

Abstract
Background
Attention-deficit/hyperactivity disorder (ADHD) is increasingly recognized as a persistent disorder requiring long-term management.
Objectives
Our objective was to evaluate the 2-year safety and efficacy of lisdexamfetamine dimesylate (LDX) in children and adolescents with ADHD.
Methods
Participants (aged 6–17 years) with ADHD received open-label, dose-optimized LDX 30, 50, or 70 mg/day for 104 weeks. Safety monitoring included treatment-emergent adverse events (TEAEs), vital signs, electrocardiography, and growth. The TEAEs decreased appetite, weight decrease, insomnia events (including insomnia, initial insomnia, middle insomnia, and terminal insomnia), headache, and psychiatric TEAEs were pre-defined as being of special interest. Efficacy was assessed as a secondary objective using the ADHD Rating Scale IV (ADHD-RS-IV), the Clinical Global Impressions-Improvement (CGI-I) scale, and the CGI-Severity (CGI-S) scale.
Results
Of 314 participants enrolled, 191 completed the study. TEAEs were reported in 89.8% of participants, led to discontinuation in 12.4%, and were reported as serious in 8.9%. TEAEs that were reported by ≥5% of participants and considered by investigators as related to LDX were decreased appetite (49.4%), weight decrease (18.2%), insomnia (13.1%), initial insomnia (8.9%), irritability (8.6%), nausea (6.7%), headache (5.7%), and tic (5.1%). The median time to first onset and duration, respectively, of TEAEs of special interest were as follows: decreased appetite, 13.5 and 169.0 days; weight decrease, 29.0 and 225.0 days; insomnia, 17.0 and 42.8 days; and headache, 22.0 and 2.0 days. Reports of decreased appetite, weight decrease, insomnia, and headache were highest in the first 4–12 weeks. Psychiatric TEAEs were infrequent: psychosis and mania (n = 1), suicidal events (suicidal ideation, n = 2; suicide attempt, n = 1), and aggression events (aggression, n = 14; anger, n = 2; hostility, n = 1). At the last on-treatment assessment (LOTA), mean increases from baseline in vital signs were as follows: pulse rate, 7.0 bpm (95% confidence interval [CI] 5.7–8.2); systolic blood pressure (SBP), 3.4 mmHg (95% CI 2.2–4.5); and diastolic blood pressure (DBP), 3.2 mmHg (95% CI 2.2–4.2). Pre-defined thresholds for a potentially clinically important (PCI) high pulse rate were met at one or more visits by 22 participants (7.0%), for PCI high SBP were met by 45 children (22.4%) and 17 adolescents (15.2%), and for PCI high DBP were met by 78 children (38.8%) and 24 adolescents (21.4%). The mean QT interval corrected using Fridericia’s formula (QTcF) decreased from baseline to LOTA (−0.6 ms [95% CI −2.3 to 1.2]; range −50 to +53). Mean changes in growth from baseline to LOTA were weight, 2.1 kg (95% CI 1.5–2.8); height, 6.1 cm (95% CI 5.6–6.7); and body mass index (BMI), −0.5 kg/m2 (95% CI −0.7 to −0.3). There was a general shift to lower z score categories for height, weight, and BMI from baseline to LOTA. The mean change in ADHD-RS-IV from baseline to LOTA was −25.8 (95% CI −27.0 to −24.5) for total score, −12.6 (95% CI −13.4 to −11.9) for the hyperactivity/impulsivity subscale score, and −13.1 (95% CI −13.8 to −12.4) for the inattention subscale score. At LOTA, 77.9% of participants had a CGI-I score of 1 or 2. In addition, 77.3 and 69.2% of participants were classified as treatment responders, based on a CGI-I score of 1 or 2 and a ≥30% or ≥50% reduction from baseline in ADHD-RS-IV total score, respectively.
Conclusions
The safety profile of LDX in this longer-term study was similar to that reported in previous studies. The efficacy of LDX was maintained throughout the 2-year study period.
ClinicalTrials.gov Identifier
Electronic supplementary material
The online version of this article (doi:10.1007/s40263-017-0443-y) contains supplementary material, which is available to authorized users.
Key Points
| This 2-year clinical study provides the most comprehensive assessment to date of the long-term safety of lisdexamfetamine dimesylate (LDX) in children and adolescents with attention-deficit/hyperactivity disorder (ADHD). |
| The observed effects of LDX on treatment-emergent adverse events, vital signs, and growth were consistent with findings from previous short-term randomized controlled trials of LDX; no new safety signals were reported. |
| LDX treatment was associated with improvements in ADHD symptoms that were maintained for the 2-year duration of the study. |
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common neurobehavioral disorder associated with high levels of functional impairment and reduced quality of life [1]. The mean worldwide prevalence of ADHD in children and adolescents is estimated to be 5.3%, and, although symptoms may ameliorate with time, impairing difficulties persist into adulthood in 50–66% of patients [2–6].
Psychostimulants, including methylphenidate and amphetamines, are commonly prescribed pharmacological treatments for ADHD [7]. Lisdexamfetamine dimesylate (LDX) is an amphetamine-based prodrug that, following oral administration, is absorbed into the blood and enzymatically hydrolyzed to release therapeutically active d-amphetamine [8–10]. The short-term efficacy and safety of LDX have been demonstrated in children, adolescents, and adults in several pivotal, randomized, double-blind, placebo-controlled clinical trials in the USA and Europe [11–16]. The safety profile of LDX has been consistent across studies, and the range of observed adverse events (AEs), including decreased appetite, weight loss, and insomnia, has been typical of those observed with other psychostimulant ADHD medications [17].
Previous longer-term, open-label studies conducted in the USA have shown that LDX reduces the core symptoms of ADHD for up to 12 months and has a longer-term safety profile similar to that observed in short-term LDX studies [18–20]. Owing to the nature of AEs commonly associated with stimulant medications, clinical practice guidelines recommend that patients receiving long-term stimulant medication are monitored regularly for height, weight, and cardiovascular parameters [21]. Here, we report primary results from the first 2-year open-label study of LDX in Europe (SPD489-404; ClinicalTrials.gov identifier: NCT01328756). This is currently the longest LDX clinical study to be performed and was designed to include a comprehensive battery of safety assessments, thereby providing the most in-depth evaluation to date of the long-term safety and efficacy of LDX in children and adolescents with ADHD.
