

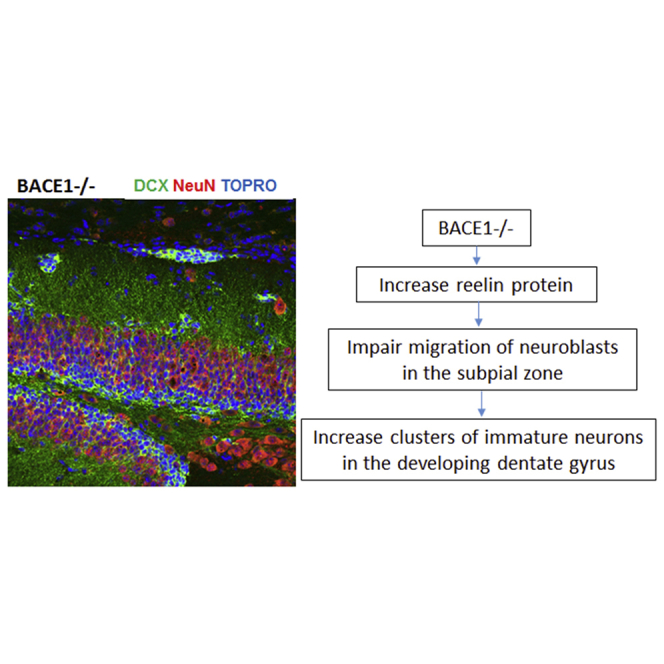
Summary
BACE1 is validated as Alzheimer's β-secretase and a therapeutic target for Alzheimer's disease. In examining BACE1-null mice, we discovered that BACE1 deficiency develops abnormal clusters of immature neurons, forming doublecortin-positive neuroblasts, in the developing dentate gyrus, mainly in the subpial zone (SPZ). Such clusters were rarely observed in wild-type SPZ and not reported in other mouse models. To understand their origins and fates, we examined how neuroblasts in BACE1-null SPZ mature and migrate during early postnatal development. We show that such neuroblasts are destined to form Prox1-positive granule cells in the dentate granule cell layer, and mainly mature to form excitatory neurons, but not inhibitory neurons. Mechanistically, higher levels of reelin potentially contribute to abnormal neurogenesis and timely migration in BACE1-null SPZ. Altogether, we demonstrate that BACE1 is a critical regulator in forming the dentate granule cell layer through timely maturation and migration of SPZ neuroblasts.
Keywords: BACE1, Alzheimer's secretase, neuronal cluster, doublecortin, neuronal migration, neurogenesis, subpial zone, meninges, subgranular zone, granule cell layer, reelin
Graphical Abstract
Highlights
-
•
BACE1 deficiency causes abnormal neuronal clusters retained in the mouse SPZ
-
•
Mis-migrated neural progenitor cells in the SPZ are destined to form granule cells
-
•
Such neural progenitor cells form excitatory neurons but not inhibitor neurons
-
•
Elevated levels of reelin contribute to abnormal neuronal maturation and migration
In this article, Yan and colleagues show that BACE1 is required for the proper formation of the granule cell layer. BACE1 deficiency in mice induces retention of clustered doublecortin-positive neuroblasts in the subpial zone (SPZ) and marginal zone (MZ). Upregulation of reelin in Cajal-Retzius cells likely causes timely migration of neuroblasts in SPZ/MZ to form Prox1-positive granule cells.
Introduction
β-Site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) initiates cleavage of APP at the β-secretase site (Vassar et al., 1999, Yan et al., 1999, Hussain et al., 1999, Sinha et al., 1999, Lin et al., 2000). The released APP C-terminal fragment is then further cleaved by γ-secretase to excise β-amyloid peptides (Aβ). In brains of patients suffering from Alzheimer's disease (AD), excessively accumulated Aβ is considered to be an early toxic event that leads to AD pathogenesis (Selkoe and Hardy, 2016). Genetic mutations surrounding the BACE1 cleavage site in APP such as the K670M671 to NL mutation in a Swedish family (which results in facilitated cleavage of APP by BACE1) can cause early onset of AD (Mullan et al., 1992), or alternatively can delay the onset of AD, as in the case of the A673 to T673 mutation (resulting in suppressed cleavage of APP by BACE1) (Jonsson et al., 2012). More strikingly, Aβ production is nearly abolished in mice deficient in BACE1, and these mice do not develop amyloid deposition, even if Swedish mutant APP is expressed (Cai et al., 2001, Luo et al., 2001, Roberds et al., 2001). Therefore, BACE1 is an important therapeutic target for reversing Aβ-mediated cognitive dysfunction in AD (Yan et al., 2016, Vassar, 2014).
Although initial examinations of BACE1-null mice in the original studies suggested no overt defects in mouse growth or fertility, subsequent morphological examinations of brains and biochemical analyses of natural substrates of BACE1 began to reveal abnormal astrogenesis, reduced neurogenesis, hyperactivities, impaired axonal growth and pathfinding, hypomyelination, altered long-term potentiation, and long-term depression, as well as defects in muscle spindles (see reviews by Barao et al., 2016, Vassar et al., 2014, Yan and Vassar, 2014, Hu et al., 2015). BACE1 is a membrane-anchored aspartic protease that is not only necessary for Aβ generation but is also indispensable for the cleavage of many other cellular substrates such as neuregulin-1 (Willem et al., 2006, Fleck et al., 2013, Hu et al., 2006, Hu et al., 2008, Luo et al., 2011), Jagged1 and Jagged2 (He et al., 2014, Hu et al., 2013), close homolog of L1 (Hitt et al., 2012, Kuhn et al., 2012, Zhou et al., 2012), seizure protein 6 (Pigoni et al., 2016), and voltage-gated sodium channel protein β subunits (Wong et al., 2005, Kim et al., 2005, Huth et al., 2011). Abrogated cleavage of these substrates may significantly contribute to many of the observed phenotypes in BACE1-null mice.
