

Abstract
To shed light on the genetic background behind the virulence and salt tolerance of Staphylococcus equorum, we performed comparative genome analysis of six S. equorum strains. Data on four previously published genome sequences were obtained from the NCBI database, while those on strain KM1031 displaying resistance to multiple antibiotics and strain C2014 causing haemolysis were determined in this study. Examination of the pan-genome of five of the six S. equorum strains showed that the conserved core genome retained the genes for general physiological processes and survival of the species. In this comparative genomic analysis, the factors that distinguish the strains from each other, including acquired genomic factors in mobile elements, were identified. Additionally, the high salt tolerance of strains enabling growth at a NaCl concentration of 25% (w/v) was attributed to the genes encoding potassium voltage-gated channels. Among the six strains, KS1039 does not possess any of the functional virulence determinants expressed in the other strains.
Introduction
Staphylococcus equorum, initially isolated from a healthy horse1, is a common component of the microbiota in the fermented foods of Europe, including fermented meat products2–4 and smear-ripened and semi-hard cheeses5, 6. This species has been reported to produce low-molecular-weight aromatic compounds, such as esters, amino acids, aldehydes and free fatty acids, in food fermentation7, 8. Recently, S. equorum has also been identified as the dominant species of jeotgal, a high-salt-fermented seafood of Korea9.
Coagulase-negative staphylococci (CNS) including S. equorum are generally known as benign bacteria, in contrast to the coagulase-producing Staphylococcus aureus. No staphylococcal food poisoning via fermented foods has been attributed to S. equorum, and no evidence of its pathogenicity has been reported. However, the emergence of strains suspected of being involved in bovine mastitis10, showing high prevalence of acquired phenotypes including antibiotic resistance and haemolysis11–13, has necessitated safety assessments of this species. In this context, safety assessments of starter candidates for traditional Spanish dry-cured sausages, dairy products, jeotgal and fermented meat products have been performed12–15.
Analyses of the genome sequences of three S. equorum strains have led to reports that the strains do not possess any of the virulence factors found in S. aureus 10, 16, 17. However, the genomic data are insufficient to identify the features of strains from different niches and those suspected of being pathogenic. In the current study, we thus extended the analyses to the genomes of two S. equorum strains showing resistance to multiple antibiotics or causing haemolysis. We also performed a comparative genomic analysis of S. equorum strains together with the four previously reported genomes to define the scale and scope of the pan-genome and the core genes and clarify the genetic background behind such antibiotic resistance and haemolysis. This study introduced intraspecific comprehensive comparative genome analysis to shed light on the genetic background behind the phenotypic characteristics.
Methods
Bacterial strains and culture conditions
Two S. equorum strains, KM1031 and C2014 isolated and characterized in our previous studies9, 13, were subjected to genomic analysis. Strain KM1031 isolated from Myeolchi-jeotgal, a Korean high-salt-fermented anchovy, exhibited resistance to chloramphenicol, erythromycin, lincomycin and penicillin G; strain C2014 from Saeu-jeotgal, a Korean high-salt-fermented tiny sea shrimp, induced haemolysis on sheep blood-supplemented tryptic soy agar (TSA; Difco, Detroit, MI, USA) plates. For experimental proof of the genomic analysis results, strains KS1039 from Saeu-jeotgal16, Mu2 from French smear-ripened cheese17, and UMC-CNS-924 from bovine mastitis10 were used. S. equorum subsp. linens separated from S. equorum in 20036 was reunified into a species in 201318. S. equorum strains were cultured in tryptic soy broth (TSB; Difco) at 30 °C for 24 h to maintain their traits13. Escherichia coli BL21(DE3) was used as the cloning host and was incubated in Luria–Bertani (LB) medium (Difco) at 37 °C for 12 h.
Genome sequencing
Genomic DNA was isolated and purified using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The concentration and purity of the extracted DNA were determined using a Quanti-iT PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA), and the contamination of DNA was checked by sequencing the 16 S rRNA gene using an ABI 3730 DNA sequencing machine (Applied Biosystems, Foster City, CA, USA). Whole-genome sequencing was performed using a combination of the Illumina Miseq system (150 bp paired end) and the PacBio Single-Molecule Real-Time (SMRT) sequencing system (20 kbp) at ChunLab, Inc. (Seoul, South Korea). Four and six contigs were generated from a hybrid assembly of reads from the Illumina system (4,663,200 reads and >341.43 coverage for KM1031 strain; 3,873,510 reads and >287.08 coverage for C2014 strain) and PacBio system (75,778 reads and >400.53 coverage for KM1031 strain; 71,229 reads and >389.10 coverage for C2014 strain) for S. equorum strains KM1031 and C2014, respectively. The reads were assembled using CLC Genomics Workbench ver. 7.5.1 (CLC Bio, Aarhus, Denmark) and CodonCode Aligner (CodonCode Co., Centerville, MA, USA). Gene prediction was performed using Glimmer 319, followed by annotation through a search against the Clusters of Orthologous Groups (COG)20 and SEED databases21.
Comparative genomics
For comparative genomic analysis within the species S. equorum, the genome sequence data of strains KS1039 (GenBank accession: CP013114.1), Mu2 (CAJL00000000.1), UMC-CNS-924 (AVBD00000000.1) and G8HB1 (LAKE00000000.1) published before July 2016 were obtained from the NCBI database (http://ncbi.nlm.nih.gov/genomes). The genome sequence data of 15 strains published after September 2016 were also used for additional comparative genomic analysis22. The average nucleotide identity (ANI), which provides a robust measurement of genetic distance among bacterial genomes, among the conserved genes of the genomes was used for comparative analysis23. To determine the rearrangements in each genome, a progressive alignment algorithm implemented in MAUVE24 was used. The Efficient Database framework for comparative Genome Analyses using BLAST score Ratios (EDGAR) was used for core genome, pan-genome and singleton analyses25; the genome of strain KS1039 was used as a reference genome for Venn diagram construction. Comparative analyses at the protein level were performed by an all-against-all comparison of the annotated genomes. The algorithm used was BLASTP and were normalized according to the best score26. The score ratio value, which shows the quality of the hit, was calculated by dividing the scores of further hits by the best hit27. Two genes were considered orthologous when revealing a bidirectional best BLAST hit with a single score ratio value threshold of at least 32% for orthology estimation.
Haemolysis activity test
TSA supplemented with 5% sheep blood (v/v) (BBL Microbiology Systems, Sparks, MD, USA) was used for β- and δ-haemolytic activity tests. β-Haemolytic activity was determined by cold shock at 4 °C for 24 h after incubation at 30 °C for 24 h; δ-haemolytic activity was determined by cross-streaking the test strains perpendicularly to S. aureus RN4220 at 30 °C for 24 h. The same method was implemented in the selection of S. equorum strains having β- and δ-haemolytic activities13. Experiments were conducted three times, on separate days.