Method
SPD489-404 was a phase IV, multicenter, open-label, 2-year study of the long-term safety and efficacy of LDX in children and adolescents with ADHD. The study was conducted in accordance with current applicable regulations, International Conference on Harmonization Good Clinical Practice Guideline E6 (1996), EU Clinical Trials Directive 2001/20/EC (2001) and its updates, and local ethical and legal requirements. The study protocol was approved by an independent ethics committee/institutional review board and regulatory agency in each center (as appropriate). Each patient’s parent/legal guardian provided written informed consent, and assent was obtained from each participant (as applicable) before they took part in the study. The study was conducted between 7 July 2011 and 30 September 2014 at 35 sites in ten European countries (Belgium, Germany, Hungary, Italy, Netherlands, Poland, Romania, Spain, Sweden, and the UK).
Participants
Children (aged 6–12 years) and adolescents (aged 13–17 years) were either enrolled directly or had taken part in a previous LDX study (ClinicalTrials.gov identifiers: NCT01106430 [22], NCT00763971 [14], and NCT00784654 [23]). Eligible individuals had a primary diagnosis of ADHD based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition—Text Revision™ (DSM-IV-TR) criteria and a baseline ADHD Rating Scale IV (ADHD-RS-IV) total score ≥28. Participants were excluded if they had been terminated from a previous LDX study for protocol non-adherence or non-compliance or had experienced an AE leading to discontinuation, a medication-related serious AE, or a clinically significant AE in a previous LDX study. Patients whose current ADHD medication provided effective control of symptoms with acceptable tolerability were also excluded. Additional inclusion and exclusion criteria are described in the Electronic Supplementary Material (ESM) 1.
Study Design
SPD489-404 consisted of three phases: 3–42-day screening and washout; 104-week open-label treatment (4 weeks of dose optimization and 100 weeks of dose maintenance); and 28–30-day safety follow-up (Fig. 1). LDX was administered as a once-daily morning dose. Dose optimization (weeks 1–4) continued until an “acceptable” response was obtained, which was defined in previous dose-optimized studies of LDX as a ≥30% reduction in ADHD-RS-IV total score from baseline and a Clinical Global Impressions-Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved) with tolerable side effects [14, 16, 20, 22–24]. Dose adjustments could be made throughout the dose-maintenance phase.
Fig. 1.
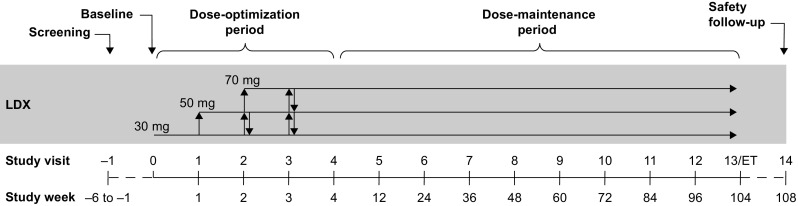
SPD489-404 study design. ET early termination, LDX lisdexamfetamine dimesylate
Safety
The primary objective of the study was to evaluate the long-term tolerability and safety of LDX based on treatment-emergent AEs (TEAEs), vital signs (single measurements of sitting systolic blood pressure [SBP], diastolic blood pressure [DBP], and pulse taken after 5 min of rest), and electrocardiogram (ECG) parameters. TEAEs were defined as AEs that started or worsened after the first dose of LDX and up to the third day after treatment cessation. TEAEs were coded using version 14.1 of the Medical Dictionary for Regulatory Activities (MedDRA). The sponsor required any new onset of seizures, loss of consciousness, or syncope to be reported as a serious TEAE, and defined psychiatric TEAEs (psychosis, mania, suicidal events, and aggression events) as being of special interest. Based on their reported association with stimulant treatment, the TEAEs decreased appetite, weight decreased (hereafter referred to as weight decrease), insomnia events (including insomnia, initial insomnia, middle insomnia, and terminal insomnia) and headache were also defined as being of special interest. AE data were collected via questions such as “have you had any health problems since your last visit” at each study visit. It should be noted that the TEAE weight decrease was based on perceived weight loss rather than actual measurements of weight.
Additional safety assessments included height, weight, and clinical laboratory measurements (biochemistry, hematology, and urinalysis). Height, weight, and body mass index (BMI) z scores were derived using the Centers for Disease Control and Prevention growth charts [25]. Potentially clinically important (PCI) thresholds for high pulse (≥110 bpm), SBP (≥125 mmHg in children and ≥135 mmHg in adolescents), DBP (≥80 mmHg in children and ≥85 mmHg in adolescents), QT interval corrected using Fridericia’s formula (QTcF; ≥450 ms), and changes in weight (≥7% change from baseline) were pre-defined by the sponsor, based on clinical experience.
Psychiatric safety was monitored using the Brief Psychiatric Rating Scale for Children (BPRS-C) and the Columbia-Suicide Severity Rating Scale (C-SSRS). The BPRS-C is an investigator-rated 21-item scale that assesses behavior disorders, depression, thinking disturbance, psychomotor excitation, withdrawal retardation, anxiety, and organicity using a 7-point Likert scale, from ‘not present’ (0 points) to ‘extremely severe’ (6 points) [26]. The C-SSRS is a semi-structured interview that captures the occurrence, severity, and frequency of suicide-related thoughts and behaviors [27]. Neurocognition and sexual development were monitored using the Cambridge Neuropsychological Test Automated Battery [28] and Tanner staging [29], respectively (Coghill DR et al. unpublished data and Banaschewski T et al. unpublished data).
Efficacy
Efficacy was assessed as a secondary objective using the investigator-administered ADHD-RS-IV, CGI-I, and CGI-Severity (CGI-S) scales [30, 31]. The ADHD-RS-IV consists of 18 items grouped into two subscales, “hyperactivity/impulsivity” and “inattention”, designed to reflect current symptomatology based on DSM-IV-TR criteria. Each item is scored from 0 (no symptoms) to 3 (severe symptoms), giving a total possible score of 0–54. The CGI-S rates ADHD severity using a 7-point scale ranging from 1 (normal, not at all ill) to 7 (among the most extremely ill). The CGI-I scale rates patient improvement, relative to baseline CGI-S, on a 7-point scale from 1 (very much improved) to 7 (very much worse). Individuals with a CGI-I score of 1 or 2 were categorized as improved.
Responder analyses were also performed, in which a clinically relevant response was defined using two standards: (1) ≥30% reduction in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2, and (2) ≥50% reduction in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2.