We recently reported that increased astrogenesis in BACE1-null dentate gyrus (DG) is evident during early postnatal development, while neurogenesis is correspondingly decreased (Hu et al., 2013), suggesting a shift in the fate determination of radial glial stem cells. To determine whether neurogenesis is altered in other brain regions, we examined brain sections with doublecortin (DCX), a protein predominantly expressed by neuronal precursor cells and immature neurons (Magavi et al., 2000, Francis et al., 1999). Surprisingly, DCX+ clustered cells were found in the BACE1-null subpial zone (SPZ) after postnatal day 10 (P10), and such clustered DCX+ cells were rarely seen in the same region of wild-type (WT) mice at this age. We further confirmed that these DCX+ cells were present in the SPZ of more mature mice and appeared to migrate toward the dentate granular cell layer during development. BACE1 deficiency appears to impair timely migration of neurons from these DCX-clustered cells. To determine the molecular mechanism, we noted that reelin protein levels were significantly elevated and that increased reelin activity can cause neuronal migration defects (Kubo et al., 2010, Pujadas et al., 2010, Jossin et al., 2007), suggesting a potential contribution of reelin to this abnormal neuronal clustering during brain development of BACE1-null mice. Thus, we provide morphological evidence that BACE1 is required for proper neuronal migration during early development.
Results
BACE1 Deficiency Produces Doublecortin-Positive Clusters in the Developing Dentate Gyrus
Altered neurogenesis in the BACE1-null mouse DG has recently been shown (Hu et al., 2013), but it is unclear whether BACE1 deficiency affects the migration, maturation, and/or differentiation of newborn cells in the DG during early developmental stages. Morphogenesis of the DG in mice is known to start at approximately embryonic day 14.5 (E14.5) and to end on approximately P7 (Li and Pleasure, 2007, Hodge et al., 2013, Yu et al., 2014). To investigate neuronal differentiation in the DG of BACE1-null mice during early postnatal development, we examined the expression of doublecortin (DCX), which is a widely used marker for immature neurons and is required for neuronal migration and differentiation (von Bohlen Und, 2011).
As expected, DCX in WT mice was highly expressed at P3 (Figure 1A), reflecting actively ongoing neurogenesis. At this time point, lamination of DG was not evident. At P6, DG cytoarchitecture and fiber lamination became clearer, while DCX expression levels were correspondingly decreased. BACE1 deficiency resulted in no obvious differences in the distribution of DCX+ immature neurons in DG at these early stages (P3–P6 in Figure 1A). Interestingly, DCX+ clustered cells were found in the transient SPZ, marginal zone (MZ), and dentate molecular layer (ML) of P8 BACE1-null DG (arrows in Figure 1A). At this age such clustered cells were also detected in WT DG, but with much lower frequency. Between P10 and P20, the sizes of DCX+ clustered cells in the SPZ/MZ were larger and more readily detected in BACE1-null DG, while the numbers of DCX+ clusters were significantly diminished in the same regions of WT littermate controls (Figure 1A). Reductions in the size and numbers of DCX+ clustered cells in SPZ/MZ were observed at P30 in BACE1-null DG. We also conducted quantification of the area occupied by DCX+ clustered cells in relation to the entire area of the DG and confirmed an abnormal increase in DCX+ clustered cells in BACE1-null SPZ and MZ compared with WT (Figure 1B; 0.303% ± 0.031% in WT versus 0.769% ± 0.056% in BACE1-null P8 DG; 0.031% ± 0.008% in WT versus 0.462% ± 0.031% in BACE1-null P20 DG; 0% in WT and 0.305% ± 0.033% in BACE1-null P30 DG; n = 5 pairs of WT and BACE1-null mice in each age group; ∗∗∗p < 0.001, Student's t test).
Figure 1.

Expression of DCX+ Clusters in the Dentate Gyrus during Mouse Development
(A) Doublecortin (DCX) is expressed by immature neurons in the dentate gyrus (DG) after birth. No DCX+ clusters were readily visible in either wild-type (WT) or BACE1-null mice at early developmental stages (postnatal day 3 [P3] and P6). At P8, scatted DCX+ cells were detected in the transient subpial zone (SPZ)/marginal zone (MZ) in WT mice as well as small DCX+ clusters in the SPZ/MZ in BACE1-null mice (arrow). From P10 to P20, more DCX+ clusters were formed in the SPZ/MZ and were even observed in the molecular layer (ML) of BACE1-null DG, which are very rarely detected in WT P10 mice. The expression of DCX gradually decreased in DG during development. DCX+ clusters were found in close proximity to the granule cell layer (GCL) (arrowhead) at P30. Nuclei were labeled with TOPRO. Scale bars, 100 μm.
(B) Areas occupied by clustered cells were quantified by ImageJ and divided by the total DG area in the field. The percentage of clustered cell area was plotted from mice at the ages of P8, P20, and P30 (n = 5 pairs; ∗∗∗p < 0.01, Student's t test).
Morphologically, DCX+ cell clusters were diversely shaped in BACE1-null DG after P20; some of the clusters were found in the ML of DG and connected to the granule cell layer (GCL) in DG (see arrowheads in Figure 1A). The reduction of DCX+ clusters in developing BACE1-null DG and the detection of these cells in the ML suggest potential migration of DCX+ neuroblasts to the DG. While the expression of DCX in DG was expectedly decreased during mouse development, this decrease was correlatively reflected in the intensity of DCX+ clusters seen in BACE1-null DG during development. In conclusion, DCX+ clustered cells were rarely observed in WT DG after P10 and normally disappeared during subsequent development. However, BACE1 deficiency caused the retention of clustered cells, which provides a unique opportunity to study DCX+ cells in SPZ/MZ during neuronal development.
DCX+ Clusters Form Mature Neurons in Developing BACE1-Null Mice
During embryonic stages, morphogenesis of the DG initiates from neural stem cells (NSCs) located in the dentate neuroepithelium (Li and Pleasure, 2007, Yu et al., 2014). Normally, NSCs leave the dentate neuroepithelium and move along the dentate migratory stream to form a transient neurogenic SPZ that is adjacent to the meninges (Li et al., 2009). At P3 to P4, neural progenitor cells populating the transient SPZ are redistributed and merge into the subgranular zone (SGZ) niche. DG is formed at P5–P7, and few, if any, neural progenitor cells are normally found in the SPZ after this stage (Li et al., 2009, Hodge et al., 2013).