Growth monitoring in the presence of antibiotics and NaCl
S. equorum strains cultured in TSB were normalized to 0.5 turbidity at OD600 and then diluted 1:100 in TSB supplemented with antibiotics to confirm the function of annotated antibiotic resistance genes. The antibiotics chloramphenicol, ciprofloxacin, erythromycin, lincomycin, methicillin, penicillin G and tetracycline were purchased from Sigma (St. Louis, MO, USA) and employed at concentrations of 30, 5, 15, 30, 8, 6 and 30 μg/ml, respectively, based on previous research results11, 13 and the guidelines of methicillin for S. aureus set out by the Clinical and Laboratory Standards Institute28. The salt tolerance of S. equorum strains was determined by examining their growth in TSB supplemented with NaCl at concentrations of 15% (w/v), 20% and 25%. To determine the salt tolerance of E. coli transformants, NaCl was employed at a final concentration of 3% or 6% in LB broth. Cell growth was monitored by measuring OD600 using a Varioskan Flash (Thermo Scientific, Waltham, MA, USA).
Cloning of the potassium voltage-gated channel genes
The potassium voltage-gated channel genes of strains KM1031 and C2014 were amplified with specific primer sets (Supplementary Table S1) designed from their genome sequences. PCR amplifications were performed using a T-3000 thermocycler (Biometra, Göttingen, Germany) and the PCR mixture consisted of the template DNA, 0.5 μM of each primer, 1.25 units of Inclone Taq polymerase (Inclone Biotech, Daejeon, South Korea), 10 mM dNTPs and 2 mM MgCl2. Samples were preheated for 5 min at 95 °C and then amplified using 30 cycles of 1 min at 95 °C, 30 s at 55 °C and 1 min at 72 °C. The amplified PCR products were cloned into pGEM-T Easy vector (Promega, Madison, WI, USA) under the control of the T7 promoter. The successful integration of the cloned fragments was confirmed by sequencing using GenoTech (Daejeon, South Korea) with the T7 primer set.
Nucleotide sequence accession numbers
The complete genome sequences and annotation data of S. equorum strains KM1031 and C2014 have been deposited in GenBank under the accession numbers CP013980–CP013983 and CP013714–CP013719, respectively.
Results and Discussion
Genome summary and general features
The general features of the genomes of the six S. equorum strains including strains KM1031 and C2014 are summarized in Table 1. The genome (2,792,213 bp) of S. equorum KM1031 consists of a single circular DNA chromosome of 2,693,398 bp with G + C content of 33.1% and three plasmids. The genome contains 2,642 predicted open reading frames (ORFs), 60 tRNAs and 22 rRNAs. The genome (2,930,519 bp) of S. equorum C2014 consists of a single circular DNA chromosome of 2,753,539 bp with G + C content of 32.9% and five plasmids. The genome contains 2,846 predicted ORFs, 59 tRNAs and 22 rRNAs. In total, 2,295 and 2,431 protein-coding sequences (CDSs) were predicted from the genome sequences of strains KM1031 and C2014 containing 2,642 and 2,846 ORFs, respectively, with 86.9% and 85.4% being assigned a COG functional classification.
Table 1.
General genomic and specific phenotypic features of six Staphylococcus equorum strains.
| Feature | KM1031 | C2014 | KS1039 | Mu2 | UMC-CNS-924 | G8HB1 |
|---|---|---|---|---|---|---|
| Size (bp) | 2,792,213 | 2,930,519 | 2,822,193 | 2,927,171 | 2,700,865 | 2,799,869 |
| Chromosome size (bp) | 2,693,398 | 2,753,539 | 2,822,193 | ND | ND | ND |
| G + C content (%) | 33.05 | 32.85 | 33.07 | 32.80 | 32.96 | 33.08 |
| No. of plasmids | 3a | 5b | 0 | ND | 4 | ND |
| Open reading frames | 2,642 | 2,846 | 2,681 | 2,745 | 2,499 | 2,621 |
| CDSs assigned by COG | 2,295 | 2,431 | 2,363 | 2,469 | 2,332 | 2,408 |
| CDSs assigned by SEED | 1,990 | 2,009 | 2,009 | 2,045 | 1,940 | 1,994 |
| No. of rRNAs | 22 | 22 | 22 | 4 | 24 | 8 |
| No. of tRNAs | 60 | 59 | 61 | 55 | 57 | 54 |
| Contigs | 4 | 6 | 1 | 30 | 39 | 22 |
| Scaffolds | 0 | 0 | 0 | 30 | 39 | 22 |
| Origin | Myeolchi-jeotgal | Saeu-jeotgal | Saeu-jeotgal | French smear-ripened cheese | Milk from Holstein cow | Human gall bladder |
| Specific phenotypic featuresc | ||||||
| Antibiotic resistance | Chlr, Eryr, Linr, Penr | − | − | − | Linr, Tetr | NT |
| Haemolysis | − | β-Haemolysis | − | − | δ-Haemolysis | NT |
| Growth on 25% NaCl | − | + | + | − | – | NT |
aPlasmids in the strain KM1031: pKM1031-1, 45.9 kb; pKM1031-2, 50.2 kb; and pSELNU3, 2.6 kb. bPlasmids in the strain C2014: pC2014-1, 80.3 kb; pC2014-2, 64.4 kb; pC2014-3, 13.0 kb; pC2014-4, 7.3 kb; and pC2014-5, 12.0 kb. cPhenotypic characteristics were reconfirmed in this study, except for strain G8HB1. Abbreviations: Chl, chloramphenicol; Ery, erythromycin; Lin, lincomycin; Pen, penicillin G; Tet, tetracycline; ND, not determined by the authors; NT, not tested in this study; +, positive; −, negative.
The average genome sequence length of the six strains is 2,828,805 bp. S. equorum UMC-CNS-924 exhibits the smallest genome (2,700,865 bp), while strain C2014 possesses the largest one (2,930,519 bp). All S. equorum strains display average G + C content of 33%.
To facilitate a coherent comparative analysis, we performed consistent ORF prediction for the six S. equorum (complete and incomplete) genome sequences. In this way, comparable numbers of genes were obtained for each genome, with an average of 2,672 ORFs per genome (Table 1). Notably, (BLAST-based) functional in silico prediction could be performed for 89.2% of the identified ORFs, while the remaining 10.8% not assigned a COG functional classification were predicted to encode hypothetical proteins.
Analysis using the SEED subsystem categorization and COG functional categorization predicted the existence of an average of 1,998 CDSs and 2,383 CDSs per genome, respectively (Table 1). Based on the SEED subsystem, over 321 CDSs accounting for 15.8–16.8% of the S. equorum genomes were allocated to the genes for amino acid biosynthesis and utilization (Fig. 1). The next most abundant subsystem category is related to carbohydrate utilization (15.5–16.7%), followed by protein metabolism. Major COG subsystems are related to amino acid transport and metabolism, as well as carbohydrate transport and metabolism. Both analyses enabled coherent conclusions to be drawn regarding the subsystem category of S. equorum.
Figure 1.
