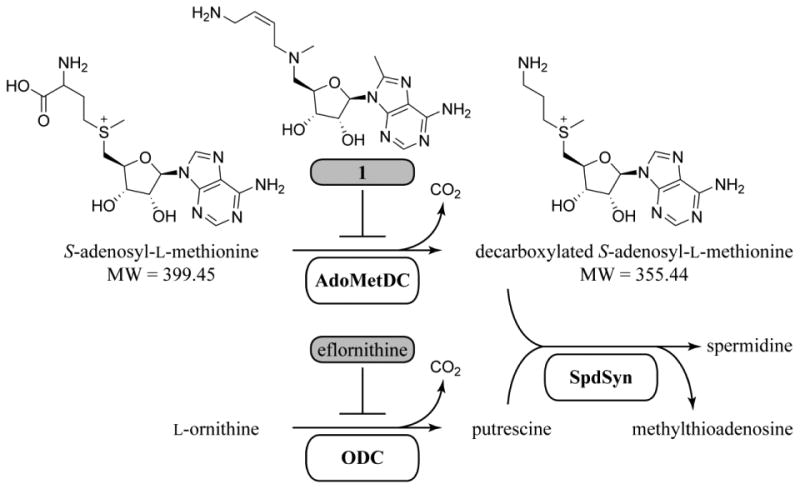
S-Adenosylmethionine decarboxylase (AdoMetDC) and ornithine decarboxylase (ODC) are inactivated by their respective mechanism-based covalent inhibitors, Genz-644131 (1) and eflornithine. Compound 1 was used as a positive control in this study. Spermidine synthase (SpdSyn) catalyzes a transfer of the aminopropyl group from dcAdoMet to putrescine with an essential triamine, spermidine, as its product.
Trypanosomatid AdoMetDC is a pyruvoyl-dependent enzyme that underwent a unique gene duplication leading to the requirement for heterodimerization with the cognate paralogous pseudoenzyme, termed prozyme, for full activity17, 24, 25. Heterodimerization of T. brucei AdoMetDC with prozyme leads to greater than 1000-fold activation, resulting in catalytic efficiency similar to that of the homodimeric mammalian enzymes. This activation results from a large conformational change that leads to displacement of an autoinhibitory peptide and stabilization of the active conformation by insertion of the N-terminus of AdoMetDC into the TbAdoMetDC/prozyme dimer interface26. This unique regulatory mechanism, as well as amino acid residue differences in the active site between human and T. brucei AdoMetDC suggests that species-selective trypanosomatid AdoMetDC inhibitors can be identified, strengthening the value of the target.
Here, we describe a novel end-point assay based on the RapidFire-mass spectrometer system (Agilent Technologies, Santa Clara, CA) that allowed for rapid and reproducible quantification of the AdoMetDC activity in a high-throughput screen (HTS) format. Using this assay, we screened two large small-molecule libraries and identified a number of new chemical scaffolds that inhibit T. brucei AdoMetDC with low-micromolar affinity. These hits were further validated in several chemical and biological activity assays to identify scaffolds that warrant hit-to-lead development. Most of the identified inhibitors showed good species selectivity, and were significantly less active against human AdoMetDC.
Results
Development of a RapidFire-Mass Spectrometry-based high-throughput AdoMetDC activity assay
Published assays for AdoMetDC activity relied on capture and detection of released radioactive carbon dioxide (CO2)27, 28 and were not suitable for HTS. We previously described an alternative decarboxylase assay that enzymatically couples CO2 production to NADH oxidation, and this assay was optimized for HTS and used for the identification of T. brucei ODC inhibitors29. However, in preliminary work we found this assay was unsuitable for detection of AdoMetDC activity in HTS format due to low signal-to-background ratio. Thus, a new HTS-compatible assay for AdoMetDC was required. To that end, we developed a direct end-point assay to quantitatively detect AdoMetDC enzymatic activity based on mass spectrometry (MS). The assay utilizes the RapidFire instrument (Agilent Technologies), a robotic liquid-handling system with in-line solid-phase extraction (SPE) for rapid mobile phase exchange, interfaced with a triple quadrupole mass spectrometer for quantitative detection of the substrate (AdoMet) and the product (dcAdoMet) of the enzymatic reaction. The RapidFire technology has been used successfully for high-throughput primary screening of enzyme targets otherwise not amenable to rapid testing30, including phosphatidylserine decarboxylase31.
In the RapidFire-MS assay, AdoMet and dcAdoMet were detected using 399.1 > 250.1 and 355.1 > 250.1 transitions, respectively. The areas under the resulting MS peaks represent relative quantities of the substrate and product. The linearity of integrated MS signal response to varying AdoMet and dcAdoMet concentrations was tested by RapidFire-MS analysis of serial dilutions of the analytes in the quenched reaction solution (Figure 2A, B). The substrate signal (S) was linear over AdoMet concentrations from 1 to 250 μM (Figure 2A, AdoMet–E). Since dcAdoMet was not readily available as a pure synthetic control, we generated dcAdoMet enzymatically and then analyzed the reaction mixture for the presence of dcAdoMet (Figure 2B, AdoMet+E) to assess the linearity. dcAdoMet concentration was determined at each point of the serial dilution curve (Figure 2B) using the Eq. 1, which assumes the concentration was equal to the amount of AdoMet consumed during the enzymatic reaction:
Figure 2. RapidFire-MS AdoMetDC assay development.
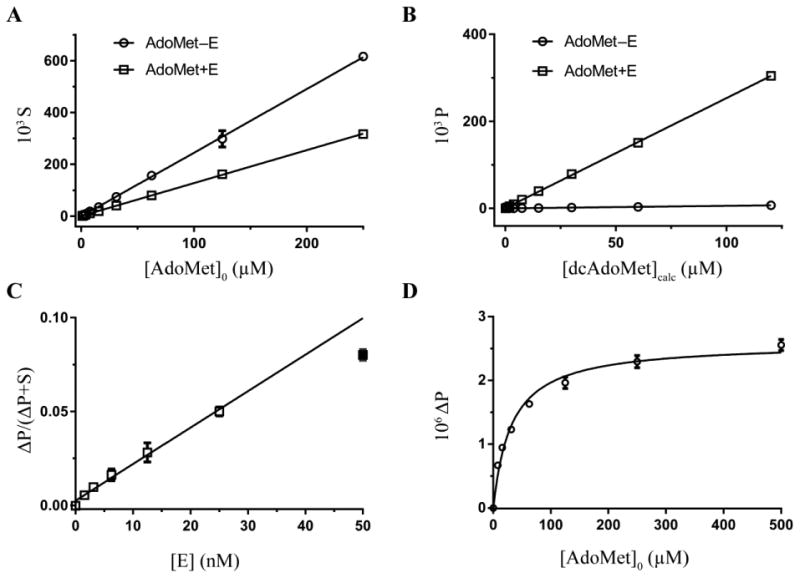
(A, B) Integrated MS signal intensities S (A) and P (B) versus ligand concentration for substrate AdoMet (399.1 > 250.1) and product dcAdoMet (355.1 > 250.1) MRM transitions, respectively. Points represent serial dilutions of substrate (AdoMet–E, circles) and a mixture of the substrate and product (AdoMet+E, squares) in a quenched enzymatic reaction, where E represents enzyme AdoMetDC. Data were fitted by linear regression analysis in Prism (GraphPad) (R2 > 0.99). (C) Fractional converted substrate, ΔP/(ΔP+S), as a function of the TbAdoMetDC/prozyme concentration, [E]. Data (accept for [E] = 50 nM, filled square) were fitted by linear regression analysis in Prism (R2 > 0.99). (D) Substrate saturation data plotted as the background-subtracted product signal, ΔP, and fitted to Michaelis-Menten model in Prism; KM = 32 ± 2 μM (mean ± standard deviation). All data were collected in triplicate, with standard deviations shown as error bars.
| (1) |
where a and b are, respectively, a slope and y-intercept of linear fit to AdoMet–E (1) and AdoMet+E (2) data (Figure 2A). This approach allowed us to determine that the product signal (P) was linear over dcAdoMet concentrations from 0.1 to 120 μM. The magnitude of the response was nearly identical for AdoMet and dcAdoMet as seen from the slopes (both 2.5) of the linear fit of the integrated MS data (Figure 2A, AdoMet–E, and Figure 2B, AdoMet+E). Thus, the P/(P+S) ratio equals fractional conversion in the end-point reaction. The integrated dcAdoMet signal P from the samples of pure AdoMet (Figure 2B, AdoMet–E) was sufficiently low to suggest there was no cross-talk between the S and P transitions, at least not in S-to-P direction.
To optimize the assay, both assay time and TbAdoMetDC/prozyme heterodimer concentration were varied to identify conditions in the linear range. The end-point assay incubation time was then fixed at 20 min. The integrated signal response to enzyme concentration was linear up to 25 nM TbAdoMetDC/prozyme but dropped off at 50 nM, thus 25 nM enzyme was chosen as the concentration for the screen (Figure 2C). The % conversion of substrate at these conditions was 5% (Figure 2C), thus, steady-state assumptions are applicable. The Michaelis-Menten constant, KM, for AdoMet was experimentally determined using the RapidFire-MS assay to be 32 μM (Figure 2D), which is lower than previously reported values (140–170 μM) obtained using the 14C-based assay24, 25. The discrepancy was likely due to differences in assay composition, although this issue was not explored in detail.