DCX+ clusters in the SPZ of BACE1-null mice likely derive from NSCs, and some cells in these clusters may retain dividing potential. To confirm this, we pulse-labeled P21 and 1-month-old mice with bromodeoxyuridine (BrdU) and newborn cells incorporated with BrdU in DG were labeled with BrdU antibody. While BrdU+ cells were mostly enriched in the SGZ as expected, we also observed some BrdU+ cells localized in SPZ/MZ, which was more evident in BACE1-null mice (Figures 2A and 2B). Consistent with a prior report (Hu et al., 2013), the number of BrdU+ cells in WT mouse SGZ was not different from that in BACE1-null SGZ, but BrdU+ cells were sparsely distributed in SPZ/MZ of WT mice at P21 or older. Further double labeling of BrdU with DCX in 1-month-old BACE1-null mice confirmed the presence of BrdU+ cells co-localized with the DCX+ clusters (Figure 2C), indicating that the increased size of DCX+ clusters is likely related to the proliferation of neural progenitor cells in SPZ/MZ.
Figure 2.

Division of DCX+ Clusters in BACE1-Null Mice
Mice were treated with BrdU (50 mg/kg, intraperitoneally) twice in 1 day and then euthanized after 24 hr. BrdU+ cells were mostly restricted to SGZ in WT DG at P21 (A). In BACE1-null brains, BrdU-labeled cells also mostly resided in the SGZ, but were also visible in the SPZ/MZ and ML (B). Dividing cells were co-localized with DCX+ clustered cells in SPZ/MZ of P30 BACE1-null mice (C). It appeared that most DCX+ cells were no longer dividing after P30. Dashed line indicates SPZ/MZ or SGZ areas. Scale bars, 100 μm.
To understand whether these clusters could mature into neurons, we co-labeled DCX+ clusters with an antibody specific to NeuN, a widely used marker for mature neurons. At P10 in BACE1-null brains, NeuN+ cells were sparsely detected within the DCX+ clusters (Figure 3A), but small NeuN+ clusters began to appear near DCX+ clusters at P20 (Figure 3B, arrows). In 2-month-old BACE1-null samples, more DCX+ clustered cells appeared to mature into NeuN+ neurons, while DCX+ immature neurons correspondingly decreased (Figure 3C); no DCX+ immature neurons were visible in the clusters of 6-month-old samples (Figure 3D). No NeuN+ clusters were ever detected in 2- (Figure 3E) or 6-month-old WT DG (Figure 3F). From monitoring DCX+ clusters during development, we conclude that DCX+ clusters are NSCs in origin and appear to migrate toward and merge into the GCL (Figures 3B and 3C, arrowheads).
Figure 3.

DCX+ Clustered Cells Become Mature Neurons in the Adult Mouse
(A–D) At P10 only a few cells in DCX+ clusters were positive for NeuN, and this number was significantly increased during development (B–D). At P30, small NeuN+ clusters were found near DCX+ clusters in BACE1-null DG (arrows in B), but all DCX+ cells appeared to be converted to NeuN+ clusters at 6 months of age (D). Some clusters were in close proximity to the GCL during development (C and D). Arrows show clusters containing NeuN+ mature neurons while arrowheads refer to clusters containing mainly DCX+ neurons. Similar to the immature neurons from SGZ, DCX+ clusters originating from SPZ differentiated into mature neurons. It is possible that DCX+ clustered cells are programmed to form DG granule cells.
(E and F) DCX+ or NeuN+ clusters in adult 2- or 6-month-old WT DG were not detected.
Scale bars, 50 μm.
DCX+ Clusters Form Dentate Granule Cells
In addition to the presence of DCX+ clusters in SPZ/MZ, we also noted the presence of DCX+ clusters adjacent to the mossy fiber side of the DG ML in some BACE1-null mouse brains (Figure S1). Examinations of serial sections of one mouse DG showed changes in the sizes of DCX+ clusters (Figures S1A–S1H), reflecting the emergence of a “pseudo DG” from this cluster, which was also positive for NeuN in most of the clustered area and appeared to be partially dissociated from the DG GCL (for an enlarged view see Figure S1I). This phenotype occurred at relatively low penetrance (6 out of 29 examined BACE1-null brains) and appeared as a pseudo DG in this case (Figures S1A–S1H). However, in another case it appeared to be breaking from GCL or to be incomplete fusion to GCL (Figure S1J), suggesting impaired GCL formation or mis-migration.
To determine whether these cells in the so-called pseudo DG are truly related to DG granule cells, we stained the adjacent sections with Prox1, which is a mature granule cell marker (Lavado and Oliver, 2007). While Prox1 predominantly labeled DG granule cells in the hippocampus, we observed clear staining of the pseudo DG (Figure 4A), which is more evident in the enlarged view in Figure 4B. Moreover, we showed that clustered cells in the SPZ were partially positive for Prox1 (clustered cells are circled in Figure 4C), suggesting an origin related to DG formation. Hence, clustered cells are neuronal precursor cells destined to form DG granular cells.
Figure 4.

Neuronal Clusters Are Destined to Form Dentate Granule Cells
(A and B) Brain sections from P11 BACE1-null mice were labeled with Prox1 antibody. Prox1+ cells were localized mainly in the DG granule cell layer, but not CA pyramidal neurons (A). The inset in (A) is shown in (B), and clustered cells near the granule layer were labeled by Prox1, indicating a similar origin.
(C–F) The clustered cells in SPZ at P20 (C and D) and 2 months (E and F) were also shown to be labeled by Prox1. It is clear that Prox1 is expressed only by DG granule cells, but not CA pyramidal cells, at different ages.
Scale bars, 100 μm.