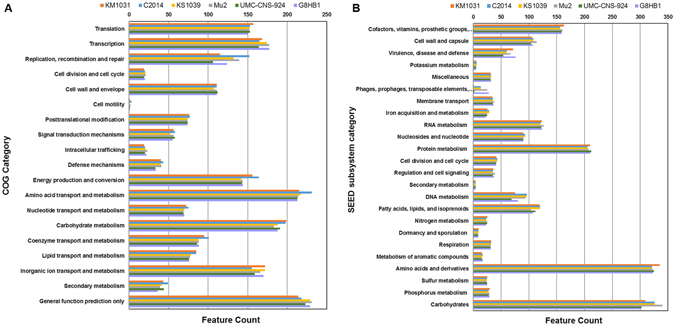
Comparison of functional categories in six S. equorum genomes based on COG (A) and SEED (B). Genome sequences of six strains KM1031, C2014, KS1039, Mu2, UMC-CNS-924 and G8HB1 were uploaded to the COG and SEED viewer servers independently. Functional roles of annotated genes were assigned and grouped in subsystem feature categories. Coloured bars indicate the number of genes assigned to each category.
Comparative analysis of S. equorum genomes
Whole-genome comparison of the six S. equorum strains showed that the genomes are highly homologous in terms of functional category (Fig. 1). MAUVE alignment of the six genomes allowed the identification of approximately 8–10 locally collinear blocks (LCBs), regions without rearrangement of the homologous backbone sequence (Supplementary Fig. S1). However, the LCBs are interspaced by specific DNA stretches of various lengths. MAUVE analysis showed an overall collinear relationship across S. equorum strains KM1031, C2014 and KS1039, which were isolated from different jeotgal samples9, 13. When the genome of strain KS1039 was established as the standard, a large-scale chromosomal reorganization by a single recombination event was found to have occurred in the genome of strain G8HB1, which resulted in the inversion of a genomic region; in addition, the results indicated that the genomes of strains Mu2 and UMC-CNS-924 might have been generated by complex rearrangements. The collinear relationship and complex rearrangement found in the six S. equorum genome structures can be explained by their differences in isolation source and geographic location. However, the contigs in genomic data for strains Mu2, UMC-CNS-924 and G8HB1 might have distorted the MAUVE analysis results.
The gene pools shared by the genomes of the five S. equorum strains KM1031, C2014, KS1039, Mu2 and UMC-CNS-924 are depicted in a Venn diagram (Fig. 2). These five strains share 2,166 CDSs in their core genome, corresponding to approximately 76.1–86.7% of their ORFs. Many of the CDSs in the core genome are assigned via COG annotation to functions relating to metabolism and the transport of amino acids and carbohydrates. The genome of strain UMC-CNS-924 has the smallest proportion (2.0%) of unique CDSs that are absent from the four other S. equorum genomes. In contrast, the proportions of unique CDSs in the genomes of strains KM1031, C2014, KS1039 and Mu2 are 4.5%, 10.9%, 6.0% and 8.1%, respectively. The majority of singleton-specific genes are associated with hypothetical proteins (Supplementary Table S2). Meanwhile, functional singletons in the genomes of strains KM1031, C2014, KS1039, UMC-CNS-924 and Mu2 are allocated to transporter, transposase, CRISPR-associated protein, tetracycline resistance and phage-related genes, respectively.
Figure 2.
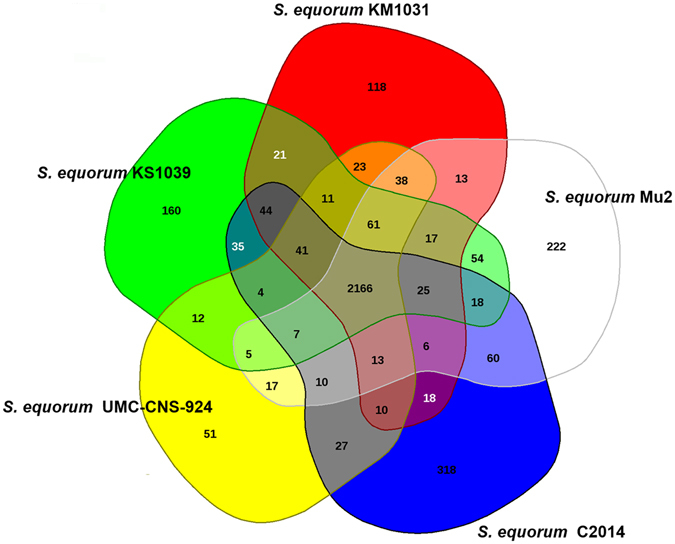
Venn diagram of five S. equorum genomes. The Venn diagram shows the pan-genome of strains KM1031, C2014, KS1039, Mu2 and UMC-CNS-924 generated using EDGAR. Overlapping regions represent common CDSs shared between the S. equorum genomes. The numbers outside the overlapping regions indicate the numbers of CDSs in each genome without homologs in the other sequenced S. equorum genomes.
Insights into virulence
The well-known food pathogen S. aureus produces several virulence factors: a clumping factor to protect against phagocytosis, an extracellular adhesion protein for adhesion, an enterotoxin, a toxic shock syndrome toxin, and cytotoxins such as α- and β-haemolysins for tissue invasion29. However, genomic analysis revealed that the six S. equorum strains do not possess any of the virulence determinants for adhesion, enterotoxins and pathogenicity islands that are found in S. aureus. Nonetheless, strain C2014 induced haemolysis on sheep blood-supplemented agar and strain KM1031 displayed resistance to chloramphenicol, erythromycin, lincomycin and penicillin G13; therefore, we focused on genetic analysis to explain the differences in phenotype among the S. equorum strains.
Haemolysis
The α- and β-haemolysins are prevailing toxins of S. aureus, but their homologs were not identified in any of the six S. equorum genomes. However, three CDSs annotated as haemolysin, haemolysin III and haemolysin activation protein genes were identified in all of the genomes of five S. equorum strains, excluding strain KS1039 (Table 2). Strain KS1039 does not possess a predicted haemolysin gene. These three CDSs also exist in all of 15 S. equorum strains from cheeses (Supplementary Table S3).
Table 2.
Potential virulence determinants identified in six S. equorum genomes.