Data Analysis
In this open-label, uncontrolled study, the target enrollment of approximately 300 participants was not based on statistical considerations. Safety analyses were performed using the safety population, defined as all enrolled participants who received at least one dose of LDX during the study.
Efficacy analyses were performed using the full analysis set (FAS), defined as all participants who had at least one on-treatment post-baseline efficacy assessment. ADHD-RS-IV and CGI-I scores were summarized for each visit and for the last on-treatment assessment (LOTA) using observed values. Changes from baseline to LOTA in ADHD-RS-IV scores were assessed using a two-sided, one-sample t test at a 0.05 significance level.
Results
Patient Disposition and Demographics
All 314 enrolled participants received at least one dose of LDX and were included in the safety population (Fig. 2). Of these, 124 (39.5%) had participated in an antecedent LDX study and 190 (60.5%) were directly enrolled. In total, 299 participants (95.2%) were included in the FAS. Of the 15 individuals excluded from the FAS, one did not have a post-baseline efficacy assessment and 14 participants were excluded on the basis of a serious breach of Good Clinical Practice (GCP) compliance at a single study site. The breach of GCP was identified by the sponsor during a monitoring visit for a different study at the site; as a precaution, the sponsor also discontinued participants from all other studies at the site, including SPD489-404. In total, 191 participants (60.8%) completed all visits for SPD489-404. The primary recorded reasons for early discontinuation were withdrawal by the participant (13.1%), AEs (12.4%), other (9.2%), lost to follow-up (1.6%), investigator-perceived lack of efficacy (1.6%), and protocol deviations (1.3%). Baseline demographic data are summarized in Table 1, and further information is provided in Table S1 in the ESM.
Fig. 2.

Patient disposition. aThe safety population comprised all enrolled participants who received at least one dose of LDX during the study. bThe FAS comprised all participants who received one dose of LDX and had at least one on-treatment post-baseline efficacy assessment; all 14 participants from a single study site were excluded from the efficacy analyses because of a serious violation of Good Clinical Practice. cThe number of participants refers to individuals in the enrolled population who completed the study. A total of 191 participants who were included in the FAS completed the study. dAccording to the protocol, lack of efficacy (in the opinion of the investigator) was to be reported as an adverse event. Five additional patients discontinued because of investigator-perceived lack of efficacy; according to the protocol, these should have been recorded as treatment-emergent adverse events. FAS full analysis set, LDX lisdexamfetamine dimesylate
Table 1.
Baseline demographics and disease characteristics (safety population)
| Characteristic | Safety population (N = 314) |
|---|---|
| Demographics | |
| Age, years | 11.4 ± 2.88 (6–19)a |
| 6–12 | 202 (64.3) |
| 13–17a | 112 (35.7) |
| Sex, male | 250 (79.6) |
| Race, White | 310 (98.7) |
| Body mass index, kg/m2b | 19.22 ± 3.389 (13.0–29.8) |
| Participants who received at least one previous ADHD medication | 271 (86.3) |
| Disease characteristics | |
| ADHD subtype | |
| Combined | 251 (79.9) |
| Predominantly inattentive | 56 (17.8) |
| Predominantly hyperactive-impulsive | 7 (2.2) |
| ADHD-RS-IV total score | 41.1 ± 7.03 (17–54)c |
| Inattention subscale score | 22.1 ± 3.52 (11–27) |
| Hyperactivity/impulsivity subscale score | 19.0 ± 5.86 (2–27) |
Data are presented as n (%) or mean ± SD (range)
ADHD attention-deficit/hyperactivity disorder, ADHD-RS-IV ADHD Rating Scale IV, LDX lisdexamfetamine dimesylate, SD standard deviation
aFour participants were aged >17 years at baseline and were included in the age category 13–17 years. These participants were enrolled because, for study eligibility purposes only, age was based on age at the time of consent for this study or for the previous LDX study if applicable
bCalculated at screening
cOne participant had a score of 17, which was lower than the protocol-specified value of ≥ 28; this was recorded as a protocol deviation/violation
Lisdexamfetamine Dimesylate Dosing and Exposure
The mean average daily dose of LDX was 51.08 mg (standard deviation [SD] 14.352), and the mean duration of exposure to LDX was 555.3 days (SD 253.50). Further information on dosing is provided in ESM 1.
Treatment-Emergent Adverse Events (TEAEs)
TEAEs were reported in 89.8% of participants, with most reported as mild (n = 112 [35.7%]) or moderate (n = 133 [42.4%]) (Table 2 and ESM 1, Table S2). No deaths were reported during the study. The overall proportion of participants experiencing TEAEs, and the frequency of commonly reported TEAEs, was generally similar in children and adolescents (ESM 1, Table S3). The proportion of participants reporting TEAEs was generally higher among those receiving higher doses of LDX (ESM 1, Table S3).
Table 2.