DCX+ Clustered Cells in SPZ/MZ Are Neuronal Precursor Cells
To further investigate the migration and differentiation of newborn cells from SPZ in BACE1-null DG, we intraperitoneally injected BrdU into P11 BACE1-null mice and WT littermate controls for 5 consecutive days and then euthanized the treated mice at P21, 2 months of age, or 6 months of age. In the example of P21 BACE1-null DG, two visible DCX+ clusters in the SPZ/MZ were identified and the clusters were partially labeled by BrdU (Figure 5B), suggesting a proliferative capacity within the cluster. One of these two clusters formed a stream and migrated toward the GCL, while the other remained in the SPZ/MZ (Movie S1). Several smaller DCX+ clusters were also found in the ML (Figure 5B), and no DCX+ clusters were visible in the same regions of WT controls. DCX+ cells in both 2-month-old WT and BACE1-null DG were significantly fewer and mostly restricted to the SGZ, reflecting the continuing differentiation into mature neurons during development (Figures 5C and 5D). Only in BACE1-null DG were BrdU+ clusters detected in the MZ and ML (Figure 5D). In 6-month-old BACE1-null mice, some BrdU+ clusters in MZ and ML were still visible (Figure 5F) but were no longer positive for DCX. This is consistent with our aforementioned observations that clusters became mature neuronal clusters (NeuN+).
Figure 5.

Differentiation of DCX+ Cells Located in SPZ
(A–F) Mice were treated with BrdU at P11 once per day (15 mg/kg, intraperitoneally) for 5 consecutive days. The treated mice were then euthanized at P21 (A and B), at 2 months of age (C and D), or at 6 months of age (E and F). At P21, two BrdU+ clusters originating from SPZ/MZ were clearly positive for DCX in BACE1-null DG (B). One of these formed a stream and appeared to migrate toward the GCL, while the other remained in the SPZ/MZ. BrdU/DCX double-labeled clusters in MZ and ML were still detected in 2-month-old BACE1-null mice (D), but no double-positive clusters were observed in 6-month-old BACE1-null DG (F).
(G) A portion of DCX+ clustered cells, mostly on the rim, was co-labeled with GFAP antibody in P20 BACE1-null SPZ.
(H) DCX-clustered cells may contain cells that differentiate and proliferate into GFAP+ astrocytes in SPZ/MZ of 1-month-old BACE1-null DG.
(I) GFAP+ cells were mostly seen on the rim of DCX+ clusters in P20 BACE1-null DG, and these were significantly reduced in 6-month-old BACE1-null DG.
Scale bars, 100 μm (A–F) and 50 μm (G–I).
We also asked whether DCX+ clusters contained stem cells that would differentiate into astrocytes. To address this, we co-labeled clustered cells with glial fibrillary acidic protein (GFAP), a protein expressed by both DG radial glial stem cells and mature astrocytes. We showed that a portion of DCX+ clustered cells, mostly on the rim, was co-labeled with GFAP antibody in P20 BACE1-null SPZ (Figure 5G). For BrdU-pulsed 1-month-old samples, GFAP co-labeled with BrdU in the SPZ (Figure 5H), suggesting a potential differentiation into astrocytes from the clusters. In older BACE1-null samples more mature astrocytes were found in the SPZ, mostly on the rim of the cluster (Figure 5I). In the SGZ, radial stem cells are recognized as multi-potent NPCs (Doetsch, 2003, Malatesta et al., 2008), and these clustered cells in the SPZ potentially contain cells that are capable of differentiating into mature astrocytes and neurons.
DCX+ Clusters Differentiate into Glutamatergic Neurons
To understand the types of neurons into which DCX+ clusters differentiate, we conducted double labeling of DCX with various neuronal markers. We found that vesicular glutamate transporter (VGlut1), which is mainly expressed by excitatory glutamatergic neurons (Kaneko and Fujiyama, 2002), partially co-labeled with DCX in clusters as early as P20 (Figure 6A) and more intensely in brain sections from older animals (Figures 6B and 6C). However, interneurons labeled by parvalbumin were not found in clusters (Figure 6D; enlarged inset in 6E). L-Glutamic acid decarboxylase (GAD) isoforms GAD65 and GAD67, predominantly expressed by inhibitory GABAergic neurons (Dupuy and Houser, 1996), were also scarcely found in the clusters (data not shown). Hence, the immature DCX+ clustered neurons will mainly differentiate into glutamatergic neurons, the major type of neuron in the DG (Crawford and Connor, 1973, Amaral et al., 2007).
Figure 6.

DCX+ Clusters Differentiate into Glutamatergic Neurons
Brain sections were co-labeled with antibodies to DCX and either VGlut1 (A–C) or parvalbumin (D and E). Clustered cells are circled in E. Comparable with the SGZ, DCX+ clusters originating from the SPZ/MZ also appeared to form VGlut1-positive glutamatergic neurons and not inhibitory neurons. Ages of mice are specified and nuclei were labeled with TOPRO. Scale bars, 100 μm.
In addition to normal differentiation, we also examined whether apoptosis would occur in the clusters. By staining brain sections with antibody to activated caspase-3 or performing TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assays, we found no significantly increased apoptotic signals in association with changes in the clustered cells in BACE1-null SPZ/MZ (data not shown).
Reelin Upregulation Potentially “Arrests” the Migration of NPCs and Causes the Formation of Neuronal Clusters in DG in BACE1-Null Mice
To explore the molecular mechanisms underlying the formation of clustered NPCs in the SPZ, we examined the expression of reelin, which is a secreted glycoprotein synthesized by early-generated Cajal-Retzius cells in the MZ that plays a critical role in neuronal migration and lamination during the development of the CNS, especially during the morphogenesis of DG (Stanfield and Cowan, 1979, Pujadas et al., 2010, Forster et al., 2010, Li et al., 2009). In relation to this study, one of the remarkable phenotypes in reelin-null mice is dispersed granule cells in the DG (Stanfield and Cowan, 1979), while increased reelin also causes neuronal migration defects (Kubo et al., 2010, Pujadas et al., 2010, Jossin et al., 2007). Ectopic expression of reelin induces neuronal aggregation in developing cortex (Kubo et al., 2010), while transgenic mice overexpressing reelin have phenotypes such as small neuronal clusters in the hippocampus, wide distribution of BrdU-labeled proliferating cells in the ML of the DG, and abnormal positioning of adult-generated neurons (Pujadas et al., 2010), similar to the phenotypes identified in our mice.