| Virulence factor | Gene locus | |||||
|---|---|---|---|---|---|---|
| KM1031 | C2014 | KS1039 | Mu2 | UMC-CNS-924 | G8HB1 | |
| Haemolysis-related | ||||||
| Haemolysin | AWC34_RS03405 | AVJ22_RS03205 | SEQMU2_RS08595 | SEQU_RS25035 | UF72_RS01895 | |
| Haemolysin III | AWC34_RS09060 | AVJ22_RS09420 | SE1039_RS09485 | SEQMU2_RS01345 | SEQU_RS26575 | UF72_RS08525 |
| Haemolysin activation protein | AWC34_RS11995 | AVJ22_RS12370 | SE1039_RS12450 | SEQMU2_RS04245 | SEQU_RS16015 | UF72_RS08300 |
| SEQU_RS16020 | ||||||
| SEQU_RS16025 | ||||||
| Haemolysin family calcium-binding region | AVJ22_RS14095* | |||||
| Antibiotic resistance | ||||||
| Efflux pump | ||||||
| Chloramphenicol resistance protein DHA1 | AWC34_RS10365 | AVJ22_RS10770 | SE1039_RS10790 | SEQMU2_RS02670 | SEQU_RS15060 | UF72_RS09825 |
| Lincomycin resistance protein LmrB | AWC34_RS12700 | AVJ22_RS13165 | SEQMU2_RS05490 | SEQU_RS20895 | UF72_RS07545 | |
| Quinolone resistance protein NorB | AWC34_RS00175 | AVJ22_RS00115 | SE1039_RS00130 | SEQMU2_RS05750 | SEQU_RS20615 | UF72_RS07265 |
| Quinolone resistance protein NorB | AWC34_RS10900 | AVJ22_RS13260 | SE1039_RS11310 | SEQMU2_RS03165 | SEQU_RS23900 | UF72_RS10355 |
| Multidrug resistance protein SepA | AWC34_RS09050 | AVJ22_RS09410 | SE1039_RS09475 | SEQMU2_RS01335 | SEQU_RS26585 | UF72_RS08515 |
| Multidrug resistance protein SMR | AWC34_RS00690 | AVJ22_RS00690 | SE1039_RS00770 | SEQMU2_RS06295 | SEQU_RS23055 | UF72_RS06755 |
| Antibiotic ABC transporter ATP-binding protein | AWC34_RS11115 | SEQU_RS24115 | UF72_RS10570 | |||
| Tetracycline resistance MFS efflux pump | SEQU_RS26615* | |||||
| Enzymatic inactivation | ||||||
| Methicillin resistance protein | AWC34_RS05640 | AVJ22_RS05905 | SE1039_RS06065 | SEQMU2_RS11115 | SEQU_RS17120 | UF72_RS04430 |
| Methicillin resistance protein | AWC34_RS05645 | AVJ22_RS05910 | SE1039_RS06070 | SEQMU2_RS11120 | SEQU_RS17125 | UF72_RS04435 |
| Methicillin resistance protein FemA | AWC34_RS11375 | AVJ22_RS11680 | SE1039_RS11790 | SEQMU2_RS03635 | SEQU_RS17305 | UF72_RS10830 |
| β-Lactamase | AWC34_RS13020* | AVJ22_RS12330 | SEQMU2_RS04220 | SEQU_RS22365 | UF72_RS12370 | |
| Lincomycin resistance protein LnuA | AWC34_RS13300* | SEQU_RS26665* | ||||
| Other | ||||||
| Antibiotic biosynthesis monooxygenase | AWC34_RS01805 | |||||
*Genes located in a plasmid.
The deduced amino acid sequence of annotated haemolysin gene (AVJ22-RS03205) from strain C2014 has 98% identity with the TlyC amino acid sequence of Streptococcus equi. TlyC was described as a haemolysin in Brachyspira hyodysenteriae and was considered to be a virulence factor contributing to the disease caused by this spirochete because tlyC-expressing E. coli displayed haemolytic activity in vitro 30. Meanwhile, Carvalho et al.31 concluded that TlyC of Leptospira does not exert a direct haemolytic effect but may contribute to Leptospira binding to the extracellular matrix during host infection. In addition, Turner and Helmann29 reported that YhdP (TlyC) in Bacillus subtilis is a multidrug efflux protein and its amino acid sequence has 67% similarity with that of a membrane protein in Bacillus isronensis. These results suggest that the haemolysin homolog may not contribute to haemolysin activity directly but may instead be a membrane protein or enhance haemolytic activity.
The integral membrane protein gene hlyIII was identified from Bacillus cereus and Vibrio vulnificus; its involvement in haemolysis was verified by its expression in a nonhaemolytic E. coli strain30, 32. However, its homolog found in Bacteroides fragilis was not linked to haemolytic activity33. Among the S. equorum strains tested for haemolytic activity, only strain C2014 exhibited β-haemolytic activity, despite all strains having the haemolysin III gene, which means that this gene is not an independent determinant of β-haemolysis.
All of the six S. equorum strains possess a gene encoding a putative haemolysin activation protein composed of 43 amino acids. However, only strain UMC-CNS-924 has three genes encoding three distinct haemolysin activation proteins under the control of a promoter (Table 2) and exhibited δ-haemolysis (Supplementary Fig. S2B). The three putative haemolysin activation proteins presented ≤45.5% amino acid sequence identities with each other and ≤37.2% sequence identity with that of strain C2014 (Supplementary Table S4). Staphylococcus lugdunensis was reported to secrete three 43-amino-acid peptides with synergistic haemolytic activity, phenotypically similar to the δ-haemolysin of S. aureus, and their genes are located in an operon30. The identification of δ-haemolysin activity only in strain UMC-CNS-924 may be attributable to these three genes, while their homologs found in the other five strains may contribute to other functions.
Among the strains tested for haemolytic activity, only strain C2014 exhibited β-haemolytic activity (Supplementary Fig. S2A). Comparative genomic analysis revealed an additional CDS only found in strain C2014, a putative haemolysin family calcium-binding region gene (AVJ22_RS14095) (Table 2) and a unique gene harboured in the plasmid named pC2014-5 (12.0 kb). The product of this gene was reported to induce stabilization of the β-sheet structure of haemolysin and be enhanced by Ca2+ binding34. Therefore, we assumed that the expression of the gene AVJ22_RS14095 may enhance the haemolytic activity of other gene products that cannot induce haemolysis on their own. Based on the SEED subsystem, the genes AVJ22_RS03205 and AVJ22_RS09420 in strain C2014 were classified as encoding magnesium/cobalt efflux protein and the membrane protein haemolysin III, respectively. Thus, we cautiously suggest that the gene AVJ22_RS09420 might be a determinant of the haemolysis activity, with the support of AVJ22_RS14095, in strain C2014.
Acquired antibiotic resistance
Based on the functional categories as determined by the SEED subsystem, an average of 65.5% of the annotated CDSs in the virulence, disease and defence category for the six S. equorum strains are predicted to be genes for resistance to antibiotics and toxic compounds (Fig. 1B). Meanwhile, the genes for resistance to antibiotics and toxic compounds in strains KM1031 and G8HB1 constitute 71.8% and 73.7% of the CDSs in this category, respectively.
Putative efflux pump genes for chloramphenicol, lincomycin, quinolone and multiple drugs were identified across the six S. equorum chromosomes as well as those of 15 S. equorum strains from cheeses (Table 2, Supplementary Table S3). KM1039 is the only strain that does not harbour the putative lincomycin-resistance gene lmrB. Conversely, the phenotype of lincomycin resistance was exhibited in strains KM1031 and UMC-CNS-924 (Fig. 3). The lincomycin-resistant S. equorum KM1031 harbours a plasmid encoding the lnuA gene, which it can transfer to Gram-positive recipients11. Strain UMC-CNS-924 also harbours the lnuA-encoding plasmid. The lincomycin resistance of both strains might thus have been acquired via horizontal transfer of the resistant plasmid. The six commonly identified putative efflux pump genes including the lmrB homolog may thus not function in lincomycin resistance.
Figure 3.