RapidFire-MS Assay Validation
Assay performance was tested for reproducibility and consistency using standard industry guidelines32. Reagent stability and effects of freezing and thawing on assay performance were evaluated. No substantial vehicle effect on the rate of the reaction in the RapidFire-MS assay was observed at up to 3.75% dimethyl sulfoxide (DMSO) (mean activity and standard deviation over 16 replicates normalized to 0% DMSO control was 93 ± 8%). Three-day plate uniformity studies were performed in interleaved-signal format as described32 using two concentrations (150 nM or 10 μM) of the known AdoMetDC inhibitor 1 (Figure 1) and 0.1% DMSO to define MID, MIN and MAX signal. Tests were evaluated using Data Analysis Templates32, and all intra- and inter-plate results met acceptance criteria (not shown).
Primary screening for TbAdoMetDC small-molecule inhibitors
Two small-molecule libraries (Genzyme and UT Southwestern) together totaling 414,124 compounds were screened at a compound concentration of 10 μM using the validated RapidFire-MS assay (Figures 3 and 4). Assays were performed in 384-well polypropylene microplates (Corning, Lowell, MA), and the P/(P+S) ratio representing fractional conversion in the end-point assay was used as a measure of progression of the enzymatic reaction. The fractional conversion was then transformed to percent inhibition by normalizing to the plate means of the vehicle and compound 1-inhibited controls (0 and 100% inhibition, respectively) as described in Methods. Genzyme library compounds with percent inhibition above 30% were considered hits (Figure 4A). For the UT Southwestern library, controls were not used for normalization but only to monitor assay performance (see below). Instead, a compound was considered a hit when the Z score expressed in standard deviations (see Methods), was ≤ –6.2 (Figure 4B). As a result, 103 and 114 compounds were identified as hits from Genzyme and UT Southwestern libraries, respectively.
Figure 3. Flowchart of high-throughput screening campaign to identify and validate TbAdoMetDC inhibitors.
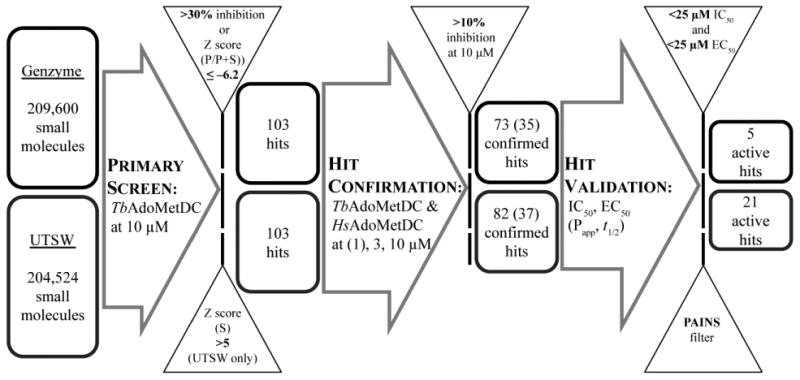
Compounds that demonstrated greater than 30% inhibition (Genzyme library) or a Z score of P/(P+S) ≤–6.2 (UT Southwestern (UTSW) library) were considered hits after the primary screening. A select set of confirmed compounds (35 for Genzyme library and 37 for UTSW library) were procured and their biological activities were tested in concentration-response studies to generate IC50 (TbAdoMetDC enzyme half-maximal inhibitory concentrations) and EC50 (in vitro parasite cells half-maximal effective growth-inhibitory concentration) values. Compounds with activity on both TbAdoMetDC (IC50 < 25 μM) and T.b. brucei cells (EC50 < 25 μM) were progressed. In vitro metabolism and distribution data (intestinal/CNS permeability, Papp, and microsomal stability, t1/2) were also generated during hit validation.
Figure 4. Primary screening results.
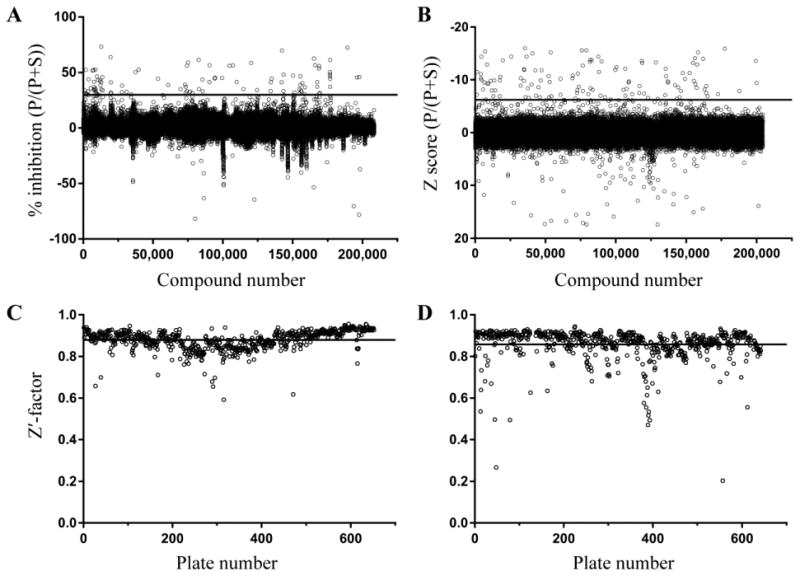
(A) Percent inhibition values of Genzyme library compounds ordered according to their position in the library. The threshold of 30% inhibition, above which a compound was considered a hit, is presented as a line. (B) Z scores of normalized P/(P+S) values for UT Southwestern library compounds; the threshold of –6.2 is shown as a line. (C, D) Z'‐factors calculated for each plate for (C) Genzyme and (D) UT Southwestern libraries. The library mean values (shown as a line) and their standard deviations were 0.88 ± 0.05 (C) and 0.86 ± 0.08 (D).
As part of the quality control of the experimental results, the average sample Z'-factor value33 was calculated for each plate based on vehicle and compound 1-inhibited controls (Figure 4C, D). Plates with Z'-factor values less than 0.45 were repeated. A concentration-response for 1 was included in each plate in column 24 for additional quality control (8 concentrations in duplicate for 1/3 serial dilutions starting from 10 μM (Genzyme library) or 3 μM (UT Southwestern library)). The average apparent IC50 (±standard deviation) for 1 across all library plates was 166 ± 63 nM for the Genzyme library (no pre-incubation) and 11 ± 3 nM for the UT Southwestern library (30 min pre-incubation). The difference in values is a result of the fact that 1 is a time-dependent inhibitor, and thus the apparent IC50 is also time dependent and is thus lower for the longer incubation time (0 vs. 30 min pre-incubation).
False-positive hits
Within both libraries, several compounds were present that had the same transitions as those used for AdoMet detection (399.1 > 250.1). In these cases, the MS signal of the compound added to the signal of AdoMet causing the compound to be falsely identified as an inhibitor if only the P/(P+S) ratio was used to evaluate reaction rate (Figure S1). Also, some carry-over of this effect was observed in up to six of the next samples (Figure S1). To eliminate this type of false-positive hits, Z scores of normalized S values were also evaluated along with P/(P+S) values. Compounds with a Z score for S of greater than 5 were considered false-positive and were excluded from further analysis. Out of 114 UT Southwestern library hit compounds, 11 were excluded, reducing the number of hits progressed to confirmation to 103. Genzyme library hits totaling 103 were not analyzed for false positives because the issue was not appreciated at the time of the screen; instead, three hit compounds (structures not shown) with molecular weight of 399 ± 1 were removed from consideration during hit validation after two failed to inhibit in the alternative 14C-based enzyme assay (described below).
Carry-over of the false-positive effect across multiple wells (Figure S1) is indicative of physical carry-over of a compound across multiple samples. A portion of a compound is likely retained on the SPE cartridge following RapidFire loading, washing, elution, and re-equilibration steps and thus is present in the cartridge material when the following sample is loaded. However, we did not notice any impact of this carry-over on the assay results besides generating false hits through the aforementioned mechanism.
Hit confirmation
The primary screen hits (103 from each library) were re-tested in a limited concentration-response format (3 and 10 μM for the Genzyme Library and 1, 3, and 10 μM for the UT Southwestern library) on both TbAdoMetDC/prozyme heterodimer (Figure S2A,B) and, to evaluate the species selectivity, on human AdoMetDC (HsAdoMetDC) (Figure S2C,D). Compounds with a mean (Genzyme library) or a median (UT Southwestern library) inhibition of TbAdoMetDC greater than 10% at 10 μM were considered confirmed hits. 73 compounds in the Genzyme library and 82 compounds in the UT Southwestern library met this criterion (Figures 3 and S2 A,B). Two of the confirmed compounds were represented in both libraries, thus 153 unique confirmed hits were identified. Notably, 61 confirmed hits showed ≥50% inhibition of the T. brucei enzyme at 10 μM, and for 42 of these hits inhibition of the human enzyme at 10 μM was ≤10% (Figure S2 C,D), suggesting that species selectivity was achievable.
The presence of the false-positive compounds negatively affected the correlation between the primary screen and confirmation screen inhibition data for the Genzyme library compounds: Pearson correlation coefficients (with 5% confidence interval) for Genzyme and UT Southwestern were 0.39 (0.18–0.57) and 0.87 (0.81–0.92), respectively (Figure S2 E,F). Even though the number of AdoMet mimics among Genzyme library hits was comparatively small (two compounds with anticipated 399.1 > 250.1 transitions), those compounds appeared to affect the values from the following wells due to substantial carry-over.
Identification of Pan-Assay Interference compounds
Libraries of compounds often contain promiscuous inhibitors that re-appear as hits in the screening campaigns of unrelated proteins34, 35. Using a Pipeline Pilot protocol based on filters described in35, we identified 45 compounds out of 153 confirmed hits with substructural motifs characteristic of these pan-assay interference compounds (PAINS), including toxoflavin, one of the better-known PAINS. Compounds identified as PAINS were not immediately removed from consideration and some were purchased for analysis of their biological activity and metabolic properties during hit validation as described below (Table S1).