Based on these observations, we tested reelin expression in mouse DG during development. Our immunostaining results showed that reelin+ cells were mostly restricted to the SPZ/MZ in both WT and BACE1-null mice at P3, and a small number of reelin+ cells were scattered in the ML region that is adjacent to the MZ (Figures 7A and 7B). This is consistent with previous observations that reelin is expressed by Cajal-Retzius cells located in the MZ (Pesold et al., 1998). The number of reelin+ cells was sharply decreased in DG at P7, and BACE1 deficiency did not alter these expression patterns (Figures 7C and 7D). At P20, only small numbers of reelin+ cells located in the SPZ/MZ were labeled (Figures 7E and 7F), and noticeably, DCX+ clusters in SPZ/MZ were in close proximity with reelin+ cells in BACE1-null DG. An enlarged view further shows no reelin+ cells in the clusters (data not shown). The numbers of reelin+ cells were also counted from each captured image, and average numbers of reelin+ cells in three age groups were used for comparison (Figure 7G). Based on our quantification, we found no significant differences in the numbers of reelin+ cells between BACE1-null and WT controls (n = 5 pairs, p > 0.05, Student's t test).
Figure 7.

Enhanced Expression of Reelin Resulting from BACE1 Deficiency
(A–F) Confocal staining of brain sections showed restricted expression of reelin by Cajal-Retzius cells located in the MZ at P3, P7, and P20. Scale bars, 50 μm.
(G) Numbers of reelin+ cells were counted from each captured image in three age groups, and average numbers of reelin+ cells per DG section were used for comparison (n = 5 pairs of mice, Student's t test).
(H) Western blotting of hippocampal lysates showed that expression of reelin in hippocampi gradually decreased after birth.
(I) Bar graphs show the relative levels of reelin over β-actin. Quantification was performed from three independent experiments (n = 6 pairs of P6, 8 pairs of P20, and 6 pairs of P30 mice; ∗p < 0.05, ∗∗p < 0.01). Reelin upregulation potentially arrests the migration of neuroprogenitor cells and may lead to the formation of neuronal clusters in DG of BACE1-null mice.
However, by using a reelin-specific antibody (G10), we showed that the active form of reelin was significantly increased in the BACE1-null hippocampus (Figure 7H). Further quantification confirmed significant upregulation of reelin in BACE1-null DG (Figure 7I). Since BACE1 is expected to cleave membrane-anchored molecules (Yan et al., 2001) and reelin is a secreted molecule (D'Arcangelo et al., 1995), it is unlikely that reelin is a natural substrate of BACE1. More likely, an indirect mechanism regulates the levels of reelin in BACE1-null brain.
Discussion
BACE1 is a critical enzyme for Aβ generation, and the use of specific BACE1 inhibitors is expected to reduce amyloid deposition in humans (Yan, 2016, Vassar, 2014). However, BACE1 is also important for various neurological functions (Yan and Vassar, 2014). Here, we show that BACE1 deficiency impairs neurogenesis and neuronal maturation in the DG. Specifically, we observed abnormal neural clusters in the BACE1-null SPZ, MZ, and ML, and these DCX+ clusters were not readily detected in corresponding WT control regions (see summary in Table S1). This observation allowed us to address the important questions of how DCX+ clusters evolve, migrate, and mature into neurons.
The DCX-expressing cells in the developing and adult brain are often multi-potential precursors, and a majority are neuron-forming cells (Francis et al., 1999, Brown et al., 2003). The appearance of DCX+ clusters in SPZ/MZ of BACE1-null mice begins at P7–P8, a time point at which the DG has just fully formed. These clustered DCX+ cells are capable of forming mature neurons during mouse development, as demonstrated in BrdU labeling experiments and co-labeling with NeuN antibody in adult brains. Shifting from immature neurons to mature neurons between 1 and 2 months of age is evident (Figures 3C and S1I), and no immature neurons are observed in 6-month-old BACE1-null DG (Figure 3D). Clustered DCX+ cells in SPZ/MZ appear to be mainly neuronal progenitor cells. However, we cannot not exclude the possibility that a small fraction might be multi-potent with the ability to differentiate into astrocytes, which are found adjacent to the cluster (Figure 5H). We did not investigate whether they would differentiate into oligodendrocytes in this study, as myelination is normally completed by 1 month of age. Nevertheless, most of these DCX+ clustered cells are theoretically neural progenitor cells residing in the SPZ/MZ, which can differentiate into newborn neurons in adult BACE1-null DG.
Collectively, our results indicate that the origin of NSCs/neuroprogenitor cells residing in SPZ and SGZ in the developing DG are likely the same, both of which migrate from embryonic dentate epithelium. While only few NPC clusters are normally found in P10 or younger WT SPZ/MZ, BACE1 deficiency significantly delays their maturation and migration, as these neuronal clusters were more readily observed in P20–P30 DG and were even detected in adult BACE1-null mice. Like NPCs in the SGZ, these DCX+ cells in SPZ/MZ and/or ML will mainly differentiate into mature granule cells in the GCL, as evidenced by Prox1 staining (Figure 4).
Where these clustered NPCs in the SPZ/MZ originate is an important neural developmental question. Although multi-potent NPCs in the subventricular zone (SVZ) migrate into adjacent gray matter and differentiate into different types of glia (oligodendrocytes and astrocytes) and neurons (Goldman, 1995), no clustered NPCs were found in SVZ and they are therefore unique to the DG. A prior study identified a transient subpial neurogenic zone by tracking nestin-GFP during development of the DG, and showed that neurogenic precursors migrate initially through a subpial migratory stream to fimbriodentate junctions in embryonic stages and then move across the hilus and reside in the subpial region in neonatal stages (Li et al., 2009). BACE1 deficiency clearly arrests NPCs in the SPZ, as they fail to migrate to SGZ in a timely manner. While most DCX+ clusters were found in the SPZ/MZ or ML on the suprapyramidal side, we also noted the presence of DCX+ clusters in the ML of the infrapyramidal side (Figure S1). NPCs in SPZ migrate to the SGZ, where they are capable of self-renewal and are long lived. In this example a pseudo DG was noted, which likely arises from the mis-migration of NPCs to SGZ in a timely manner and from failure to form an intact granule cell pool. Hence, BACE1 deficiency impairs normal neuronal migration and timely maturation. This finding also raises the question as to whether BACE1 inhibition causes defects in adult neuronal migration, which will be answered by future studies.