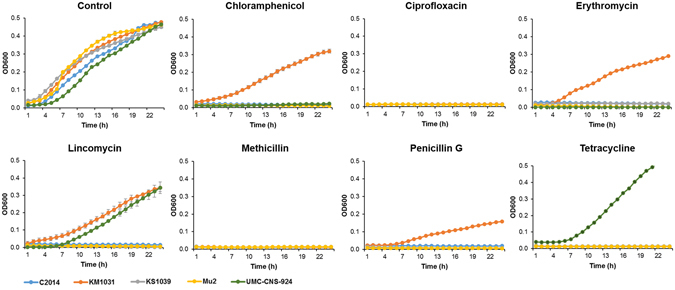
Growth of five S. equorum strains in the presence of antibiotics.
Resistance to ciprofloxacin, a kind of quinolone, was not identified in any of the test strains. In our previous antibiotic resistance test of 126 S. equorum strains, two strains showed independent ciprofloxacin and ofloxacin resistance and both strains also exhibited multidrug resistance14. Their quinolone resistance might be an acquired trait and the chromosomal quinolone resistance homologs may be weakly related to the phenotypic quinolone resistance of S. equorum.
Among the test strains, only the strain KM1031 exhibited resistance to chloramphenicol as well as erythromycin. Comparative genomic analysis has highlighted the putative antibiotic ABC transporter ATP-binding protein gene (AWC34_RS11115) and the antibiotic biosynthesis monooxygenase gene (AWC34_RS01805) identified in KM1031 as the possible determinants of chloramphenicol and erythromycin resistance. Nguyen and Nguyen35 reported that an E. coli transformant containing antibiotic ABC transporter ATP-binding protein homolog showed resistance to cefalotin, kanamycin, ampicillin, erythromycin and chloramphenicol. In addition, the antibiotic ABC transporter ATP-binding protein was reported to confer resistance to several antibiotics via a ribosomal protection mechanism36. Therefore, we assumed that the antibiotic ABC transporter ATP-binding protein gene may confer the chloramphenicol and erythromycin resistance to strain KM1031. However, the existence of an antibiotic ABC transporter ATP-binding protein gene (SEQU_RS24115) does not confer resistance to chloramphenicol and erythromycin to the strain UMC-CNS-924. The putative antibiotic ABC transporter ATP-binding protein of strain KM1031 showed 100% sequence identity to that of UMC-CNS-924 (Supplementary Table S4), which means that the homologs do not contribute to the chloramphenicol and erythromycin resistance if their transcriptional regulators function properly. Uniquely, an antibiotic biosynthesis monooxygenase gene (AWC34_RS01805) was identified only in strain KM1031; this gene was reported to alter polyketide antibiotics such as macrolide antibiotics through oxidation37, 38. Meanwhile, strain-specific possession of ABC transporter ATP-binding protein or antibiotic biosynthesis monooxygenase homologs in the 15 strains from cheeses hampered clarification of which gene determines the resistance (Supplementary Table S3). Interestingly, two IS6 family transposase genes (COG AWC34_RS01810 and AWC34_RS01800) were identified in the flanking regions of the antibiotic biosynthesis monooxygenase gene for strain KM1031. Antibiotic biosynthesis monooxygenase gene was also found in the contigs of strains RE2.35 and 908_10 and an IS6 family transposase gene (A4A32_12915) was identified upstream of the homolog in strain RE2.35. The contig of strain RE2.35 composed of a pseudogene exists between the transposase and antibiotic biosynthesis monooxygenase genes. The contig of strain 908_10 composed of a pseudogene and antibiotic biosynthesis monooxygenase gene suggests the existence of transposase in the flanking region. Therefore, we cautiously assumed that AWC34_RS01805 in KM1031 inserted by the action of transposase conferred the chloramphenicol and erythromycin resistance in this strain.
The penicillin G-resistant strain KM1031 harbours a plasmid encoding a putative β-lactamase gene (Table 2, Fig. 3). β-Lactamase homologs are found in the chromosomes of the other four strains, except strain KS1039. The putative amino acid sequence of the plasmid-encoded β-lactamase gene (AWC34_RS13020) shows 79.7–95.3% identity with those from homologs encoded chromosomally (Supplementary Table S4). Meanwhile, the plasmid-encoded β-lactamase has 97% identity with the Zn-dependent hydrolase of S. aureus (Supplementary Table S5). This suggests that the chromosomal β-lactamase homologs contribute not to penicillin G resistance but to other functions. The absence of a β-lactamase homolog in strain KS1039 implies that this gene is not critical to the survival of this strain.
Strain UMC-CNS-924 was resistant to tetracycline and was shown to contain a plasmid-encoded gene whose product has 99% sequence identity with tetracycline resistance MFS (major facilitator superfamily) efflux pumps of Staphylococcus species (Table 2, Fig. 3, Supplementary Table S5). This gene may thus encode an efflux pump that directly promotes resistance to tetracycline.
It is already well known that the mecA gene, which encodes the low-affinity penicillin-binding protein PBP 2A, confers methicillin resistance39. The mecA gene was not identified in the six strains, and methicillin resistance was also not identified. Meanwhile, three genes annotated to encode methicillin resistance proteins were commonly identified in the six strains. Two putative methicillin resistance proteins show ≥88% sequence identity with the aminoacyltransferase of Staphylococcus species (Supplementary Table S5). For these two genes, there is the possibility of mis-annotation. The third gene has ≥66% sequence identity with FemA of the femAB operon. FemA and FemB are known as additional components for methicillin resistance, which enhance the methicillin resistance of MecA. Therefore, these three genes may not confer methicillin resistance to S. equorum strains.
In the current study, all plasmid-mediated antibiotic resistance genes were linked to antibiotic resistance. However, most of the putative antibiotic resistance genes encoded chromosomally were not linked to their expected resistance. The potential antibiotic resistance genes encoded chromosomally may not contribute to resistance owing to their low activity40, 41. It is already well known that plasmid-mediated antibiotic resistance genes confer higher resistance than genes on chromosomes42, 43. However, antibiotic-resistant strains with chromosome-generated adaptation have been reported to survive at a higher rate than strains without antibiotic resistance genes upon exposure to antibiotics41, 44. Therefore, antibiotic resistance genes on chromosomes may allow organisms to survive under various conditions.
Two-component systems
The success of S. aureus as a pathogen is in part due to the precise regulation of genes for survival in various environments. Two-component systems (TCSs) serve as a basic stimulus-response coupling mechanism to allow organisms to sense and respond to changes in the environment. S. aureus has been reported to possess 16 TCSs within its relatively small genome, two of which, saeRS and agrCA, are known to regulate virulence45–47. In the genomes of S. equorum strains, three types of putative TCS involved in the regulation of ion acquisition, cell-wall synthesis and nitrate reduction have been identified (Table 3). arlSR (yhcSR) was reported to be essential for survival under various environmental conditions48 but was not reported to be involved in the regulation of virulence factors. arlSR TCS was also reported to regulate the biofilm formation of Staphylococcus epidermidis in an ica-dependent manner49. However, the ica gene involved in biofilm formation was not identified in the six S. equorum genomes. These results imply that S. equorum arlSR is involved not in the regulation of virulence genes but in adaptation to the environment. As another example, the vraSR TCS system was reported to be a positive modulator of cell wall biosynthesis50. Other TCSs in S. equorum have not been reported to be related to virulence factors. In this context, S. equorum may have little possibility of expressing the pathogenicity seen in S. aureus, but it can exhibit virulence simply by acquiring virulence determinants including those for haemolysis and antibiotic resistance.