Hit validation
Confirmed compounds were grouped into classes based on substructure searches. Classes of compounds with preliminarily evidence of either structure-activity relationship (SAR), or tractable chemistry, or both, were prioritized for purchasing from commercial sources. A total of 72 compounds (35 from Genzyme and 37 from UT Southwestern library) were procured and tested (Figure 3). Identity and purity of the purchased compounds was verified by LC/MS analysis (Table S2). In total, 13 classes of compounds, including 7 classes comprising more than one hit in our screens, were confirmed in the in vitro assays to be inhibitors of both TbAdoMetDC/prozyme activity and parasite growth (Table 1 and Figure 5). This total is excluding compounds identified by the PAINS filter. Individual hit validation assays are described below.
Table 1. In vitro biological data for verified compounds.
| Activity | Selectivity | Alternative assay (14C) | On-target mechanism of action | |||||
|---|---|---|---|---|---|---|---|---|
| T. brucei cell EC50, μMa | T. brucei AdoMetDC IC50, μMb | Human AdoMetDC IC50, μMc | T. brucei AdoMetDC IC50, μMd | Prozyme inductione | Spd/Put ratiof | T. brucei cell EC50, μMg | ||
| – Spd | + 1 mM Spd | |||||||
| 1 | 0.0018 (0.0015–0.0022) | 0.23 (0.18–0.30) | 0.11 (0.09–0.13) | NDh | yes | 0.21 | 0.00030 (0.00028–0.00033) | >0.25 |
| 2a | 0.27 (0.21–0.35) | 0.6 (0.4–0.9) | >50 | ND | yes | 1.0 | 0.52 (0.44–0.61) | 0.43 (0.36–0.52) |
| 2b | 0.34 (0.25–0.46) | 1.5 (1.2–1.8) | >50 | 1.4 (1.0–2.1) | yes | ND | ND | ND |
| 3a | 5.4 (4.5–6.4) | 0.4 (0.26–0.61) | 13 (7–25) | 0.7 (0.5–1.1) | yes | 0.09 | 8.6 (5.7–12.9) | >25 |
| 3b | 3.7 (3.1–4.7) | 40% at 1.7 μM | >50 | ND | yes | 0.20 | ND | ND |
| 3c | 7.6 (6.2–9.3) | 5.7 (3.9–8.3) | >50 | ND | ND | ND | ND | ND |
| 3d | 25 (17–37) | 33 (27–41) | >50 | ND | ND | ND | ND | ND |
| 4a | 1.0 (0.9–1.1) | 1.4 (1.2–1.7) | 29 (23–38) | 1.1 (0.6–1.9) | yes | 0.9 | 2.3 (0.76–6.7) | 2.4 (1.8–3.2) |
| 4b | 2.0 (1.6–2.6) | 11 (8.0–15) | 25% at 17 | ND | yes | ND | ND | ND |
| 4c | 3.2 (2.8–3.6) | >50 | >50 | ND | no | ND | ND | ND |
| 5a | 9.9 (8.5–12) | 8.6 (7.9–9.3) | >50 | 1.3 (0.6–3.3) | yes | 0.13 | 16 (12–20) | 37 (32–43) |
| 5b | 11 (9.8–12) | 9.2 (7.6–11) | >50 | ND | ND | ND | ND | ND |
| 6a | 16 (13–19) | 3.0 (2.7–3.4) | >50 | 4 (1–19) | yes | ND | 14 (12–17) | 14 (11–17) |
| 6b | 37% at 25 μM | 4.2 (3.7–4.9) | >50 | ND | ND | ND | ND | ND |
| 6c | 25 (22–29) | 5.2 (4.7–5.9) | >50 | ND | ND | ND | ND | ND |
| 6d | 33% at 25 | 8.0 (6.6–9.7) | >50 | ND | ND | ND | ND | ND |
| 7a | 1.5 (0.5–4.3) | 3.5 (2.8–4.3) | >50 | 6.8 (5.0–9.2) | ND | ND | ND | ND |
| 7b | 1.8 (1.6–2.0) | 5.0 (4.3–5.7) | >50 | ND | ND | ND | ND | ND |
| 7c | 3.2 (2.5–4.1) | 5.5 (4.5–6.9) | >50 | ND | ND | ND | ND | ND |
| 7d | 13 (11–15) | 12 (9–15) | >50 | ND | ND | ND | ND | ND |
| 8a | 6.2 (5.3–7.2) | 1.3 (1.1–1.7) | >50 | 41% at 6 | no | ND | ND | ND |
| 8b | 1.4 (1.1–1.9) | 12 (9.1–15) | >50 | 5.0 (3.4–7.3) | no | 0.9 | 2.6 (2.3–2.9) | 2.7 (2.4–3.0) |
| 9 | 9.5 (8.0–11) | 4.6 (3.1–6.8) | 47% at 50 | 3 (0.4–17) | yes | ND | 23 (16–32) | >25 |
| 10 | 9.8 (7.6–13) | 5.8 (4.7–7.1) | >50 | 19 (15–25) | yes | 0.6 | ND | ND |
| 11 | 11 (9.8–13) | 11 (9.0–13.3) | >50 | 3.1 (2.3–4.0) | no | 0.5 | 17 (14–20) | 24 (21–27) |
| 12 | 8.6 (7.7–9.6) | 26 (22–32) | >50 | 11 (7–16) | yes | 0.2 | 16 (13–21) | 15 (14–16) |
| 13 | 6.1 (5.2–7.2) | 46% at 17 | >50 | ND | no | 0.7 | 16 (14–19) | 22 (20–25) |
| 14 | 41% at 25 | 3 (2.2–4.2) | >50 | 2.3 (0.8–6.5) | ND | ND | ND | ND |
Half-maximal inhibitory concentration for T. brucei cells showing the mean for triplicate data with 95% confidence interval (CI) (in parentheses).
Half-maximal inhibitory concentration for T. brucei AdoMetDC/prozyme complex obtained without pre-incubation showing the mean for triplicate data with 95% CI (in parentheses).
Half-maximal inhibitory concentration for human AdoMetDC showing the mean for triplicate data with 95% CI (in parentheses).
Half-maximal inhibitory concentration for T. brucei AdoMetDC/prozyme complex showing the mean for unreplicated data with 95% CI (in parentheses).
Increased amount of prozyme in T. brucei cells grown in the presence of 1–2 × EC50 concentrations of compound as compared to the vehicle control as determined by western blotting.
Fractional spermidine/putrescine ratio (Spd/Put) in T. brucei cells grown in the presence of near-EC50 concentrations of a compound, with spermidine-to-putrescine ratio for a vehicle control set to 1. Values are either the mean of two technical replicates or the mean of two technical replicates of biological triplicates.
Half-maximal inhibitory concentration for T. brucei cells grown in chicken-serum containing medium without or with 1 mM Spd showing the mean for triplicate data with 95% CI (in parentheses).
ND, not determined.
Figure 5. Structures of validated hits.
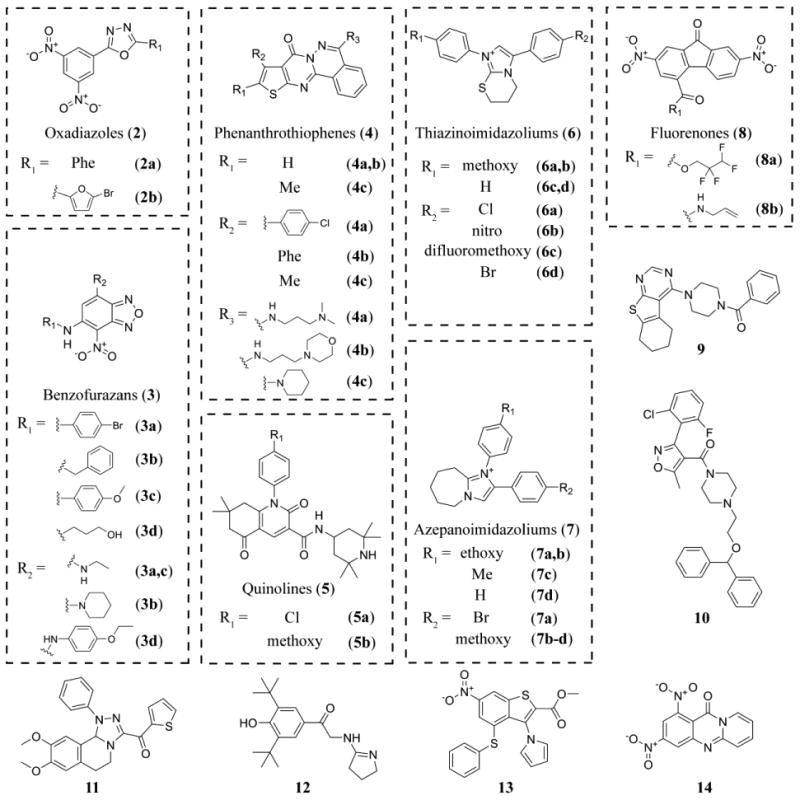
Compound 4c was not a primary screening hit (Z score = 0.5) and was used in the study as an inactive control.