Treatment-emergent adverse events (safety population)
| TEAE—preferred term | Safety population (N = 314) | |
|---|---|---|
| Participants | Events | |
| Any TEAE | 282 (89.8) | 1803 |
| Severe TEAEa | 37 (11.8) | 52 |
| Serious TEAEb | 28 (8.9) | 36 |
| TEAEs considered related to study drugc | 232 (73.9) | 785 |
| TEAEs leading to treatment discontinuation | 39 (12.4) | 59 |
| TEAEs resulting in death | 0 | 0 |
| TEAEs reported in ≥5% of participants | ||
| Decreased appetite | 170 (54.1) | 214 |
| Nasopharyngitis | 73 (23.2) | 129 |
| Headache | 68 (21.7) | 139 |
| Weight decrease | 63 (20.1) | 68 |
| Insomnia | 60 (19.1) | 75 |
| Initial insomnia | 38 (12.1) | 46 |
| Irritability | 36 (11.5) | 39 |
| Pyrexia | 32 (10.2) | 41 |
| Nausea | 31 (9.9) | 41 |
| Abdominal pain | 30 (9.6) | 38 |
| Abdominal pain upper | 28 (8.9) | 34 |
| Vomiting | 27 (8.6) | 33 |
| Cough | 22 (7.0) | 25 |
| Depressed mood | 19 (6.1) | 20 |
| Gastroenteritis | 18 (5.7) | 21 |
| Oropharyngeal pain | 18 (5.7) | 24 |
| Tic | 18 (5.7) | 25 |
| Pharyngitis | 16 (5.1) | 18 |
| TEAEs considered related to study drug reported in ≥5% of patientsc | ||
| Decreased appetite | 155 (49.4) | |
| Weight decreased | 57 (18.2) | |
| Insomnia | 41 (13.1) | |
| Initial insomnia | 28 (8.9) | |
| Irritability | 27 (8.6) | |
| Nausea | 21 (6.7) | |
| Headache | 18 (5.7) | |
| Tic | 16 (5.1) | |
Data are presented as n or n (%)
LDX lisdexamfetamine dimesylate, TEAE treatment-emergent adverse event
aA severe TEAE was defined as an adverse event that interrupted usual activities of daily living, significantly affected clinical status, or may require intensive therapeutic intervention
bA serious TEAE was defined as any untoward medical occurrence that resulted in death, was life-threatening, required inpatient hospitalization or prolonged existing hospitalization, resulted in persistent or significant disability/incapacity, was a congenital abnormality/birth defect, or was an important medical event. Important medical events may have been considered as serious TEAEs when, based upon medical judgement, they may have jeopardized the patient and may have required medical or surgical intervention to prevent one of the outcomes listed above. Any new onset of seizures, syncope, or loss of consciousness was required by the sponsor to be reported as a serious TEAE
cAs determined by the investigator
In total, 36 serious TEAEs were reported in 28 participants, four of which were considered by the investigator to be related to LDX (three syncope events and one severe arrhythmia event) (ESM 1, Table S4). Serious TEAEs reported in more than one participant were syncope (n = 6, seven events), appendicitis (n = 3, three events), and pyelonephritis (n = 2, two events). One syncope event was recorded as severe and related to LDX treatment and resulted in treatment interruption. The remaining six syncope events did not result in treatment interruption and were recorded as moderate (n = 4, two events considered related to LDX) or mild (n = 2) in intensity. All syncope events resolved without pharmacological intervention. The arrhythmia event was considered to be related to a pre-existing heart defect (patent foramen ovale) that was unknown at screening (ESM 1, Table S4).
TEAEs resulted in early discontinuation from the study for 39 (12.4%) participants; of these, two TEAEs were recorded as serious: arrhythmia (n = 1) and suicide attempt (n = 1; described in Sect. 3.4) (ESM 1, Tables S4 and S5). The most commonly reported TEAEs leading to discontinuation were decreased appetite (n = 7 [2.2%]), drug ineffective (n = 6 [1.9%]), depressed mood (n = 4 [1.3%]), irritability (n = 4 [1.3%]), tic (n = 3 [1.0%]), insomnia (n = 3 [1.0%]), aggression (n = 2 [0.6%]), apathy (n = 2 [0.6%]), tachycardia (n = 2 [0.6%]), and weight decrease (n = 2 [0.6%]). Five additional patients discontinued because of investigator-perceived lack of efficacy; according to the protocol, these should have been recorded as TEAEs.
TEAEs of Special Interest
Based on their reported association with stimulant treatment, decreased appetite, weight decrease, insomnia, headache, and psychiatric TEAEs were pre-selected as being of special interest in this study [17]. Of these, decreased appetite and weight decrease were reported by 54.1 and 20.1% of participants, respectively (Table 2). Of the 170 participants who reported decreased appetite, 28.2% also experienced weight decrease TEAEs (details of actual weight changes are presented in Sect. 3.6). The incidence of reported decreased appetite and weight decrease TEAEs peaked at weeks 1 and 12, respectively, and declined thereafter (Fig. 3a, b). The median time to the first report of decreased appetite was 13.5 days (range 1–653), and the median duration was 169.0 days (range 1–749). For weight decrease TEAEs, the median time to first report was 29.0 days (range 1–677), with a median duration of 225.0 days (range 26–724). Most decreased appetite (210/214) and weight decrease (66/68) TEAEs were rated as being mild or moderate in severity. The dose of LDX was adjusted as a result of decreased appetite or weight decrease TEAEs in 14.9 and 17.6% of cases, respectively. Seven participants discontinued the study because of decreased appetite, and two discontinued as a result of weight decrease. At the end of the study, 24.3% of decreased appetite and 17.6% of weight decrease TEAEs were ongoing.
Fig. 3.

Incidence of treatment-emergent adverse events identified by the sponsor as of special interest: a decreased appetite, b weight decrease, c insomnia, and d headache (safety population). Percentages are based on the number of participants in the safety population who received lisdexamfetamine dimesylate for the given week. TEAE treatment-emergent adverse event
In total, 124 insomnia TEAEs (75 insomnia events, 46 initial insomnia events, two middle insomnia events, one terminal insomnia event) were reported in 98 (31.2%) participants. The median time to first report was 17.0 days (range 1–729), and the median duration was 42.8 days (range 1–739). The incidence of insomnia TEAEs peaked at week 1 and then rapidly decreased (Fig. 3c). Most insomnia TEAEs (122/124) were mild or moderate; the dose of LDX required adjustment in 15.3% of cases. Four patients discontinued because of insomnia TEAEs. At the end of the study, 17 insomnia events (13.7%) were ongoing.
Of 139 headache TEAEs reported in 68 participants (21.7%), two were severe in intensity. The incidence of headache was highest at week 1, decreased by week 2, and remained stable at subsequent visits (Fig. 3d). The median time to the first reported headache was 22.0 days (range 1–718), and the median duration was 2.0 days (range 1–729). The dose of LDX remained unchanged in 97.8% of cases of headache. One patient discontinued as a result of headache, and one (0.7%) headache event was ongoing at the end of the study.
Psychiatric TEAEs of special interest were psychosis and mania (n = 1), suicidal events (suicidal ideation, n = 2; suicide attempt, n = 1), and aggression events (aggression, n = 14; anger, n = 2; hostility, n = 1). The suicide attempt was reported as a serious, severe AE. No medical event occurred, no treatment was given, and the event resolved on the same day. The suicide attempt was considered by the investigator to be not related to LDX treatment; however, LDX was discontinued (ESM 1, Table S2). The time-course of psychiatric TEAEs was not assessed.