We further explored how BACE1 deficiency causes abnormal neurogenesis. Our previous studies suggested that BACE1 cleaves Jag1 and regulates Jag/Notch signaling during neurogenesis (He et al., 2014, Hu et al., 2013). However, this pathway appears to be restricted in the SGZ region and is not directly involved in controlling neuronal migration, as Jag1 was not detected in the cluster (data not shown). During embryonic development of brain laminated structures, the protein reelin is known to be synthesized by Cajal-Retzius cells located in the MZ and regulates formation of the SPZ. Reelin is normally secreted into the extracellular matrix of the cortex and hippocampus and then binds to its receptor on cortical and hippocampal neurons that do not synthesize reelin. This trans-signaling between cells contributes to the regulation of migration and the positioning of targeted neurons. Reelin is a large protein with a molecular weight of approximately 385 kDa, and is composed of eight consecutive repeat domains (R1–R8); R3–R6 are necessary for its activity (D'Arcangelo et al., 1995). Previous studies have shown that reelin is cleaved by metalloproteinases at two sites after repeat domains 2 and 6 to produce an R3–R6 fragment (Jossin et al., 2007, Lambert de et al., 1999). Although reelin is unlikely to be a substrate of BACE1 due to the lack of a transmembrane domain, we observed a clear increase in total full-length reelin (Figure 7H). The active form of reelin (full-length and N-R6) was also correspondingly increased, and this increase likely allows broader diffusion to affect neighboring cells. While it is not clear at this time how BACE1 deficiency increases reelin protein levels, one possibility is via the enhanced transcription of reelin by CREB, as CREB binding sites are found in the reelin promoter (Chen et al., 2002). In a previous study, BACE1 was found to interact with adenylate cyclase via its transmembrane domain, resulting in reduction of cellular cyclic AMP levels, thus leading to protein kinase A inactivation and reduced CREB phosphorylation (Chen et al., 2012). BACE1 deficiency will increase CREB activation and subsequent reelin expression. Alternatively, BACE1 deficiency may regulate the translation of reelin. All of these possibilities will be investigated in future studies. In mice overexpressing reelin, BrdU+ cells are distributed more widely along the GCL and in the ML (Figure 4A in Pujadas et al., 2010), and this phenotype is similar to our results shown in Figures 5D and 5F. Hence, elevated reelin levels potentially contribute to the abnormal distribution of NPCs in the hippocampus.
How impaired neuronal migration affects brain function remains to be established. We have previously observed epileptic seizures in BACE1-null mice, and abnormal neurogenesis has been linked to epileptic seizures (Jessberger and Parent, 2015, Cho et al., 2015). It is likely that abnormal neuronal migration and neurogenesis may also contribute to seizure activity in BACE1-null mice. In conclusion, we have demonstrated abnormal neuronal clusters present in BACE1-null SPZ/MZ, and these clustered cells likely form granule cells in the DG. Thus BACE1-null mice will be useful tools for future studies of neuronal migration and maturation from SPZ to SVZ, as such phenotypes are not seen during normal neuronal development. It will be important to determine how such abnormal changes lead to changes in brain functions and whether sustained inhibition of BACE1 will significantly affect neuronal migration.
Experimental Procedures
Animals
BACE1-null mice were generated as described in the original publication (Cai et al., 2001) and maintained in a C57BL/6 background. All animal use and procedures were first described and submitted for approval by the Institutional Animal Care and Use Committee at the Lerner Research Institute and in compliance with the guidelines established by the Public Health Service Guide for the Care and Use of Laboratory Animals.
BrdU Administration
To test neurogenesis in our experiments, we injected BACE1-null mice and littermate controls with BrdU (50 mg/kg, intraperitoneally) twice on the same day (at 6-hr intervals) and euthanized them after 24 hr. For neuronal differentiation experiments, mice were treated with BrdU (15 mg/kg/day, intraperitoneally) once per day for 5 consecutive days and euthanized after 3 weeks. For the investigation of neuronal migration in the DG during development, the BrdU administration protocol was identical to that of the neuronal differentiation experiments, beginning at P11. Mice were then euthanized at P21, at 2 months of age, or at 6 months of age.
Western Blotting and Antibodies
Proteins were extracted from WT and BACE1-null mice in RIPA buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail [Roche]). Equal amounts of protein (30 μg) were resolved on a NuPAGE Bis-Tris Gel (Invitrogen) and transferred onto nitrocellulose membranes (Invitrogen) for western blot analysis. Horseradish peroxidase-conjugated secondary antibodies were used and visualized using enhanced chemiluminescence (Thermo Scientific). Reelin (1:1,000) antibody was purchased from Millipore; BACE1 (1:1,000) was purchased from Proteintech; and β-actin (1:10,000) was purchased from Sigma.
Morphological Analyses
For confocal microscopy, the indicated genotypes of mice were euthanized at various ages (see Results for details) during development. The right halves of the brains were fixed in 4% paraformaldehyde for 24 hr, followed by 20% sucrose. The left halves of the brains were used for protein analyses. Sagittal brain sections were cut on a cryostat (Microm). The thicknesses of serial sections were 14 μm or 50 μm (used for Figures 2C, S1G–S1I, and 5A–5F) according to different aims in this study. The 14-μm brain sections were bonded on glass slides for regular immunostaining, whereas the 50-μm sections were used as free-floating sections. Doublecortin (DCX; 1:500; #4604; RRID: AB_561007) was purchased from Cell Signaling; NeuN (1:500; MAB377; RRID: AB_2298772), parvalbumin (1:1,000; #MAB1572; RRID:AB_2174013), and Reelin antibody (1:300; #MAB5364; RRID: AB_2179313) were purchased from EMD Millipore; Prox1 (1:100; #ab101851; RRID: AB_10712211) and BrdU (1:250; ab6326; RRID: AB_305426) antibodies were from Abcam; GFAP (1:1,000; SMI22R; RRID: AB_509980) antibody was from Covance; and VGlut1 (1:5; #73-066, RRID: AB_10673111) antibody was from NeuroMab. The sections were incubated with primary antibody at 4°C overnight. After washing with PBS three times, sections were incubated with the secondary antibody goat anti-mouse or anti-rabbit immunoglobulin G conjugated with Alexa Fluor 488 or 568 (Molecular Probes). All images were captured with a Leica SP5 confocal microscope.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 4.0 (GraphPad Software). All data were analyzed for statistical significance using an F test for equal variance followed by a two-tailed Student's t test or a two-way ANOVA. Significant p values are denoted by the use of asterisks in the figures (∗p < 0.05, ∗∗p < 0.01). All data values are expressed as mean ± SEM.