Inhibition of AdoMetDC enzymatic activity
Compounds were assessed over multiple concentrations for their ability to inhibit TbAdoMetDC/prozyme or HsAdoMetDC using end-point assay on the RapidFire-MS instrument, either with or without pre-incubation of the enzyme with a compound. The IC50 values on TbAdoMetDC/prozyme ranged from 0.4 to 25 μM for the tested series. None of the non-PAINS hit compounds (Figure 5 and Table 1) exhibited a difference in IC50 when comparing assays done with or without pre-incubation (data not shown) consistent with reversible binding. There were, however, apparent time-dependent inhibitors among compounds that were identified by the PAINS filter including all tested phenylpiperazines (15a–e) and compound 18 (Table S1). The UT Southwestern library, as opposed to Genzyme library, was assessed after pre-incubating the enzyme with library compounds. Thus, it was expected that a higher fraction of time-dependent inhibitors would have been identified from the UT Southwestern library hits. However, this expectation was not supported by our data, as there were an equal number of classes of validated time-dependent inhibitors in the two libraries: 15a–e originated from UT Southwestern library and 18 – from Genzyme library (Table S1).
HsAdoMetDC inhibition was assessed without pre-incubation. The majority of the validated hits did not show any detectable inhibition up to 50 μM (Table 1). Out of four compounds that did demonstrate measurable inhibition of HsAdoMetDC, 3a, 4a, and 9 were still 30-, 20-, and 10-fold selective relative to TbAdoMetDC, respectively.
Finally, TbAdoMetDC/prozyme inhibition was assessed using an alternative assay based on the detection of the radioactive CO2 to rule out any assay artifacts (Table 1). With the exception of 8a, all tested compounds were confirmed to be inhibitors with similar activity by both methods. Compound 8a was >6-fold less potent in the 14C-based assay than in the RapidFire assay, suggesting a possible RapidFire assay artifact.
Inhibition of T. brucei cell growth in vitro
Confirmed hits were tested to determine if they inhibited T. brucei growth in suspended culture in a 48 h growth assay at multiple concentrations. Concentration-response curves were fitted to yield EC50 values that ranged from 0.3 to 25 μM for most of the tested hits (Table 1 and Figure S3). Overall, there was a good correlation (Pearson correlation coefficient = 0.5 for 21 compounds with numerical IC50 and EC50 values) between potency on T. brucei AdoMetDC/prozyme (IC50) and inhibition of cell growth (EC50). Exceptions included compounds that were more potent on the parasite than on the enzyme, suggestive of off-target inhibition (e.g. a non-hit analog 4c) and compounds with better potency on the enzyme than the parasite suggestive of potential uptake issues (e.g. 6a–d and 14). The control inhibitor 1 showed potent cell killing activity with an EC50 that was 100-fold below the measured IC50 on the enzyme. The difference is due to 1 being a mechanism-based inhibitor. The apparent IC50 measured without pre-incubation at a fixed time point does not reflect the full inhibitory potential of the compound at that concentration, i.e., given sufficient time, it would fully inactivate the enzyme. Indeed, 1 appears to be fully on-target in its trypanocidal mechanism based on the biological assays that are described below, and served as a control for mechanism-of-action studies on compounds identified in the screen.
Prozyme induction in treated cells
We previously reported that prozyme protein levels were upregulated in T. brucei cells when AdoMetDC activity was either reduced by genetic knockdown or chemically inhibited17, 36. We exploited this parasite-specific regulatory phenomenon as one of several markers to assess whether compounds were affecting parasite growth through AdoMetDC inhibition, i.e. through the on-target mechanism. We reasoned that if TbAdoMetDC activity in T. brucei cells was inhibited following the treatment with hit compounds, then minimally prozyme should be induced. Compound 1- and vehicle-treated samples were used as positive and neutral controls, respectively. Cells were treated with compounds at 1–2 × EC50 concentrations for 24 h, followed by cell lysis and Western blot analysis of prozyme levels (Figures 6A, S4 and Table 1).
Figure 6. Mechanism-of-action studies in T. brucei parasite cells.
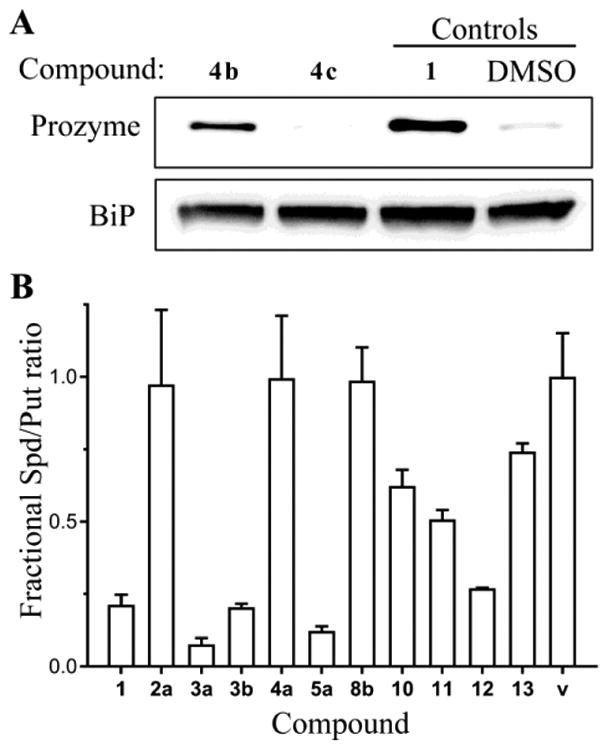
(A) Western blot analysis of prozyme levels in the T. brucei cells grown for 24 h in culture in the presence of EC50 concentrations of the active hit 4b and its inactive library analog 4c, which was not inhibitive in the TbAdoMetDC assay but suppressed parasite cell growth (see Table 1). Positive (1) and neutral (DMSO) controls were also included on the gel. A section of the blot was probed with anti-BiP antibodies as a loading control. The blot is a representative experiment of two biological replicates. (B) Levels of polyamines putrescine and spermidine as measured by HPLC in T. brucei cells grown for 72 h in culture in the presence of near-EC50 concentrations of hit compounds. The spermidine-to-putrescine ratio was normalized to the DMSO vehicle control (v). Data from cells treated with 1 was used as a positive control. The number of biological replicates (each done in technical duplicates) was five for controls (1 and v); three for compounds 2a, 3a, and 5a; and one for other compounds. Data are shown as the mean, and error bars represent the range.
Induction of prozyme protein levels equivalent to that observed for 1 was evident after incubation with all tested oxadiazoles (2a and 2b), benzofurazans (3a and 3b), phenanthrothiophenes (4a and 4b), representative quinolone 5a, thiazinoimidazolium 6a, and singletons 9, 10, and 12. In contrast, neither of the tested fluorenones (8a and 8b) nor singletons 11 and 13 showed prozyme upregulation suggesting an off-target mechanism of action for these compounds. The data on the phenanthrothiophenes 4a–c suggest a mixed picture with both some on- and off-target effects in evidence. Compounds 4a and 4b are both T. brucei AdoMetDC inhibitors that induced prozyme, while 4c is not a significant inhibitor of AdoMetDC, and consistent with the lack of inhibition does not induce prozyme. However, 4c inhibits parasite growth with an EC50 that is similar to 4a and 4b suggestive of an off-target mechanism of action (Figure 6A and Table 1).
Polyamines levels in treated cells
The metabolic response of T. brucei cells to AdoMetDC inhibition was previously shown to include increased putrescine and decreased spermidine levels17, 36. Thus, the spermidine-to-putrescine ratio normalized to a vehicle control in treated T. brucei cells was used to estimate the degree to which the polyamine levels in cells were affected by treatment with ∼EC50 concentrations of each of our TbAdoMetDC inhibitors (Figure 6B, Spd/Put ratio in Table 1). Normalized ratios of <0.25, based on compound 1 control, suggest that AdoMetDC activity was inhibited in cells due to treatment with a compound. Conversely, normalized ratios close to 1 are suggestive of no evident intracellular AdoMetDC inhibition and an off-target mechanism. Putrescine and spermidine levels were measured for ten hit compounds representing nine scaffolds. Four compounds (benzofurazans 3a and 3b, quinolone 5a, and singleton 12) demonstrated low ratios suggestive of AdoMetDC inhibition in cells (Figure 6B and Table 1). Three compounds (2a, 4a, and 8b) exhibited no evidence of perturbation in polyamine levels, and three singletons (10, 11, and 13) showed evidence for AdoMetDC inhibition, but the Spd/Put ratio was not as strongly lowered as for 1.
Rescue of cell growth inhibition with spermidine
It has been previously shown that cell growth inhibition caused by TbAdoMetDC RNAi knockdown can be fully rescued by supplying 100 μM spermidine in the growth media17. Here, we demonstrate that cell growth inhibition caused by over 100× EC50 concentrations of compound 1 can also be rescued in concentration-dependent manner by spermidine supplementation; 1 mM spermidine yields a full rescue (Figure 7A). Thus, spermidine rescue of compound-induced cell growth phenotype was used as a third marker for demonstrating on-target mechanism of action. To this end, we collected EC50 data for ten classes of compounds (one representative per class) with or without 1 mM spermidine (Table 1). In the majority of cases, spermidine did not have an appreciable effect on EC50 values, suggesting that these inhibitors are predominantly affecting cell growth by acting on targets other than TbAdoMetDC. Notably, spermidine did confer limited resistance to 5a (2.3-fold shift in EC50), and there was evidence of resistance to 3a and 9 at tested concentrations (Figure 7B-D). However, due to solubility limits, we were unable to titrate the two latter compounds to high enough concentrations for the results to be conclusive.
Figure 7. Rescue of compound-mediated parasite cell growth inhibition with a downstream metabolite.