Table 3.
Putative two-component systems identified in six S. equorum genomes.
| Function | Product | Gene | Gene locus | |||||
|---|---|---|---|---|---|---|---|---|
| KM1031 | C2014 | KS1039 | Mu2 | UMC-CNS-924 | G8HB1 | |||
| Ion acquisition | Histidine kinase | arlS | AWC34_RS05855 | AVJ22_RS06100 | SE1039_RS06290 | SEQMU2_RS11400 | SEQU_RS19660 | UF72_RS04635 |
| Response regulator | arlR | AWC34_RS05860 | AVJ22_RS06105 | SE1039_RS06295 | SEQMU2_RS11405 | SEQU_RS19665 | UF72_RS04640 | |
| Histidine kinase | arlS | AWC34_RS12210 | SEQMU2_RS04475 | SEQU_RS18135 | UF72_RS08085 | |||
| Response regulator | arlR | AWC34_RS12205 | SEQMU2_RS04470 | SEQU_RS18130 | UF72_RS08090 | |||
| Cell wall synthesis | Sensor kinase | vraS | AWC34_RS08090 | AVJ22_RS08380 | SE1039_RS08510 | SEQMU2_RS00385 | SEQU_RS24695 | UF72_RS12815 |
| Response regulator | vraR | AWC34_RS08085 | AVJ22_RS08375 | SE1039_RS08505 | SEQMU2_RS00380 | SEQU_RS24690 | UF72_RS12810 | |
| Nitrate reduction | Histidine kinase | AWC34_RS07785 | AVJ22_RS08060 | SE1039_RS08200 | SEQMU2_RS13330 | SEQU_RS26235 | UF72_RS13400 | |
| Response regulator | AWC34_RS07780 | AVJ22_RS08055 | SE1039_RS08195 | SEQMU2_RS13325 | SEQU_RS26230 | UF72_RS13405 | ||
Salt tolerance of C2014 and KS1039
Bacteria respond to hyperosmotic stress either by controlling the flux of ions across their cellular membrane or by accumulating osmolytes called compatible solutes51. In ion homeostasis, potassium plays a pivotal role and is the most abundant ion in the cytoplasm of bacteria52. Although CNS are frequently identified in foods with a high salt concentration, the mechanism behind their salt tolerance is not well understood. Conversely, a number of characteristics allowing S. aureus to survive osmotic stress have been reported. For example, osmoprotectants such as choline, glycine, betaine and proline accumulate in S. aureus in response to osmotic stress53, 54. In addition, multiple genes, including the branched-chain amino acid transporter gene brnQ 55 and the arsenic operon regulatory gene arsR 56, have been reported to cooperate in conferring salt tolerance to S. aureus. Furthermore, the involvement of a very large cell-wall protein, Ebh, in the tolerance to transient hyperosmotic pressure was reported57. The phospholipid cardiolipin, an important component of the cell membrane, was also reported to be necessary for the prolonged survival of S. aureus in high-salt conditions58, 59. The six S. equorum strains also possess two cardiolipin synthetase genes (Supplementary Table S6), as well as a sodium/potassium transport system (Supplementary Table S7), and all strains exhibited growth on TSA plates supplemented with 15% (w/v) NaCl. Meanwhile, two strains, KS1039 and C2014, exhibited growth at a NaCl concentration of 25%; the growth rate of KS1039 was slightly higher than that of C2014 under these conditions (Supplementary Fig. S3).
Strain C2014 possesses an ortholog (AVJ22_RS01775) of an ion transporter that encodes a protein having a ball domain and a potassium voltage-gated channel in its ORF (Supplementary Fig. S4, Supplementary Table S7). The ball domain was shown to be responsible for the inactivation of voltage-gated ion channels60, 61. DNA sequences encoding ball domains have been widely identified in Staphylococcus species as well as in a Bacillus species and its relatives; the potassium voltage-gated channel-encoding sequences are located in the downstream parts of these ball domains (Supplementary Fig. S4). Strain KS1039 possesses two unique orthologs, SE1039_RS01900 and SE1039_RS01905, which encode a potassium voltage-gated channel and a protein with a ball domain, respectively; we supposed that these genes contribute to the high salt tolerance of strain KS103916. The ion selectivity of potassium voltage-gated channels was reported to be associated with a conserved sequence motif TVGYG located in a re-entrant loop present in-between two predicted transmembrane regions62. This conserved sequence was identified in both AVJ22_RS01775 and SE1039-RS01900 genes.
To investigate the effect of the potassium voltage-gated channel and ball domain on salt tolerance, the SE1039_RS01900/RS01905 and AVJ22_RS01775 genes were amplified and then cloned into the pGEM-T easy vector under the control of the T7 promoter. The resulting plasmids were designated as p1039P for the gene SE1039_RS01900, p1039B for the gene SE1039_RS01905, p1039BP for the genes SE1039_RS01905/RS01900 and p2014BP for the gene AVJ22_RS01775 (Supplementary Fig. S4). Under IPTG induction, the effect of NaCl on the growth was pronounced when NaCl was applied at a concentration of 6% (Fig. 4). The transformant harbouring p1039P showed the highest growth, followed by the transformant harbouring p1039BP. E. coli harbouring p1039B showed much slower growth than the control containing pGEM-T easy vector. These results suggest that the ball domain downregulates the activity of voltage-gated ion channels. Higher salt tolerances were exhibited in strain KS1039 and the transformant harbouring p1039BP than in strain C2014 and the recombinant harbouring p2014BP, implying that salt tolerance can be increased when potassium voltage-gated channels and ball domains exist in separate ORFs.
Figure 4.
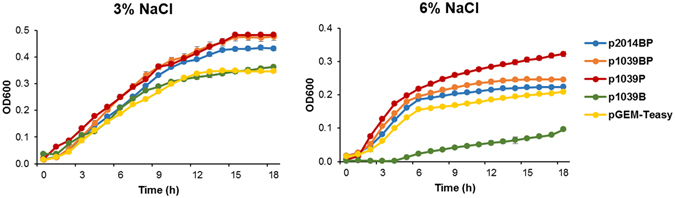
Effect of two types of potassium voltage-gated channel genes on the growth of E. coli cells under salt stress.
The flanking regions of the potassium voltage-gated channel genes for strains C2014 and KS1039 do not provide any clues about if or where any insertion might have occurred. However, transposase genes are found at distant loci on both sides of the potassium voltage-gated channel genes (COG AVJ22_RS01765 and AVJ22_RS03035 in strain C2014; SE1039_RS03265 and SE1039_RS01160 in strain KS1039). We thus assume that strains C2014 and KS1039 acquired the potassium voltage-gated channel gene by a random insertion event, enabling them to survive in the high-salt conditions of jeotgal. The potassium voltage-gated channel gene allows S. equorum strains to survive in high-salt fermented foods with a NaCl concentration of over 15%.