Author Contributions
H.H. contributed to the results, experimental design, and discussions; Q.F. and W.H. contributed to the experimental results; H.S. contributed to the discussion during this study; X.H. contributed to the results and discussion; R.Y. contributed to the experimental design and writing of the manuscript.
Acknowledgments
This study was supported by grants from the NIH to R.Y. (NS074256 and AG046929). We thank Dr. Chris Nelson for critical reading of the manuscript.
Published: June 29, 2017
Footnotes
Supplemental Information includes one figure, one table, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.05.030.
Supplemental Information
{kind=link}
References
- Amaral D.G., Scharfman H.E., Lavenex P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies) Prog. Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barao S., Moechars D., Lichtenthaler S.F., De Strooper B. BACE1 physiological functions may limit its use as therapeutic target for Alzheimer's disease. Trends Neurosci. 2016;39:158–169. doi: 10.1016/j.tins.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Brown J.P., Couillard-Despres S., Cooper-Kuhn C.M., Winkler J., Aigner L., Kuhn H.G. Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Cai H., Wang Y., McCarthy D., Wen H., Borchelt D.R., Price D.L., Wong P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chen Y., Sharma R.P., Costa R.H., Costa E., Grayson D.R. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30:2930–2939. doi: 10.1093/nar/gkf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Huang X., Zhang Y.W., Rockenstein E., Bu G., Golde T.E., Masliah E., Xu H. Alzheimer's beta-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of beta-amyloid. J. Neurosci. 2012;32:11390–11395. doi: 10.1523/JNEUROSCI.0757-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K.O., Lybrand Z.R., Ito N., Brulet R., Tafacory F., Zhang L., Good L., Ure K., Kernie S.G., Birnbaum S.G. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 2015;6:6606. doi: 10.1038/ncomms7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford I.L., Connor J.D. Localization and release of glutamic acid in relation to the hippocampal mossy fibre pathway. Nature. 1973;244:442–443. doi: 10.1038/244442a0. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G., Miao G.G., Chen S.C., Soares H.D., Morgan J.I., Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Doetsch F. The glial identity of neural stem cells. Nat. Neurosci. 2003;6:1127–1134. doi: 10.1038/nn1144. [DOI] [PubMed] [Google Scholar]
- Dupuy S.T., Houser C.R. Prominent expression of two forms of glutamate decarboxylase in the embryonic and early postnatal rat hippocampal formation. J. Neurosci. 1996;16:6919–6932. doi: 10.1523/JNEUROSCI.16-21-06919.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck D., van B.F., Colombo A., Galante C., Schwenk B.M., Rabe L., Hampel H., Novak B., Kremmer E., Tahirovic S. Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J. Neurosci. 2013;33:7856–7869. doi: 10.1523/JNEUROSCI.3372-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster E., Bock H.H., Herz J., Chai X., Frotscher M., Zhao S. Emerging topics in Reelin function. Eur. J. Neurosci. 2010;31:1511–1518. doi: 10.1111/j.1460-9568.2010.07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis F., Koulakoff A., Boucher D., Chafey P., Schaar B., Vinet M.C., Friocourt G., McDonnell N., Reiner O., Kahn A. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Goldman J.E. Lineage, migration, and fate determination of postnatal subventricular zone cells in the mammalian CNS. J. Neurooncol. 1995;24:61–64. doi: 10.1007/BF01052660. [DOI] [PubMed] [Google Scholar]
- He W., Hu J., Xia Y., Yan R. beta-site amyloid precursor protein cleaving enzyme 1(BACE1) regulates Notch signaling by controlling the cleavage of Jagged 1 (Jag1) and Jagged 2 (Jag2) proteins. J. Biol. Chem. 2014;289:20630–20637. doi: 10.1074/jbc.M114.579862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt B., Riordan S.M., Kukreja L., Eimer W.A., Rajapaksha T.W., Vassar R. beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 2012;287:38408–38425. doi: 10.1074/jbc.M112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge R.D., Garcia A.J., III, Elsen G.E., Nelson B.R., Mussar K.E., Reiner S.L., Ramirez J.M., Hevner R.F. Tbr2 expression in Cajal-Retzius cells and intermediate neuronal progenitors is required for morphogenesis of the dentate gyrus. J. Neurosci. 2013;33:4165–4180. doi: 10.1523/JNEUROSCI.4185-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Hicks C.W., He W., Wong P., Macklin W.B., Trapp B.D., Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu X., He W., Diaconu C., Tang X., Kidd G.J., Macklin W.B., Trapp B.D., Yan R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008;22:2970–2980. doi: 10.1096/fj.08-106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., He W., Luo X., Tsubota K.E., Yan R. BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep. 2013;4:40–49. doi: 10.1016/j.celrep.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Fan Q., Hou H., Yan R. Neurological dysfunctions associated with altered BACE1-dependent Neuregulin-1 signaling. J. Neurochem. 2015;136:234–249. doi: 10.1111/jnc.13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain I., Powell D., Howlett D.R., Tew D.G., Meek T.D., Chapman C., Gloger I.S., Murphy K.E., Southan C.D., Ryan D.M. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Huth T., Rittger A., Saftig P., Alzheimer C. beta-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel beta4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflugers Arch. 2011;461:355–371. doi: 10.1007/s00424-010-0913-2. [DOI] [PubMed] [Google Scholar]
- Jessberger S., Parent J.M. Epilepsy and adult neurogenesis. Cold Spring Harb. Perspect. Biol. 2015;7 doi: 10.1101/cshperspect.a020677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Jossin Y., Gui L., Goffinet A.M. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007;27:4243–4252. doi: 10.1523/JNEUROSCI.0023-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T., Fujiyama F. Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci. Res. 2002;42:243–250. doi: 10.1016/s0168-0102(02)00009-3. [DOI] [PubMed] [Google Scholar]