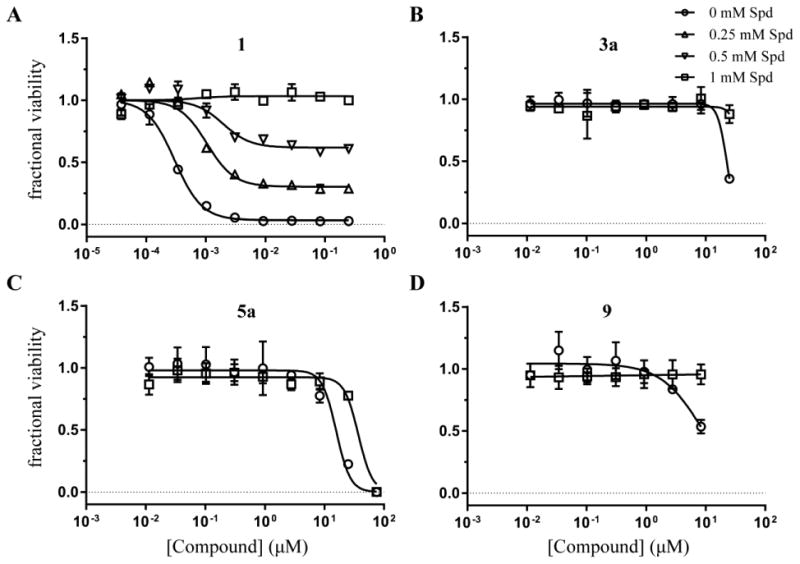
(A) T. brucei cell viability concentration-response curves for compound 1 grown in the presence of 0 (circles), 0.25 (triangles), 0.5 (inverse triangles), and 1 mM (squares) spermidine (Spd). (B-D) T. brucei cell viability concentration-response curves for hit compounds 3a (B), 5a (C), and 9 (D) in the presence of 0 and 1 mM Spd. Data are shown as fractional viability based on CellTiter-Glo assay results after 48 h growth in the medium supplemented with chicken serum. Error bars represent standard deviation of the mean of three replicates.
In vitro metabolic stability
The in vitro metabolic stability data were collected using mouse and human liver microsomes for representative inhibitors from 10 classes of confirmed hits and reported as half-life (t1/2) values in Table 2. Compounds 3a, 4a, 5a, and 12 demonstrated high microsomal stability (t1/2 > 90 min) in at least one of the two tested species. Compounds 11, 13, and 14 were not tested due to limiting material.
Table 2. In vitro ADME data for select verified compounds.
| 1 | 2a | 2b | 3a | 4a | 5a | 6a | 7a | 8b | 9 | 10 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Microsomal stability t1/2, min | ||||||||||||
|
| ||||||||||||
| human | >90 | 31 | 11 | >90 | >90 | 40 | 35 | 11 | <2.5 | <2.5 | <2.5 | >90 |
|
| ||||||||||||
| mouse | >90 | 31 | 3 | 22 | 79 | >90 | 4 | 7 | <2.5 | <2.5 | <2.5 | >90 |
|
| ||||||||||||
| Permeability Papp, nm/s | ||||||||||||
|
| ||||||||||||
| – GF918a | 11 | 415 | 540 | 89 | 46 | 13 | 6 | 2 | 270 | 494 | 288 | 95 |
|
| ||||||||||||
| + GF918b | 11 | 341 | 357 | 70 | 33 | 21 | 7 | 32 | 265 | 433 | 310 | 328 |
The apparent permeability values in the absence (a) or presence (b) of Pgp inhibitor GF120918
Estimation of CNS permeability
The in vitro MDCKII-hMDR1 monolayer assay37 was used to estimate how predisposed a hit compound was to cross the BBB. The apparent permeability values in the absence of P-glycoprotein 1 (Pgp) inhibitor GF120918 (Papp (– GF918)) of greater than 150 nm/s are indicative of good prospects for CNS delivery38. Out of eleven hit compounds tested representing ten classes of novel inhibitors, five compounds (2a, 2b, 8b, 9, and 10) demonstrated Papp values consistent with high predicted BBB penetration (Table 2). Compound 12 demonstrated high Papp values only in the presence of GF120918 (Papp (+ GF918)) and is likely a substrate to Pgp-mediated efflux.
Evidence of cytotoxicity
UT Southwestern library historical data were available from 26 phenotypical assays that have been run using CellTiter-Glo as a measure of cell viability were used to evaluate toxicity of the hit compounds in human cells (data not shown). The compound concentrations were generally in the range of 2.5–5 μM in these assays with a single replicate per compound. Only a small subgroup of compounds (4a and 7a–c) significantly inhibited cell growth (>30% inhibition with Z score greater than 5) in six or more out of 26 independent primary screens. The rest of compounds with data available (2a, 3a–c, 5a–b, 6a–d, 7d, 9, 13, and 14) significantly inhibited cell growth only in three or fewer out of 23–26 primary screens.
We also considered the activity of a subset of compounds that were tested in a non-small cell lung cancer (NSCLC) confirmation study (data available via the Cancer Target Discovery and Development Network data portal, https://ocg.cancer.gov/programs/ctd2/data-portal). In this study, compounds were dosed at 2.5 μM across 12 NSCLC cell lines and an immortalized human bronchial epithelial cell line (HBEC30KT) with 3 replicates per compound. In this context, 4a also demonstrated 92% inhibition of HBEC30KT, whereas 3a, 4b, 7b–c, and 13 did not show substantial inhibition (2–21% inhibition). Taken together, the data suggest there is no indication of pervasive cytotoxicity for a majority of confirmed hit compounds, 4a being an exception.
Discussion and Conclusions
TbAdoMetDC and polyamine synthesis are well-established and validated molecular targets for drug discovery and development for the treatment of HAT. However, previously discovered TbAdoMetDC inhibitors, such as CGP 40215A and MDL 73811, were never developed into therapies at least in part because they were hindered by poor efficacy in CNS models of HAT in mice20, 22. To search for new chemotypes of TbAdoMetDC inhibitors, we developed a new end-point AdoMetDC activity assay that relied on direct, mass spectrometric quantitative detection of the substrate and product. Integrated robotic sample-handling and rapid in-line analyte cleanup lends itself to high-throughput applications, not feasible with previously available radioactive isotope-based methods. The assay reported here was robust, yielding on average Z'-factors >0.8. This enabled us to screen inhibitory potencies of more than 400,000 small molecules in two libraries. The number of compounds with inhibition above the selected thresholds for the two libraries constituted 0.05% of all compounds screened, which is below the oft-cited range of 0.1–2% for a well-behaved assay on a druggable target39. The low hit rate suggests that the libraries contained limited chemical space that was a good match for the TbAdoMetDC active site.
The identified, confirmed, and validated hits represent new chemical scaffolds previously unknown to have TbAdoMetDC inhibitory properties. The hits exhibited tractable chemotypes and good biological activities (half-maximal inhibitory concentrations on TbAdoMetDC and T. brucei cells in the low- to mid-μM range of 0.4–25 μM). Importantly, a number of scaffolds demonstrated high propensity to cross the BBB in the monolayer assay (permeability Papp > 150 nm/s), suggesting CNS exposure is achievable for oxadiazoles (2a and 2b) and fluorenones (8b) as well as singletons 9 and 10. Benzofurazan compound 3a showed intermediate Papp values ∼80 nm/s that perhaps can be increased through medicinal chemistry.
Notably, all of the validated hits (except 4b, for which the exact value could not be determined) showed at least 10-fold differences in inhibitory potencies between the T. brucei and human enzymes. This is an encouraging result as previously described reversible inhibitors of AdoMetDC showed the opposite trend, e.g. CGP 40215A was 500-fold more potent against the HsAdoMetDC40. Our study shows that selective TbAdoMetDC inhibition is achievable. Selectivity may be important to avoid potential toxicity arising from the host AdoMetDC inhibition.
As part of the hit validation, we sought to explore whether hit compounds inhibit the T. brucei parasite cell growth primarily through an on-target mechanism of action. Compounds were tested for inhibition of intracellular TbAdoMetDC/prozyme by two independent approaches: prozyme induction and measurement of polyamine levels. While for most of the tested compounds the two assays demonstrated similar results (i.e. either prozyme induction and normalized spermidine-to-putrescine ratio <0.25 or no prozyme induction and unperturbed spermidine-to-putrescine levels), compounds 2a and 4a induced prozyme upregulation but did not decrease the spermidine-to-putrescine ratio. The apparent discrepancy may be explained by the difference in the time scale between the two assays (24 h for prozyme induction vs. 72 h for polyamine levels). The prozyme induction causes a profound activation of TbAdoMetDC. Unperturbed polyamine levels after 72 h might indicate that prozyme activation leads to partial resistance to chemical inhibition. Alternatively, compound stability over the course of the 72 h assay may be a factor.
While the prozyme induction and polyamine assays do provide an indication of AdoMetDC inhibition, they do not conclusively demonstrate that this inhibition is what leads to growth arrest. A third approach, spermidine rescue, allowed direct testing for on-target cell growth inhibition. Notably, cells were partially resistant to compounds 3a, 5a, and 9 in the presence of spermidine suggesting that intracellular on-target TbAdoMetDC inhibition (demonstrated in the prozyme induction assay and, in the case of 3a and 5a, by the polyamine ratio analysis) is a considerable component of their mechanism of action. However, this assay suggested that the remainder of the tested compounds were at least partially off-target in their killing mechanisms because spermidine was unable to rescue the observed growth inhibition. This conclusion assumes that the compounds themselves did not block spermidine uptake. While not desirable, this result might be expected given the relatively modest, micromolar potencies of the hit compounds. The off-target component can likely be reduced in parallel with IC50 optimization through medicinal chemistry during hit-to-lead expansion.