Conclusions
The previously reported analyses of the three genomes of S. equorum strains Mu2, UMC-CNS-924 and KS1039 revealed that these three strains do not possess any clear virulence factors found in the well-known pathogen S. aureus 10, 16, 17. Genomic analysis should help to confirm the lack of known safety hazard determinants in strains, but cannot guarantee the safety of candidate industrial strains. This comparative genomic analysis identified the strain-specific determinants of S. equorum strains, which are linked to phenotypic characteristics including virulence and salt tolerance. The identification of factors related to phenotypic safety hazards should help in guaranteeing the safety of industrial strains. In this context, this study strongly supports the safety of strain KS1039, which was selected as a starter candidate for jeotgal fermentation13 because it does not possess any of the functional virulence determinants identified in the other strains. Additionally, this study identified the gene that contributes to the strain-specific high-salt tolerance of S. equorum strains. This study supports the usefulness of comparative genomics in the safety assessment and functional analysis of industrial strains including starter candidates for food fermentation.
Electronic supplementary material
Acknowledgements
S. equorum Mu2 was kindly provided by Dr. Francoise Irlinger at the Institut National de la Recherche Agronomique in France. S. equorum UMC-CNS-924 was kindly provided by Dr. John R. Middleton at the University of Missouri in the USA. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1D1A1B01011421 and NRF-2016R1D1A1B03930239).
Author Contributions
D.W.J. and J.H.L. designed the experiment, analysed the data and wrote the paper. D.W.J. and S.H. performed the experiments. S.R. and J.B. provided assistance in the experiment. J.H.L. supervised the project.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05918-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schleifer KH, Kilpperbalz R, Devriese LA. Staphylococcus arlettae sp. nov., Staphylococcus equorum sp. nov. and Staphylococcus kloosii sp. nov.: three new coagulase-negative, novobiocin-resistant species from animals. Syst Appl Microbiol. 1984;5:501–509. doi: 10.1016/S0723-2020(84)80007-7. [DOI] [Google Scholar]
- 2.Blaiotta G, et al. Diversity and dynamics of communities of coagulase-negative staphylococci in traditional fermented sausages. J Appl Microbiol. 2004;97:271–284. doi: 10.1111/j.1365-2672.2004.02298.x. [DOI] [PubMed] [Google Scholar]
- 3.Mauriello G, Casaburi A, Blaiotta G, Villani F. Isolation and technological properties of coagulase negative staphylococci from fermented sausages of Southern Italy. Meat sci. 2004;67:149–158. doi: 10.1016/j.meatsci.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Corbiere Morot-Bizot S, Leroy S, Talon R. Staphylococcal community of a small unit manufacturing traditional dry fermented sausages. Int J Food Microbiol. 2006;108:210–217. doi: 10.1016/j.ijfoodmicro.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Bockelmann W, Willems KP, Neve H, Heller KH. Cultures for the ripening of smear cheeses. Int Dairy J. 2005;15:719–732. doi: 10.1016/j.idairyj.2004.08.022. [DOI] [Google Scholar]
- 6.Place RB, Hiestand D, Gallmann HR, Teuber M. Staphylococcus equorum subsp. linens, subsp. nov., a starter culture component for surface ripened semi-hard cheeses. Syst Appl Microbiol. 2003;26:30–37. doi: 10.1078/072320203322337281. [DOI] [PubMed] [Google Scholar]
- 7.Deetae P, Bonnarme P, Spinnler HE, Helinck S. Production of volatile aroma compounds by bacterial strains isolated from different surface-ripened French cheeses. Appl Microbiol Biotechnol. 2007;76:1161–1171. doi: 10.1007/s00253-007-1095-5. [DOI] [PubMed] [Google Scholar]
- 8.Fulladosa E, et al. Volatile profile and microbiological characterization of hollow defect in dry-cured ham. Meat sci. 2010;86:801–807. doi: 10.1016/j.meatsci.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Guan L, Cho KH, Lee JH. Analysis of the cultivable bacterial community in jeotgal, a Korean salted and fermented seafood, and identification of its dominant bacteria. Food Microbiol. 2011;28:101–113. doi: 10.1016/j.fm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Calcutt MJ, et al. Genome sequence analysis of Staphylococcus equorum bovine mastitis isolate UMC-CNS-924. Genome Announc. 2013;1:e00840–13. doi: 10.1128/genomeA.00840-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JH, Jeong DW. Characterization of Mobile Staphylococcus equorum plasmids isolated from fermented seafood that confer lincomycin resistance. PloS one. 2015;10:e0140190. doi: 10.1371/journal.pone.0140190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marty E, et al. Prevalence of antibiotic resistance in coagulase-negative staphylococci from spontaneously fermented meat products and safety assessment for new starters. Int J Food Microbiol. 2012;159:74–83. doi: 10.1016/j.ijfoodmicro.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 13.Jeong DW, Han S, Lee JH. Safety and technological characterization of Staphylococcus equorum isolates from jeotgal, a Korean high-salt-fermented seafood, for starter development. Int J Food Microbiol. 2014;188:108–115. doi: 10.1016/j.ijfoodmicro.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 14.Cachaldora A, Fonseca S, Franco I, Carballo J. Technological and safety characteristics of Staphylococcaceae isolated from Spanish traditional dry-cured sausages. Food Microbiol. 2013;33:61–68. doi: 10.1016/j.fm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Irlinger F. Safety assessment of dairy microorganisms: coagulase-negative staphylococci. Int J Food Microbiol. 2008;126:302–310. doi: 10.1016/j.ijfoodmicro.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Jeong DW, Na H, Ryu S, Lee JH. Complete genome sequence of Staphylococcus equorum KS1039 isolated from Saeu-jeotgal, Korean high-salt-fermented seafood. J Biotechnol. 2016;219:88–89. doi: 10.1016/j.jbiotec.2015.12.025. [DOI] [PubMed] [Google Scholar]
- 17.Irlinger F, et al. Genome sequence of Staphylococcus equorum subsp. equorum Mu2, isolated from a french smear-ripened cheese. J Bacteriol. 2012;194:5141–5142. doi: 10.1128/JB.01038-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong DW, Kim HR, Han S, Jeon CO, Lee JH. A proposal to unify two subspecies of Staphylococcus equorum: Staphylococcus equorum subsp. equorum and Staphylococcus equorum subsp. linens. Anton Leeuw. 2014;106:795–808. doi: 10.1007/s10482-014-0249-6. [DOI] [PubMed] [Google Scholar]
- 19.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 21.Disz T, et al. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics. 2010;11:319. doi: 10.1186/1471-2105-11-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kastman EK, et al. Biotic Interactions shape the ecological distributions of Staphylococcus Species. mBio. 2016;7:01157–16. doi: 10.1128/mBio.01157-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goris J, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 24.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blom J, et al. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blom J, et al. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016;44:W22–28. doi: 10.1093/nar/gkw255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lerat E, Daubin V, Moran NA. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-Proteobacteria. PLoS biology. 2003;1:E19. doi: 10.1371/journal.pbio.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.CLSI, Performance standards for antimicrobial susceptibility testing; seventeenth informational supplement. Wayne, PA: CLSI (2007).