Vital Signs and Electrocardiogram Parameters
LDX treatment was associated with increases from baseline to LOTA in mean pulse rate (7.0 bpm [95% confidence interval [CI] 5.7–8.2]; range −32 to +41), SBP (3.4 mmHg [95% CI 2.2–4.5]; range −26 to +40), and DBP (3.2 mmHg [95% CI 2.2–4.2]; range −24 to +25). Following commencement of LDX treatment, means for pulse rate, SBP, and DBP gradually increased before reaching a plateau at approximately week 36, 60, and 24, respectively (ESM 1, Fig. S1). Based on pre-specified thresholds, PCI high pulse rates (≥110 bpm) were observed in 22 (7.0%) participants, four of whom met this PCI criterion at more than one post-baseline visit. PCI high SBP readings (≥125 mmHg in children and ≥135 mmHg in adolescents) were recorded in 45 children (22.4%; 23 at more than one post-baseline visit) and 17 adolescents (15.2%; seven at more than one post-baseline visit). PCI high DBP readings (≥80 mmHg in children and ≥85 mmHg in adolescents) were recorded in 78 children (38.8%) and 24 adolescents (21.4%), with 49 children and nine adolescents meeting the criterion at more than one post-baseline visit. Additional data regarding participants meeting pre- and post hoc-defined PCI thresholds for pulse, SBP, and DBP are reported in ESM 1, Table S6. The mean QTcF decreased from baseline to LOTA (−0.6 ms [95% CI −2.3 to 1.2]; range −50 to +53), and a PCI high QTcF interval (≥450 ms) was recorded for one participant. Mean changes from baseline and proportions of participants meeting PCI thresholds for additional ECG parameters are reported in ESM 1, Tables S7 and S8.
Weight, Height, and Body Mass Index
From baseline to LOTA, a mean increase in weight (2.1 kg [95% CI 1.5–2.8]; range −20 to +34), and height (6.1 cm [95% CI 5.6–6.7]; range −1 to +20), and a mean decrease in BMI (−0.5 kg/m2 [95% CI −0.7 to −0.3]; range −7 to +10) was observed. Z-scores for weight, height, and BMI were within 1 SD of the mean (≥−1 and <1) at baseline for 197 (62.7%), 169 (53.8%), and 193 (61.5%) participants, respectively, and at LOTA for 210 (67.1%), 180 (59.8%), and 189 (60.4%) participants, respectively. However, there was a general shift to lower z score categories for height, weight, and BMI from baseline to LOTA (ESM 1, Table S9). A PCI decrease or increase in body weight (change of ≥7% from baseline) was reported overall in 112 (35.8%) and 129 (41.2%) participants, respectively, at baseline and in 33 (10.5%) and 119 (38.0%) participants, respectively, at LOTA. More in-depth analyses of growth over the 2-year study period, in addition to measures of sexual maturation, will be the subject of future investigations.
Psychiatric Assessments
Mean BPRS-C scores decreased (indicating improvement) from baseline (19.1 [SD 11.14]) to week 4 (8.4 [SD 7.52]) and then remained stable to the end of the study (week 104; 7.1 [SD 7.12]). At LOTA, the mean change from baseline in BPRS-C total score was −10.3 (SD 9.64). BPRS-C item 6 (suicidal ideation—thoughts, threats, or attempts of suicide) and item 9 (hallucinations—visual, auditory, or other hallucinatory experiences or perceptions) were considered of particular interest. Responses to item 6 were reported as moderately severe in two participants, neither of whom had a reported TEAE of suicidal ideation. In a third participant, the response to item 6 was reported as extremely severe; this participant had a suicide attempt reported as a serious severe TEAE (ESM 1, Tables S2 and S4). No responses of moderately severe, severe, or extremely severe were reported on item 9 of the BPRS-C.
Using the C-SSRS, suicidal ideation was reported in seven participants at any post-baseline visit (two participants at visit 4, two at visit 5, two at visit 7, and one participant at early termination [day 61]), two of whom also reported suicidal behavior (ESM 1, Tables S10 and S11). One participant reported suicidal behavior at their early termination visit. This individual, who had no previous positive responses reported on the C-SSRS, reported an actual suicide attempt, an interrupted attempt, and the presence of suicidal behavior in addition to an inaccurate report of a completed suicide; this was the same participant who had a suicide attempt reported as a serious severe TEAE (described in Sect. 3.4).
In the second participant, who also reported a TEAE of suicidal ideation, the reported suicide attempt was considered erroneous; this patient self-inflicted a superficial scratch that the patient knew was not dangerous (ESM 1, Table S10). A single participant reported non-suicidal self-injurious behavior on the C-SSRS. In total, across all three measures of psychiatric safety used (TEAE reporting, BPRS-C, and C-SSRS), nine participants reported suicidal ideation and/or behavior on one or more measures.
Efficacy
Attention-Deficit/Hyperactivity Disorder Rating Scale IV Total and Subscale Scores
In the FAS, the baseline mean ADHD-RS-IV total score was 41.2 (SD 7.01) and the mean scores for the subscales hyperactivity/impulsivity and inattention were 19.0 (SD 5.89) and 22.1 (SD 3.46), respectively. Total and subscale scores decreased from baseline to week 4 (total, 16.6 [SD 9.94]; hyperactivity/impulsivity, 7.4 [SD 5.47]; inattention, 9.2 [SD 5.70]) and then continued to decrease gradually before stabilizing at approximately week 48 (total, 13.9 [SD 8.95]; hyperactivity/impulsivity, 5.8 [SD 4.88]; inattention, 8.1 [SD 5.32]) (Fig. 4). At week 104, the mean ADHD-RS-IV total score was 12.8 [SD 8.47] and hyperactivity/impulsivity and inattention subscale scores were 5.0 (SD 4.53) and 7.8 (SD 5.08), respectively. The mean change from baseline to LOTA in ADHD-RS-IV total score and hyperactivity/impulsivity and inattention subscale scores was −25.8 (95% CI −27.0 to −24.5), −12.6 (95% CI −13.4 to −11.9), and −13.1 (95% CI −13.8 to −12.4), respectively (all p < 0.001).
Fig. 4.

a Mean ADHD-RS-IV total score, b inattention subscale score, and c hyperactivity/impulsivity subscale score by visit (full analysis set). ADHD-RS-IV ADHD Rating Scale IV, BL baseline, LOTA last on-treatment assessment, n number of participants with an ADHD-RS-IV score at that week, SD standard deviation
Clinical Global Impressions-Improvement Scale
The proportion of participants categorized as improved based on a CGI-I score of 1 (very much improved) or 2 (much improved) increased from 42.3% at week 1 to 83.0% at week 4 and remained above 80% for the remainder of the study (week 104; 89.1%). At LOTA, 233 of 299 (77.9%) participants were improved based on CGI-I scores (141 participants [47.2%] very much improved and 92 [30.8%] much improved).