Preliminary microsomal metabolic stability data were not used to prioritize any class of compounds as there is a widely recognized lack of correlation between in vitro t1/2 data and in vivo clearance data, even within one series of compounds41. Yet, these data may become informative later in hit-to-lead expansion when complemented by other ADME analyses.
Overall, our target-based high-throughput screening campaign yielded 13 new chemotypes with validated TbAdoMetDC/prozyme inhibition. Importantly, these classes possess an array of properties that make them attractive starting points for future lead development. These include: correlated low-micromolar potencies on both the enzyme and the parasite cells; selectivity compared with the host enzyme; and reversible mode of the enzyme inhibition. Additionally, eight of these classes inhibit AdoMetDC activity in parasite cells (i.e. are at least partially on-target based on one or more of the three assays), and four classes are predicted to cross the BBB. Oxadiazoles (2a–b) exhibit all of the aforementioned properties; however, substitution of the nitro groups should be considered when developing this series further due to toxicity and mutagenicity of nitroaromatics42-44.
Our recently reported crystal structure of TbAdoMetDC/prozyme heterodimer (PDB ID: 5TVM and 5TVF)26 will greatly facilitate hit-to-lead expansion. The molecular space around the identified scaffolds can be efficiently explored through in silico ligand docking. Additionally, the reported crystallization conditions can be applied to co-crystallize TbAdoMetDC/prozyme in complex with compounds to assist in hit optimization.
Materials and Methods
Materials
General reagents were purchased from Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography (HPLC)-grade water and acetonitrile, Gibco phosphate buffered saline (PBS), and Gibco Iscove's Modified Dulbecco's Medium (IMDM) were purchased from Thermo Fisher Scientific (Waltham, MA); ammonium formate (Alfa Aesar, 99% purity) from VWR International (Radnor, PA); S-adenosyl-L-methionine (AdoMet) sulfate p-toluenesulfonate salt from Affymetrix (Santa Clara, CA); S-adenosyl-L-[carboxy-14C]-methionine (14C-AdoMet) from American Radiolabeled Chemicals (St. Louis, MO); Fetal Bovine Serum (FBS) from Atlanta Biologicals (Flowery Branch, GA); Chicken Serum from Gemini Bio-Products (West Sacramento, CA).
TbAdoMetDC/prozyme protein expression and purification
Expression and purification of recombinant TbAdoMetDC/prozyme heterodimer complex and HsAdoMetDC were carried out as previously described25, 40.
Mass spectrometry-based high-throughput AdoMetDC assay
An end-point 384-well plate assay for AdoMetDC activity was developed using the BIOCIUS Life Sciences (presently, Agilent Technologies) RapidFire 200 (primary screen and hit confirmation) or RapidFire 300 (hit validation) high-throughput microfluidic system with integrated SPE interfaced with the Agilent 6430 triple quadrupole mass spectrometer, which allowed the conversion of substrate to product to be followed directly by their respective m/z ratios. Two compound libraries (Genzyme and UT Southwestern), each containing approximately 200,000 compounds, were screened at an initial concentration of 10 μM using this assay to identify TbAdoMetDC inhibitors.
Assays were performed in base buffer (100 mM Hepes, pH 7.7, 50 mM NaCl), which was used to make both enzyme solution (50 nM purified TbAdoMetDC/prozyme complex, 5 mM putrescine, 2 mM dithiothreitol (DTT), 0.1% bovine serum albumin and substrate solution (80 μM AdoMet, 5 mM putrescine, 0.02% Nonidet P40 substitute), both of which were prepared fresh. AdoMet sulfate p-toluenesulfonate (Affymetrix) stocks (20 mM) were prepared in 5 mM H2SO4, 10% glycerol and stored at –20 °C.
For primary screening of the Genzyme library, as well as for the confirmation and validation of both Genzyme and UT Southwestern library hits, DMSO stocks of library compounds and, if necessary, DMSO backfill were dispensed (0.04 μL constant total volume, 0.1% of the final assay volume, for Genzyme library or 0.8 μL constant total volume, 2% of the final assay volume, for the UT Southwestern library) into dry 384-well assay plates using the Echo 555 acoustic dispenser (Labcyte, Sunnyvale, CA). For the primary screening and confirmation of the Genzyme library, the substrate solution (20 μL) was added to pre-dispensed compounds first, and then the addition of the enzyme solution (20 μL) initiated the reaction. The same order of addition was used in the hit validation studies where pre-incubation was not indicated. When pre-incubation was indicated (the UT Southwestern library hit confirmation, and the hit validation assays where specified), the enzyme solution (20 μL) was added to the plates containing predispensed compounds first. After 30 min of pre-incubation, the reaction was initiated by the addition of the substrate solution (20 μL).
For the primary screening of the UT Southwestern library, the enzyme solution (20 μL) was added to the plates first, allowing accurate dispensing of compounds (0.8 μL) on the Biomek FX automated liquid handler (Beckman Coulter, Indianapolis, IN). Following the dispensing of the compounds, the enzyme was allowed to pre-incubate with compounds for 30 min before the reaction was initiated by addition of the substrate solution (20 μL).
In all cases, the enzymatic reaction was allowed to run for 20 min at room temperature and then was quenched with 1 M HCl (40 μL). Both the enzyme and substrate solutions and the quenching reagent were added to plates using either the Microflo liquid dispenser (BioTek, Winooski, VT) with a 5-μL cassette or the Multidrop 384 reagent dispenser (Thermo Fisher Scientific).
Plates were centrifuged at 1000 × g for 1 min after every addition and sealed using the PlateLoc thermal microplate sealer (Agilent Technologies) before being stored at –80 °C until analysis on the RapidFire.
AdoMetDC assay: RapidFire-MS sample analysis
Optimal RapidFire and MS settings for detection of S-adenosyl-L-methionine (AdoMet) and decarboxylated AdoMet (dcAdoMet) were developed by BIOCIUS Life Sciences under a pay for service contract. Quenched enzyme assay samples (prepared in 384-well plates as described above) were loaded on the RapidFire system by aspiration for 600 ms. The sample was then automatically loaded onto a graphitic carbon Type D SPE cartridge (Agilent Technologies), and buffer salts and protein matrix were removed from the sample by washing the cartridge with the load solution (water containing 0.1% trifluoroacetic acid (TFA)) at a flow rate of 1.5 mL/min for 2500 ms. The retained and purified analytes were eluted from the cartridge with the elution solution (acetonitrile:water (3:7, v/v) containing 0.1% TFA) at 1.25 mL/min for 3000 ms and directed to the mass spectrometer. The cartridge was re-equilibrated with load solution at 1.5 mL/min for 500 ms.
Both AdoMet and dcAdoMet were assessed using multiple selected reaction monitoring (MRM) transitions of 399.1 > 250.1 amu for AdoMet and 355.1 > 250.1 amu for dcAdoMet. Dwell time was 50 ms for each transition. The fragmentor voltage was set to 50 V, the collision energy to 5 V, the cell accelerator voltage to 4 V, and the delta EMV to 350 V. The mass spectrometer was operated with a gas temperature of 350 °C, gas flow rate of 11 L/min, nebulizer pressure of 40 psi, and a capillary voltage of 3000 V. The areas under the daughter ion peaks of AdoMet and dcAdoMet were quantified using RapidFire Integrator software (Agilent Technologies). The peak areas for AdoMet and dcAdoMet were used directly as relative quantities of the substrate (S) and product (P), respectively, from which the fractional conversion of substrate to product (P/(P+S)) was calculated. The P/(P+S) value was used in the data analysis to arrive at the degree of completion of the end-point enzymatic reaction. The S value was used separately to monitor for false-positive hits.
Primary screen data analysis
384‐well assay plates contained the test population of 320 library compounds at 10 μM final concentration located in columns 3 through 22. Compound 1 (10 μM final for the Genzyme screen or 0.2 μM for the UT Southwestern screen), was included on the assay plate (column 1) as a positive control, and DMSO was used as a neutral control (columns 2 and 23). The S, P, and P/(P+S) raw values for every well in the test population were normalized using Equation 2 (Genzyme library) or 3 (UT Southwestern library):
| (2) |
| (3) |
For the Genzyme library, the normalized value of P/(P+S), defined as the normalized percent inhibition value, was used to rank-order the compounds after the primary screen. For the UT Southwestern library, the compounds were rank-ordered based on Z score of the normalized P/(P+S) value calculated for each compound using GeneData Screener software v. 10.1 (GeneData, San Francisco, CA)45 according to:
| (4) |
where run is defined as combined test populations of the plates that were assessed in the end-point assay on the same day (24 to 46 plates). This approach assumes that hits are infrequent, structurally unrelated, and randomly distributed on individual library plates. The Z score of the normalized S values was used to detect false-positive compounds that mimic the substrate in MS (detailed in Results).
Hit confirmation data analysis
Hit compounds identified from both libraries were assessed at multiple concentrations for Genzyme library hits (3 and 10 μM) or UT Southwestern library hits (1, 3 and 10 μM) to obtain preliminary concentration-response using the AdoMetDC RapidFire assay as described above. Data were collected in triplicate. For both libraries, raw P/(P+S) values for each assay well were normalized using Equation 2. The mean (Genzyme library) or median (UT Southwestern library) of three replicates of the normalized P/(P+S) values at each concentration was considered to represent the percent inhibition at this concentration and used to rank-order the compounds.