- 29.Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DY. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol. 2010;125:39–49. doi: 10.1016/j.jaci.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ter Huurne AA, et al. Characterization of three putative Serpulina hyodysenteriae hemolysins. Microb Pathog. 1994;16:269–282. doi: 10.1006/mpat.1994.1028. [DOI] [PubMed] [Google Scholar]
- 31.Carvalho E, et al. Leptospiral TlyC is an extracellular matrix-binding protein and does not present hemolysin activity. FEBS letters. 2009;583:1381–1385. doi: 10.1016/j.febslet.2009.03.050. [DOI] [PubMed] [Google Scholar]
- 32.Chen YC, Chang MC, Chuang YC, Jeang CL. Characterization and virulence of hemolysin III from Vibrio vulnificus. Current microbiology. 2004;49:175–179. doi: 10.1007/s00284-004-4288-5. [DOI] [PubMed] [Google Scholar]
- 33.Robertson KP, Smith CJ, Gough AM, Rocha ER. Characterization of Bacteroides fragilis hemolysins and regulation and synergistic interactions of HlyA and HlyB. Infec Immun. 2006;74:2304–2316. doi: 10.1128/IAI.74.4.2304-2316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linhartova I, et al. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev. 2010;34:1076–1112. doi: 10.1111/j.1574-6976.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu Nguyen HN. Functional analysis of ATP-binding cassette transporter of Streptomyces coelicolor. J Appl Pharm Sci. 2014;4:50–53. [Google Scholar]
- 36.Wilson DN. The ABC of ribosome-related antibiotic resistance. MBio. 2016;7:e00598–16. doi: 10.1128/mBio.00598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sciara G, et al. The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis. EMBO J. 2003;22:205–215. doi: 10.1093/emboj/cdg031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemieux MJ, et al. The crystal structure of Rv0793, a hypothetical monooxygenase from M. tuberculosis. J Struct Funct Genomics. 2005;6:245–257. doi: 10.1007/s10969-005-9004-6. [DOI] [PubMed] [Google Scholar]
- 39.Brakstad OG, Maeland JA. Mechanisms of methicillin resistance in staphylococci. APMIS. 1997;105:264–276. doi: 10.1111/j.1699-0463.1997.tb00568.x. [DOI] [PubMed] [Google Scholar]
- 40.Card RM, et al. Application of microarray and functional-based screening methods for the detection of antimicrobial resistance genes in the microbiomes of healthy humans. PloS one. 2014;9:e86428. doi: 10.1371/journal.pone.0086428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev: MMBR. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harding I, Simpson I. Fluoroquinolones: is there a different mechanism of action and resistance against Streptococcus pneumoniae? J Chemother. 2000;12(Suppl 4):7–15. doi: 10.1080/1120009X.2000.11782307. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L, Li XZ, Poole K. SmeDEF multidrug efflux pump contributes to intrinsic multidrug resistance in Stenotrophomonas maltophilia. Antimicrob Agents Chemother. 2001;45:3497–3503. doi: 10.1128/AAC.45.12.3497-3503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li XZ. Quinolone resistance in bacteria: emphasis on plasmid-mediated mechanisms. Int J Antimicrob Agents. 2005;25:453–463. doi: 10.1016/j.ijantimicag.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Novick RP, Jiang D. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiol. 2003;149:2709–2717. doi: 10.1099/mic.0.26575-0. [DOI] [PubMed] [Google Scholar]
- 46.Giraudo AT, Calzolari A, Cataldi AA, Bogni C, Nagel R. The sae locus of Staphylococcus aureus encodes a two-component regulatory system. FEMS Microbiol Lett. 1999;177:15–22. doi: 10.1111/j.1574-6968.1999.tb13707.x. [DOI] [PubMed] [Google Scholar]
- 47.Jeong DW, et al. Identification of the P3 promoter and distinct roles of the two promoters of the SaeRS two-component system in Staphylococcus aureus. J. Bacteriol. 2011;193:4672–4684. doi: 10.1128/JB.00353-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun J, Zheng L, Landwehr C, Yang J, Ji Y. Identification of a novel essential two-component signal transduction system, YhcSR, in Staphylococcus aureus. J Bacteriol. 2005;187:7876–7880. doi: 10.1128/JB.187.22.7876-7880.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, et al. The two-component signal transduction system ArlRS regulates Staphylococcus epidermidis biofilm formation in an ica-dependent manner. PloS one. 2012;7:e40041. doi: 10.1371/journal.pone.0040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuroda M, et al. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol. 2003;49:807–821. doi: 10.1046/j.1365-2958.2003.03599.x. [DOI] [PubMed] [Google Scholar]
- 51.Shabala S, Shabala L. Ion transport and osmotic adjustment in plants and bacteria. Biomolecular concepts. 2011;2:407–419. doi: 10.1515/BMC.2011.032. [DOI] [PubMed] [Google Scholar]
- 52.Epstein W. The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol. 2003;75:293–320. doi: 10.1016/S0079-6603(03)75008-9. [DOI] [PubMed] [Google Scholar]
- 53.Amin US, Lash TD, Wilkinson BJ. Proline betaine is a highly effective osmoprotectant for Staphylococcus aureus. Arch Microbiol. 1995;163:138–142. doi: 10.1007/BF00381788. [DOI] [PubMed] [Google Scholar]
- 54.Peddie BA, Lever M, Randall K, Chambers ST. Osmoprotective activity, urea protection, and accumulation of hydrophilic betaines in Escherichia coli and Staphylococcus aureus. Antonie van Leeuwenhoek. 1999;75:183–189. doi: 10.1023/A:1001701400801. [DOI] [PubMed] [Google Scholar]
- 55.Vijaranakul U, Xiong A, Lockwood K, Jayaswal RK. Cloning and nucleotide sequencing of a Staphylococcus aureus gene encoding a branched-chain-amino-acid transporter. Appl Environ Microbiol. 1998;64:763–767. doi: 10.1128/aem.64.2.763-767.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scybert S, et al. NaCl-sensitive mutant of Staphylococcus aureus has a Tn917-lacZ insertion in its ars operon. FEMS Microbiol Lett. 2003;222:171–176. doi: 10.1016/S0378-1097(03)00312-4. [DOI] [PubMed] [Google Scholar]
- 57.Kuroda M, et al. Staphylococcus aureus giant protein Ebh is involved in tolerance to transient hyperosmotic pressure. Biochem Biophys Res Commun. 2008;374:237–241. doi: 10.1016/j.bbrc.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 58.Kanemasa Y, Yoshioka T, Hayashi H. Alteration of the phospholipid composition of Staphylococcus aureus cultured in medium containing NaCl. Biochim Biophys Acta. 1972;280:444–450. doi: 10.1016/0005-2760(72)90251-2. [DOI] [PubMed] [Google Scholar]
- 59.Tsai M, et al. Staphylococcus aureus requires cardiolipin for survival under conditions of high salinity. BMC Microbiol. 2011;11:13. doi: 10.1186/1471-2180-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Varshney A, Chanda B, Mathew MK. Arranging the elements of the potassium channel: the T1 domain occludes the cytoplasmic face of the channel. Eur Biophys J. 2004;33:370–376. doi: 10.1007/s00249-003-0372-1. [DOI] [PubMed] [Google Scholar]
- 62.Doyle DA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Sci. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.