Responder Analyses
Based on both of the definitions of clinically relevant response, the proportion of treatment responders increased during weeks 1–4 and then continued to increase steadily before stabilizing at approximately week 72 (ESM 1, Fig. S2). At LOTA, 77.3% of participants had a reduction of at least 30% in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2, and 69.2% had a reduction of at least 50% in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2.
Discussion
ADHD is increasingly recognized as a persistent disorder requiring long-term management [21, 32]. Stimulants are the cornerstone of ADHD pharmacotherapy, and their safety has been characterized in numerous clinical trials [7, 11–16]. However, the short duration of most previous trials limits their value in understanding the longer-term safety of these medications. Here, we report data from the longest safety and efficacy study to date of the prodrug stimulant LDX.
Several aspects of the study design should be considered when interpreting the findings presented here. Strengths of the study include the 2-year duration, the large number of participants enrolled at multiple sites across Europe, and the extensive safety assessments. Limitations include the open-label design and the lack of a placebo control arm, which limit interpretation of results. Individuals with a comorbid psychiatric diagnosis were excluded, which may limit applicability to patient populations seen in clinical practice. In addition, a sizeable proportion of participants (39.5%) were recruited from antecedent studies, which may have enriched the study population with individuals who tolerated LDX. In previous open-label LDX studies of 6–12 months’ duration, substantially higher proportions of patients were recruited from antecedent studies (86–100%) [18–20, 23]. However, this enriched population does reflect the patients who would be expected to receive long-term LDX treatment in clinical practice, because patients who do not tolerate a medication are much less likely to remain on it long term. Finally, patients were excluded if they were well-managed on their existing medication. While this may have influenced the findings, this study population does reflect patients likely to receive LDX in clinical practice, particularly in Europe where LDX is licensed as a second-line treatment in children and adolescents [33].
In SPD489-404, 89.8% of participants receiving LDX reported at least one TEAE, with most reported as mild (35.7%) or moderate (42.4%). Despite its longer duration, the incidence of TEAEs in the present 2-year study was similar to that observed in previously reported open-label studies of 6–12 months’ duration (78–88%) [18–20, 23] and only slightly greater than observed in the LDX treatment arms of randomized controlled trials of 4–9 weeks’ duration (69–79%) [11, 13–15, 22, 34]. These results suggest that the relationship between the incidence of TEAEs is not linearly related to the duration of LDX treatment. The incidence of serious TEAEs was modest (8.9%; 4 of 36 serious events were considered related to LDX) despite the length of the study and the sponsor’s requirement to record any onset of seizures, loss of consciousness, or syncope as a serious event. These findings are consistent with previous 2-year open-label studies of stimulant medications in children with ADHD [35, 36]. In a study of extended-release amphetamines, 92% of children reported at least one TEAE, 3% reported serious TEAEs, and 15% discontinued the study as a result of TEAEs [35]. Similarly, 89.2% of children reported at least one TEAE in a 2-year study of osmotic-release oral system methylphenidate and 7.6% discontinued as a result of TEAEs; the number of serious TEAEs was not reported [36].
The TEAEs most frequently reported by clinical trial participants receiving ADHD stimulant medications included decreased appetite, weight decrease, insomnia, and headache; these were identified as TEAEs of special interest for the present study. Over the 2-year duration of the study, decreased appetite was the most common TEAE, reported in over 50% of participants (in the 14.1 version of MedDRA, the term “decreased appetite” incorporates reports that would have been labelled as “anorexia” in previous versions), and weight decrease was reported as a TEAE for approximately 20% of participants. This compares with reports of decreased appetite, anorexia, and weight decrease TEAEs in 21–33, <5–15, and 16–18% of participants, respectively, in previous LDX studies of 6–12 months’ duration [18, 20, 23]. In addition to cases of weight decrease reported as TEAEs, assessment of all participants’ weight revealed that, although mean weight increased over the course of the study, z scores for weight and BMI did decrease (as did z scores for height), and approximately one-third of participants reported a decrease in weight of PCI. Relative losses in weight, height, and BMI, and slowing down in growth, compared with population norms, were observed previously in children with ADHD receiving LDX for 15 months [37]. Similarly, at the 3-year follow-up of the National Institute of Mental Health Multimodal Treatment Study of ADHD, methylphenidate-treated children were 2.7 kg lighter than un-medicated controls, with the greatest reductions in growth velocity occurring in the first year of treatment [38]. Clinical practice guidelines recommend the ongoing monitoring of height and weight in pediatric patients receiving stimulant ADHD therapies [21], and the effects of long-term LDX treatment on weight, growth, and maturation in study SPD489-404 will be the focus of further detailed analyses.
To avoid dilution of any association of LDX with sleep difficulties, multiple MedDRA terms related to sleep TEAEs (insomnia, initial insomnia, middle insomnia, and terminal insomnia) were combined and analyzed as a single group. When aggregated, insomnia events were reported in approximately 30% of participants. While the incidence of insomnia TEAEs in LDX trials based on aggregated terms has not previously been reported, the single term “insomnia” was reported in 12–17% of children and adolescents in long-term studies [18, 20, 23]. Although stimulants have often been associated with disturbed sleep, the relationship between ADHD, stimulant medications, and sleep is not straightforward. Indeed, ADHD itself is reported to be associated with sleep disturbances [39], and while some data suggest stimulants can have a negative impact on sleep, other data also indicate that some patients may experience no impact, or even a positive impact, on sleep [40–43].
Currently, few data have been published describing the time-course of TEAEs during prolonged stimulant exposure. In SPD489-404, the emergence and duration of TEAEs of special interest were monitored throughout the 2-year study. The incidence of decreased appetite, weight decrease, insomnia, and headache TEAEs peaked early in the study, and the median duration of events ranged from 2 to 225 days. For all TEAEs of special interest, most cases had resolved by the conclusion of the study. This is consistent with the previous observation that overall rates of TEAEs were similar in LDX clinical trials of varying duration (as previously discussed). These results confirm those of a 12-month open-label study of LDX in children and a 12-month study of LDX in adults [18, 19]. Furthermore, a 24-month open-label study of extended-release amphetamines in children with ADHD revealed that over 50% of all TEAEs were reported during the first 6 months of the study [35]. While these findings may suggest an adaptation to LDX-induced TEAEs over time, the possibility of participant withdrawal for reasons of poor tolerability leading to the gradual enrichment of the study population with individuals with good tolerability to LDX cannot be excluded. In addition, some individuals did experience TEAEs that did not start until late in the study, or that continued over long periods of time, highlighting the importance of careful dose titration and individualized and ongoing patient management.