Hit validation AdoMetDC assays
Compound stock solutions were made in DMSO at 25 mM or the highest achievable concentration (not less than 2.5 mM) and all stocks and DMSO stock dilutions were stored at –20 °C. Hit validation assays were performed using the RapidFire assay to determine an IC50 from a full concentration-response curve (range 0.0026 – 50 μM). Compound 1 (at 100 μM to insure full inhibition in the absence of pre-incubation with this time-dependent inhibitor) and DMSO were included on plates as the positive and neutral controls, respectively. The assay was run in triplicate, with each replicate positioned on a separate plate. The percent activity at each compound concentration was calculated as follows:
| (5) |
where P/(P+S) fractional conversion was used as the raw values. Percent activity data were fitted to the log(inhibitor) vs. normalized response equation using nonlinear regression analysis in Prism (GraphPad Software, La Jolla, CA) to determine the relative IC50. Four-parameter fits were used to fit the data, unless the concentrations of an inhibitor were not high enough to reach plateau, in which case the assumption was made that the curve would reach 0% activity at higher concentrations, i.e. the high concentration asymptote was constrained to 0% activity.
AdoMetDC 14C enzyme activity assay
The assay was run as previously described24 by capturing 14CO2 released during decarboxylation of 14C-AdoMet in a form of barium carbonate on filter paper. Briefly, reactions contained TbAdoMetDC/prozyme complex (75–200 nM), 14C-AdoMet diluted with cold AdoMet (specific activity of 10–20 μCi/μmol, final substrate concentration of 100 μM) and assay buffer (50 mM HEPES, pH 7.7, 100 mM NaCl, 5 mM putrescine, 1 mM DTT). Concentration-response data (raw values in cpm) normalized to DMSO control (100% activity) in Prism were fitted to the log(inhibitor) vs. normalized response equation using non-linear regression analysis in Prism to determine the IC50 values.
T. brucei cell viability growth assay
T. brucei brucei Lister 427 bloodstream-form (BSF) cells were maintained at densities supporting logarithmic growth rate (i.e., not exceeding 1.5 × 106 cells/mL) in HMI-19 medium46 supplemented with 10% FBS at high humidity, 37 °C, and 5% CO2. Media containing spermidine was supplemented with chicken serum to avoid generation of cell-toxic products by polyamine oxidases present in FBS47. Cell densities were monitored by counting cells in 0.1 mm3 volume using a Bright-Line hemacytometer (Hausser, Horsham, PA).
Cell viability assay employed ATP-bioluminescence detection48 and is described in Supplemental Methods. The control-normalized concentration-response data (DMSO control is 1 and no-cell medium control is 0 fractional cell viability) were fitted to the log(inhibitor) vs. normalized response equation using nonlinear regression analysis in Prism.
Western blotting analysis of prozyme expression levels
T. brucei BSF cells grown in the presence of 1- to 2-fold EC50 concentrations of a compound were harvested, lysed, separated by SDS-PAGE, and analyzed by western blotting with the rabbit polyclonal antibody raised against T. brucei prozyme (1000-fold dilution in Tris-buffered saline with 5% blotting-grade blocker (Bio-Rad, Hercules, CA)) as previously described17. Membranes were sectioned according to molecular weight marker after transfer, and the top part (≥50 kDa) was probed with the rabbit anti-BiP antibody (100,000-fold dilution) as a loading control. Horseradish peroxidase (HRP)-linked donkey anti-rabbit antibody (10,000-fold dilution) (Jackson ImmunoResearch, West Grove, PA) was used for secondary detection. Protein bands were visualized using SuperSignal West Pico chemiluminescent substrate for HRP (Thermo Fisher Scientific) and imaged on ImageQuant LAS 4000 imager (GE Healthcare Life Sciences, Pittsburgh, PA).
HPLC analysis of polyamine levels
T. brucei polyamine levels were measured using the AccQ-Fluor Reagent Kit (Waters, Milford, MA) as previously described49, 50 and detailed in Supplemental Methods. Spermidine and putrescine fluorescence peak areas were divided by external cadaverine control to correct for run-to-run variability in the integrated signal intensity. The spermidine/putrescine ratio for each compound is presented as a fraction of the DMSO control.
In vitro metabolic stability in hepatic microsomes
Metabolic stability was evaluated with male CD-1 mouse and mixed-gender human liver microsomes (XenoTech, Lenexa, KS). Test articles (1 μM) were incubated with pooled liver microsomes (0.5 mg protein/mL) for 0, 5, 10, 20, and 30 min at 37 °C in the presence of NADPH. Aliquots (50 μL) were taken at each sampling time point and extracted with 150 μL ice-cold acetonitrile containing the internal standard (labetalol). Following centrifugation, supernatants were diluted five-fold into 35/65 A/B Mobile Phase and analyzed for the parent compound by LC/MS/MS as described in Supplemental Methods. The metabolic competency of microsomal preparations was established using the control compounds: 7-ethoxycoumarin, propranolol, and verapamil. Values for half-life (t1/2) were determined with microsomes from each species by plotting the ln((compound peak area)/(internal standard peak area)) vs. time.
In vitro prediction of BBB permeability and Pgp-mediated efflux transport
The propensity of test compounds to cross the BBB was examined using an in vitro MDCKII-hMDR1 monolayer assay37 in the presence or absence of GF120918 (+/– GF918), a potent Pgp inhibitor as detailed in Supplemental Methods. Values for mass balance and apparent permeability, PappA-B (– GF918) and PappA-B (+ GF918), were calculated for each compound51, 52. Acceptance criterion for mass balance was 70–120%.
Pan-Assay Interference Compounds (PAINS)
PAINS compounds are defined as promiscuous structures that hit in multiple screens35. Family A and B PAINS category compounds were available as SMARTS35, 53, which were incorporated into a Pipeline Pilot (v. 9.0.2, Biovia, San Diego, CA) protocol that was used to identify PAINS in the dataset for this screen.
Compound purity by LC/MS
Compounds (0.2–1 mg) were dissolved in methanol or acetonitrile and run on Agilent 1290 Infinity or 1100 series liquid chromatograph with Agilent ZORBAX Eclipse XDB C18 column coupled to the ESI Mass Spectrometer in positive mode. The purity (in %) was defined within 254 nm absorption chromatogram as percent peak area ratio of the peak that corresponds to m/z of the compound to the total area under the curve.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants 2R37AI034432 and R01AI090599 (to MAP and JKDB). MAP and JKDB also acknowledge the support of the Welch Foundation (grants I-1257 and I-1422). MAP holds the Sam G. Winstead and F. Andrew Bell Distinguished Chair in Biochemistry. The authors would like to thank Sara Schock of Scynexis for performing in vitro permeability studies.
Abbreviations
- AdoMet
S-adenosyl-L-methionine
- AdoMetDC
S-adenosylmethionine decarboxylase
- dcAdoMet
decarboxylated S-adenosyl-L-methionine
- HAT
human African trypanosomiasis
- Hs
Homo sapiens
- BBB
blood-brain barrier
- BSF
bloodstream form
- CNS
central nervous system
- ODC
ornithine decarboxylase
- Pgp
P-glycoprotein 1
- SAR
structure-activity relationship
- Spd
spermidine
- SPE
solid-phase extraction
- Tb
T. brucei
Footnotes
Supporting Information. Supporting Information includes additional Methods, biological data for hits flagged as PAINS (Table S1) and LC/MS purity data for all validated active hits (Table S2). Also included are: detailed explanation of how false-positive hits manifested themselves in the primary screen (Figure S1); graphical representation of hit confirmation results (Figure S2); validation of the T. brucei cell viability assay (Figure S3); and representative western blots used to test on-target mechanism of action (Figure S4). This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of human African trypanosomiasis. Clin Epidemiol. 2014;6:257–275. doi: 10.2147/CLEP.S39728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/s0140-673(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy PGE. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness) Lancet Neurol. 2013;12:186–194. doi: 10.1016/s1474-442(12)70296-x. [DOI] [PubMed] [Google Scholar]
- 4.Hasker E, Lumbala C, Mbo F, Mpanya A, Kande V, Lutumba P, Boelaert M. Health care-seeking behaviour and diagnostic delays for Human African Trypanosomiasis in the Democratic Republic of the Congo. Trop Med Int Health. 2011;16:869–874. doi: 10.1111/j.1365-3156.2011.02772.x. [DOI] [PubMed] [Google Scholar]
- 5.WHO. Lowest caseload recorded as the world prepares to defeat sleeping sickness 2016 [Google Scholar]