- Kim D.Y., Ingano L.A., Carey B.W., Pettingell W.H., Kovacs D.M. Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J. Biol. Chem. 2005;280:23251–23261. doi: 10.1074/jbc.M412938200. [DOI] [PubMed] [Google Scholar]
- Kubo K., Honda T., Tomita K., Sekine K., Ishii K., Uto A., Kobayashi K., Tabata H., Nakajima K. Ectopic Reelin induces neuronal aggregation with a normal birthdate-dependent “inside-out” alignment in the developing neocortex. J. Neurosci. 2010;30:10953–10966. doi: 10.1523/JNEUROSCI.0486-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn P.H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31:3157–3168. doi: 10.1038/emboj.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert de R.C., de Bergeyck V., Cortvrindt C., Bar I., Eeckhout Y., Goffinet A.M. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp. Neurol. 1999;156:214–217. doi: 10.1006/exnr.1998.7007. [DOI] [PubMed] [Google Scholar]
- Lavado A., Oliver G. Prox1 expression patterns in the developing and adult murine brain. Dev. Dyn. 2007;236:518–524. doi: 10.1002/dvdy.21024. [DOI] [PubMed] [Google Scholar]
- Li G., Pleasure S.J. Genetic regulation of dentate gyrus morphogenesis. Prog. Brain Res. 2007;163:143–152. doi: 10.1016/S0079-6123(07)63008-8. [DOI] [PubMed] [Google Scholar]
- Li G., Kataoka H., Coughlin S.R., Pleasure S.J. Identification of a transient subpial neurogenic zone in the developing dentate gyrus and its regulation by Cxcl12 and reelin signaling. Development. 2009;136:327–335. doi: 10.1242/dev.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Koelsch G., Wu S., Downs D., Dashti A., Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Bolon B., Kahn S., Bennett B.D., Babu-Khan S., Denis P., Fan W., Kha H., Zhang J., Gong Y. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Luo X., Prior M., He W., Hu X., Tang X., Sheng W., Yadav S., Kiryu-Seo S., Miller R., Trapp B.D., Yan R. Cleavage of neuregulin-1 by BACE1 or ADAM10 produces differential effects on myelination. J. Biol. Chem. 2011;286:23967–23974. doi: 10.1074/jbc.M111.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magavi S.S., Leavitt B.R., Macklis J.D. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- Malatesta P., Appolloni I., Calzolari F. Radial glia and neural stem cells. Cell Tissue Res. 2008;331:165–178. doi: 10.1007/s00441-007-0481-8. [DOI] [PubMed] [Google Scholar]
- Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Pesold C., Impagnatiello F., Pisu M.G., Uzunov D.P., Costa E., Guidotti A., Caruncho H.J. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigoni M., Wanngren J., Kuhn P.H., Munro K.M., Gunnersen J.M., Takeshima H., Feederle R., Voytyuk I., De Strooper B., Levasseur M.D. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol. Neurodegener. 2016;11:67. doi: 10.1186/s13024-016-0134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujadas L., Gruart A., Bosch C., Delgado L., Teixeira C.M., Rossi D., de L.L., Martinez A., Delgado-Garcia J.M., Soriano E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010;30:4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberds S.L., Anderson J., Basi G., Bienkowski M.J., Branstetter D.G., Chen K.S., Freedman S.B., Frigon N.L., Games D., Hu K. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum. Mol. Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Anderson J.P., Barbour R., Basi G.S., Caccavello R., Davis D., Doan M., Dovey H.F., Frigon N., Hong J. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Stanfield B.B., Cowan W.M. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 1979;185:393–422. doi: 10.1002/cne.901850302. [DOI] [PubMed] [Google Scholar]
- Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer's disease. Alzheimers Res. Ther. 2014;6:89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Vassar R., Kuhn P.H., Haass C., Kennedy M.E., Rajendran L., Wong P.C., Lichtenthaler S.F. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 2014;130:4–28. doi: 10.1111/jnc.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen Und H.O. Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res. 2011;345:1–19. doi: 10.1007/s00441-011-1196-4. [DOI] [PubMed] [Google Scholar]
- Willem M., Garratt A.N., Novak B., Citron M., Kaufmann S., Rittger A., DeStrooper B., Saftig P., Birchmeier C., Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wong H.K., Sakurai T., Oyama F., Kaneko K., Wada K., Miyazaki H., Kurosawa M., De Strooper B., Saftig P., Nukina N. beta subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J. Biol. Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Yan R. Stepping closer to treating Alzheimer's disease patients with BACE1 inhibitor drugs. Transl. Neurodegener. 2016;5:13. doi: 10.1186/s40035-016-0061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13:319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Bienkowski M.J., Shuck M.E., Miao H., Tory M.C., Pauley A.M., Brashier J.R., Stratman N.C., Mathews W.R., Buhl A.E. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yan R., Han P., Miao H., Greengard P., Xu H. The transmembrane domain of the Alzheimer's beta-secretase (BACE1) determines its late Golgi localization and access to beta -amyloid precursor protein (APP) substrate. J. Biol. Chem. 2001;276:36788–36796. doi: 10.1074/jbc.M104350200. [DOI] [PubMed] [Google Scholar]
- Yan R., Fan Q., Zhou J., Vassar R. Inhibiting BACE1 to reverse synaptic dysfunctions in Alzheimer's disease. Neurosci. Biobehav. Rev. 2016;65:326–340. doi: 10.1016/j.neubiorev.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.X., Marchetto M.C., Gage F.H. How to make a hippocampal dentate gyrus granule neuron. Development. 2014;141:2366–2375. doi: 10.1242/dev.096776. [DOI] [PubMed] [Google Scholar]
- Zhou L., Barao S., Laga M., Bockstael K., Borgers M., Gijsen H., Annaert W., Moechars D., Mercken M., Gevaert K. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 2012;287:25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.