Typical of stimulant ADHD medications, prescribing information for LDX warns of the risk of serious cardiovascular reactions, including sudden death, and recommends that its use is avoided in individuals with pre-existing cardiac abnormalities [33, 44]. In addition, clinical practice guidelines recommend that patients receiving stimulants are regularly monitored for cardiovascular changes [21]. However, large-scale epidemiological studies have not established a strong link between stimulant medications and an increase in the risk of serious cardiac events [45–47]. Cardiovascular-related TEAEs were uncommon in SPD489-404, although increases in mean pulse and blood pressure were observed. The proportion of participants who exceeded pre-specified PCI thresholds ranged from 7.0% (for pulse rate) to 38.8% (for DBP in children), demonstrating that some participants did experience substantial and PCI changes. In many cases, PCI criteria were met at only one post-baseline visit, suggesting that the event was transitory rather than indicative of persistent elevation. Considering the numbers of participants meeting PCI thresholds and the reported range of the mean changes in vital signs, it is perhaps unexpected that clinicians reported so few TEAEs related to changes in blood pressure or heart rate.
Symptomatic improvements were demonstrated throughout this 2-year open-label study, indicating that the efficacy of LDX is maintained in the long term. Symptoms improved rapidly over the first 4 weeks of the study, with a slower rate of improvement observed thereafter. Based on responder analyses, the majority of individuals experienced improvements in symptoms that are likely to be clinically meaningful. Notably, the degree of improvement, based on ADHD-RS-IV and/or CGI-I scores, was similar to that observed in several double-blind, placebo-controlled trials [13–15].
Conclusions
This is the first 2-year study of LDX, and the findings represent an important addition to the pool of long-term clinical data available to enable accurate and informed treatment choices for children and adolescents with ADHD. TEAEs were as expected for the stimulant class of ADHD medication, with decreased appetite, weight decrease, insomnia, and headache among the most commonly reported. Detailed analyses of the incidence and time-course of these commonly reported TEAEs indicated that they peaked early in the study and declined thereafter. Some individuals did experience potentially clinically significant changes in cardiovascular parameters or weight, supporting the recommendations for regular monitoring of patients in clinical practice [21]. LDX treatment was associated with symptomatic improvements that were observed for the 2-year duration of the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank patients and investigators who took part in this study. Under the direction of the authors and funded by Shire International GmbH, Dr. Tamzin Gristwood and Dr. Jim Purvis of Oxford PharmaGenesis, Oxford, UK, provided writing assistance for this publication in addition to editorial assistance in formatting, proofreading, copy-editing, and fact-checking. Dr. David Duesenberg from Shire Development LLC and Dr. Tamara Werner-Kiechle from Shire International GmbH reviewed and edited the manuscript for scientific accuracy. Although employees of the sponsor were involved in the design, collection, analysis, interpretation, and fact-checking of information, the content of this manuscript, the interpretation of the data, and the decision to submit the manuscript for publication in CNS Drugs was made by the authors independently. Dr. Yan served as the statistical expert for this research.
Compliance with Ethical Standards
Funding
The study was funded by Shire Development LLC. Writing and editing assistance for this paper was funded by Shire International GmbH. Shire International GmbH also funded open access.
Conflicts of interest
BC and BY are employees of Shire and own stock or stock options. The following authors have received compensation for serving as consultants or speakers for, or they or the institutions they work for have received research support or royalties from, the companies or organizations indicated: TB (Actelion, Hexal Pharma, Lilly, Medice, Novartis, Otsuka, Shire, and Vifor Pharma); DRC (Eli Lilly, Janssen-Cilag, Medice, Novartis, Oxford University Press, Shire, and Vifor Pharma); PN (Lilly, Otsuka, and Shire); IHO (Alicia Koplowitz Foundation, Eli Lilly, Forest, Janssen-Cilag, Junta de Andalucía, Roche, Shire, Shire Pharmaceuticals Iberica S.L., and Sunovion); CAS (Alicia Koplowitz Foundation, Editorial Médica Panamericana, Eli Lilly, EUNSA [University of Navarra Press], Fundación Caja Navarra, Janssen, Mayo Eds, Medice/Juste, NeuroTech Solutions, Rubiò, Shire, Sociedad Vasco-Navarra de Psiquiatría, University of Navarra Research Projects [PIUNA], and Wolters Kluwer); AZ (Angelini, EduPharma, Lilly, Lundbeck, Otsuka, Oxford University Press, Roche, Shire, Takeda and Vifor Pharma).
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Each patient’s parent/legal guardian provided written, informed consent, and assent was obtained from each participant (as applicable) before they took part in the study.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s40263-017-0443-y) contains supplementary material, which is available to authorized users.