- 6.Holmes P. On the road to elimination of Rhodesiense human African trypanosomiasis: First WHO meeting of stakeholders. PLoS Negl Trop Dis. 2015;9:e0003571. doi: 10.1371/journal.pntd.0003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hide G. History of sleeping sickness in East Africa. Clin Microbiol Rev. 1999;12:112–125. doi: 10.1128/cmr.12.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamonneau V, Ilboudo H, Kabore J, Kaba D, Koffi M, Solano P, Garcia A, Courtin D, Laveissiere C, Lingue K, Buscher P, Bucheton B. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl Trop Dis. 2012;6:e1691. doi: 10.1371/journal.pntd.0001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berthier D, Breniere SF, Bras-Goncalves R, Lemesre JL, Jamonneau V, Solano P, Lejon V, Thevenon S, Bucheton B. Tolerance to trypanosomatids: A threat, or a key for disease elimination? Trends Parasitol. 2016;32:157–168. doi: 10.1016/j.pt.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Giordani F, Morrison LJ, Rowan TG, HP DEK, Barrett MP. The animal trypanosomiases and their chemotherapy: a review. Parasitology. 2016:1–28. doi: 10.1017/S0031182016001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobs RT, Nare B, Phillips MA. State of the art in African trypanosome drug discovery. Curr Top Med Chem. 2011;11:1255–1274. doi: 10.2174/156802611795429167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, Gaukel E, Owens J, Parham R, Randolph R, Beaudet B, Bacchi CJ, Yarlett N, Plattner JJ, Freund Y, Ding C, Akama T, Zhang YK, Brun R, Kaiser M, Scandale I, Don R. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazue G, Bray MA, Pecoul B. Fexinidazole--a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis. 2010;4:e923. doi: 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairlamb AH. Chemotherapy of human African trypanosomiasis: current and future prospects. Trends Parasitol. 2003;19:488–494. doi: 10.1016/j.pt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegg AE. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009;46:25–45. doi: 10.1042/bse0460003. [DOI] [PubMed] [Google Scholar]
- 17.Willert EK, Phillips MA. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 2008;4:e1000183. doi: 10.1371/journal.ppat.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bitonti AJ, Byers TL, Bush TL, Casara PJ, Bacchi CJ, Clarkson AB, McCann PP, Sjoerdsma A. Cure of Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense infections in mice with an irreversible inhibitor of S-adenosylmethionine decarboxylase. Antimicrob Agents Chemother. 1990;34:1485–1490. doi: 10.1128/aac.34.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacchi CJ, Barker RH, Jr, Rodriguez A, Hirth B, Rattendi D, Yarlett N, Hendrick CL, Sybertz E. Trypanocidal activity of 8-methyl-5′-{[(Z)-4-aminobut-2-enyl]-(methylamino)}adenosine (Genz-644131), an adenosylmethionine decarboxylase inhibitor. Antimicrob Agents Chemother. 2009;53:3269–3272. doi: 10.1128/AAC.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacchi CJ, Brun R, Croft SL, Alicea K, Buhler Y. In vivo trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob Agents Chemother. 1996;40:1448–1453. doi: 10.1128/aac.40.6.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacchi CJ, Nathan HC, Yarlett N, Goldberg B, McCann PP, Bitonti AJ, Sjoerdsma A. Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob Agents Chemother. 1992;36:2736–2740. doi: 10.1128/AAC.36.12.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker RH, Jr, Liu H, Hirth B, Celatka CA, Fitzpatrick R, Xiang Y, Willert EK, Phillips MA, Kaiser M, Bacchi CJ, Rodriguez A, Yarlett N, Klinger JD, Sybertz E. Novel S-adenosylmethionine decarboxylase inhibitors for the treatment of human African trypanosomiasis. Antimicrob Agents Chemother. 2009;53:2052–2058. doi: 10.1128/AAC.01674-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brockway AJ, Cosner CC, Volkov OA, Phillips MA, De Brabander JK. Improved synthesis of MDL 73811 - a potent AdoMetDC inhibitor and anti-trypanosomal compound. Synthesis (Stuttg) 2016;48:2065–2068. doi: 10.1055/s-0035-1561608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willert EK, Fitzpatrick R, Phillips MA. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci USA. 2007;104:8275–8280. doi: 10.1073/pnas.0701111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velez N, Brautigam CA, Phillips MA. Trypanosoma brucei S-adenosylmethionine decarboxylase N terminus is essential for allosteric activation by the regulatory subunit prozyme. J Biol Chem. 2013;288:5232–5240. doi: 10.1074/jbc.M112.442475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volkov OA, Kinch L, Ariagno C, Deng X, Zhong S, Grishin N, Tomchick DR, Chen Z, Phillips MA. Relief of autoinhibition by conformational switch explains enzyme activation by a catalytically dead paralog. eLife. 2016;5 doi: 10.7554/eLife.20198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pegg AE, Williams-Ashman HG. On the role of S-adenosyl-L-methionine in the biosynthesis of spermidine by rat prostate. J Biol Chem. 1969;244:682–693. [PubMed] [Google Scholar]
- 28.Kinch LN, Scott JR, Ullman B, Phillips MA. Cloning and kinetic characterization of the Trypanosoma cruzi S-adenosylmethionine decarboxylase. Mol Biochem Parasitol. 1999;101:1–11. doi: 10.1016/s0166-685(98)00181-9. [DOI] [PubMed] [Google Scholar]
- 29.Smithson DC, Shelat AA, Baldwin J, Phillips MA, Guy RK. Optimization of a non-radioactive high-throughput assay for decarboxylase enzymes. Assay Drug Dev Technol. 2010;8:175–185. doi: 10.1089/adt.2009.0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozbal CC, LaMarr WA, Linton JR, Green DF, Katz A, Morrison TB, Brenan CJ. High throughput screening via mass spectrometry: a case study using acetylcholinesterase. Assay Drug Dev Technol. 2004;2:373–381. doi: 10.1089/adt.2004.2.373. [DOI] [PubMed] [Google Scholar]
- 31.Forbes CD, Toth JG, Ozbal CC, Lamarr WA, Pendleton JA, Rocks S, Gedrich RW, Osterman DG, Landro JA, Lumb KJ. High-throughput mass spectrometry screening for inhibitors of phosphatidylserine decarboxylase. J Biomol Screen. 2007;12:628–634. doi: 10.1177/1087057107301320. [DOI] [PubMed] [Google Scholar]
- 32.Iversen PW, Beck B, Chen YF, Dere W, Devanarayan V, Eastwood BJ, Farmen MW, Iturria SJ, Montrose C, Moore RA, Weidner JR, Sittampalam GS. HTS assay validation. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Iversen PW, Li Z, McGee J, McManus O, Minor L, Napper A, Peltier JM, Riss T, Trask OJ Jr, Weidner J, editors. Assay Guidance Manual. Bethesda (MD): 2004. [Google Scholar]
- 33.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 34.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 35.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 36.Xiao Y, Nguyen S, Kim SH, Volkov OA, Tu BP, Phillips MA. Product feedback regulation implicated in translational control of the Trypanosoma brucei S-adenosylmethionine decarboxylase regulatory subunit prozyme. Mol Microbiol. 2013;88:846–861. doi: 10.1111/mmi.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polli JW, Wring SA, Humphreys JE, Huang LY, Morgan JB, Webster LO, Serabjit-Singh CS. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 38.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 39.Lipinski CA. Overview of hit to lead: The medicinal chemist's role from HTS retest to lead optimization hand off. Top Med Chem. 2009;5:125–140. doi: 10.1007/7355_2009_4. [DOI] [Google Scholar]
- 40.Beswick TC, Willert EK, Phillips MA. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2006;45:7797–7807. doi: 10.1021/bi0603975. [DOI] [PubMed] [Google Scholar]
- 41.Masimirembwa CM, Bredberg U, Andersson TB. Metabolic stability for drug discovery and development: pharmacokinetic and biochemical challenges. Clin Pharmacokinet. 2003;42:515–528. doi: 10.2165/00003088-200342060-00002. [DOI] [PubMed] [Google Scholar]
- 42.Patterson S, Wyllie S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol. 2014;30:289–298. doi: 10.1016/j.pt.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 44.Tang H, Zhu Y, Teumelsan N, Walsh SP, Shahripour A, Priest BT, Swensen AM, Felix JP, Brochu RM, Bailey T, Thomas-Fowlkes B, Pai LY, Hampton C, Corona A, Hernandez M, Metzger J, Forrest M, Zhou X, Owens K, Tong V, Parmee E, Roy S, Kaczorowski GJ, Yang L, Alonso-Galicia M, Garcia ML, Pasternak A. Discovery of MK-7145, an oral small molecule ROMK inhibitor for the treatment of hypertension and heart failure. ACS Med Chem Lett. 2016;7:697–701. doi: 10.1021/acsmedchemlett.6b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu Z, Liu D, Sui Y. Quantitative assessment of hit detection and confirmation in single and duplicate high-throughput screenings. J Biomol Screen. 2008;13:159–167. doi: 10.1177/1087057107312628. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Leija C, Rijo-Ferreira F, Chen J, Cestari I, Stuart K, Tu BP, Phillips MA. GMP synthase is essential for viability and infectivity of Trypanosoma brucei despite a redundant purine salvage pathway. Mol Microbiol. 2015;97:1006–1020. doi: 10.1111/mmi.13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebong S, Farkas WR. Absence of plasma amine oxidase in some frequently used animal models. Comp Biochem Physiol C, Comp Pharmacol Toxicol. 1993;106:483–487. doi: 10.1016/0742-841(93)90167-J. [DOI] [PubMed] [Google Scholar]
- 48.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR, Williams J, McKerrow JH. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem Biol Drug Des. 2006;67:355–363. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 49.Osterman AL, Brooks HB, Jackson L, Abbott JJ, Phillips MA. Lysine-69 plays a key role in catalysis by ornithine decarboxylase through acceleration of the Schiff base formation, decarboxylation, and product release steps. Biochemistry. 1999;38:11814–11826. doi: 10.1021/bi9906221. [DOI] [PubMed] [Google Scholar]
- 50.Pratt C, Nguyen S, Phillips MA. Genetic validation of Trypanosoma brucei glutathione synthetase as an essential enzyme. Eukaryotic Cell. 2014;13:614–624. doi: 10.1128/EC.00015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiel-Demby VE, Tippin TK, Humphreys JE, Serabjit-Singh CJ, Polli JW. In vitro absorption and secretory quotients: practical criteria derived from a study of 331 compounds to assess for the impact of P-glycoprotein-mediated efflux on drug candidates. J Pharm Sci. 2004;93:2567–2572. doi: 10.1002/jps.20166. [DOI] [PubMed] [Google Scholar]
- 52.Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm Res. 2003;20:1210–1224. doi: 10.1023/A:1025001131513. [DOI] [PubMed] [Google Scholar]
- 53.Lagorce D, Sperandio O, Baell JB, Miteva MA, Villoutreix BO. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015;43:W200–207. doi: 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.