References
- 1.Coghill D. Pragmatic measures in paediatric psychopharmacology—are we getting it right? Eur Neuropsychopharmacol. 2011;21(8):571–583. doi: 10.1016/j.euroneuro.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Faraone SV, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med. 2006;36(2):159–165. doi: 10.1017/S003329170500471X. [DOI] [PubMed] [Google Scholar]
- 3.Lara C, Fayyad J, de Graaf R, Kessler RC, Aguilar-Gaxiola S, Angermeyer M, et al. Childhood predictors of adult attention-deficit/hyperactivity disorder: results from the World Health Organization World Mental Health Survey Initiative. Biol Psychiatry. 2009;65(1):46–54. doi: 10.1016/j.biopsych.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942–948. doi: 10.1176/ajp.2007.164.6.942. [DOI] [PubMed] [Google Scholar]
- 5.Polanczyk GV, Willcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol. 2014;43(2):434–442. doi: 10.1093/ije/dyt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkley RA, Fischer M, Smallish L, Fletcher K. The persistence of attention-deficit/hyperactivity disorder into young adulthood as a function of reporting source and definition of disorder. J Abnorm Psychol. 2002;111(2):279–289. doi: 10.1037/0021-843X.111.2.279. [DOI] [PubMed] [Google Scholar]
- 7.Seixas M, Weiss M, Muller U. Systematic review of national and international guidelines on attention-deficit hyperactivity disorder. J Psychopharmacol. 2012;26(6):753–765. doi: 10.1177/0269881111412095. [DOI] [PubMed] [Google Scholar]
- 8.Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317–327. doi: 10.2147/NDT.S9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharman J, Pennick M. Lisdexamfetamine prodrug activation by peptidase-mediated hydrolysis in the cytosol of red blood cells. Neuropsychiatr Dis Treat. 2014;10:2275–2280. doi: 10.2147/NDT.S70382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ermer JC, Pennick M, Frick G. Lisdexamfetamine dimesylate: prodrug delivery, amphetamine exposure and duration of efficacy. Clin Drug Investig. 2016;36:341–356. doi: 10.1007/s40261-015-0354-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adler LA, Goodman DW, Kollins SH, Weisler RH, Krishnan S, Zhang Y, et al. Double-blind, placebo-controlled study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69(9):1364–1373. doi: 10.4088/JCP.v69n0903. [DOI] [PubMed] [Google Scholar]
- 12.Biederman J, Boellner SW, Childress A, Lopez FA, Krishnan S, Zhang Y. Lisdexamfetamine dimesylate and mixed amphetamine salts extended-release in children with ADHD: a double-blind, placebo-controlled, crossover analog classroom study. Biol Psychiatry. 2007;62(9):970–976. doi: 10.1016/j.biopsych.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Biederman J, Krishnan S, Zhang Y, McGough JJ, Findling RL. Efficacy and tolerability of lisdexamfetamine dimesylate (NRP-104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther. 2007;29(3):450–463. doi: 10.1016/S0149-2918(07)80083-X. [DOI] [PubMed] [Google Scholar]
- 14.Coghill D, Banaschewski T, Lecendreux M, Soutullo C, Johnson M, Zuddas A, et al. European, randomized, phase 3 study of lisdexamfetamine dimesylate in children and adolescents with attention-deficit/hyperactivity disorder. Eur Neuropsychopharmacol. 2013;23(10):1208–1218. doi: 10.1016/j.euroneuro.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Findling RL, Childress AC, Cutler AJ, Gasior M, Hamdani M, Ferreira-Cornwell MC, et al. Efficacy and safety of lisdexamfetamine dimesylate in adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(4):395–405. doi: 10.1016/j.jaac.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Wigal SB, Kollins SH, Childress AC, Squires L, Study G. A 13-hour laboratory school study of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. Child Adolesc Psychiatry Ment Health. 2009;3(1):17. doi: 10.1186/1753-2000-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coghill DR, Caballero B, Sorooshian S, Civil R. A systematic review of the safety of lisdexamfetamine dimesylate. CNS Drugs. 2014;28(6):497–511. doi: 10.1007/s40263-014-0166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Findling RL, Childress AC, Krishnan S, McGough JJ. Long-term effectiveness and safety of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. CNS Spectr. 2008;13(7):614–620. doi: 10.1017/S1092852900016898. [DOI] [PubMed] [Google Scholar]
- 19.Weisler R, Young J, Mattingly G, Gao J, Squires L, Adler L, et al. Long-term safety and effectiveness of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. CNS Spectr. 2009;14(10):573–585. doi: 10.1017/S1092852900024056. [DOI] [PubMed] [Google Scholar]
- 20.Findling RL, Cutler AJ, Saylor K, Gasior M, Hamdani M, Ferreira-Cornwell MC, et al. A long-term open-label safety and effectiveness trial of lisdexamfetamine dimesylate in adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2013;23(1):11–21. doi: 10.1089/cap.2011.0088. [DOI] [PubMed] [Google Scholar]
- 21.National Institute for Health and Clinical Excellence (NICE). The NICE guideline on diagnosis and management of ADHD in children, young people and adults. Londong: NICE; 2013. http://www.nice.org.uk/guidance/cg72. Accessed 17 Sept 2015.
- 22.Dittmann RW, Cardo E, Nagy P, Anderson CS, Bloomfield R, Caballero B, et al. Efficacy and safety of lisdexamfetamine dimesylate and atomoxetine in the treatment of attention-deficit/hyperactivity disorder: a head-to-head, randomized, double-blind, phase IIIb study. CNS Drugs. 2013;27(12):1081–1092. doi: 10.1007/s40263-013-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coghill DR, Banaschewski T, Lecendreux M, Johnson M, Zuddas A, Anderson CS, et al. Maintenance of efficacy of lisdexamfetamine dimesylate in children and adolescents with attention-deficit/hyperactivity disorder: randomized-withdrawal study design. J Am Acad Child Adolesc Psychiatry. 2014;53(6):647 e1–657 e1. doi: 10.1016/j.jaac.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Findling RL, Ginsberg LD, Jain R, Gao J. Effectiveness, safety, and tolerability of lisdexamfetamine dimesylate in children with attention-deficit/hyperactivity disorder: an open-label, dose-optimization study. J Child Adolesc Psychopharmacol. 2009;19(6):649–662. doi: 10.1089/cap.2008.0165. [DOI] [PubMed] [Google Scholar]
- 25.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, et al. 2000 CDC growth charts for the United States: methods and development. Vital Health Stat 11. 2002;246:1–190. [PubMed] [Google Scholar]
- 26.Hughes CW, Rintelmann J, Emslie GJ, Lopez M, MacCabe N. A revised anchored version of the BPRS-C for childhood psychiatric disorders. J Child Adolesc Psychopharmacol. 2001;11(1):77–93. doi: 10.1089/104454601750143500. [DOI] [PubMed] [Google Scholar]
- 27.Posner K. Suicidality issues in clinical trials: Columbia suicidal adverse event identification in FDA safety analyses. 2007. http://www.fda.gov/ohrms/dockets/ac/07/slides/2007-4306s1-01-CU-Posner.ppt. Accessed March 24.
- 28.Chamberlain SR, Robbins TW, Winder-Rhodes S, Muller U, Sahakian BJ, Blackwell AD, et al. Translational approaches to frontostriatal dysfunction in attention-deficit/hyperactivity disorder using a computerized neuropsychological battery. Biol Psychiatry. 2011;69(12):1192–1203. doi: 10.1016/j.biopsych.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Morris NM, Udry JR. Validation of a self-administered instrument to assess stage of adolescent development. J Youth Adolesc. 1980;9(3):271–280. doi: 10.1007/BF02088471. [DOI] [PubMed] [Google Scholar]
- 30.DuPaul GJ, Power TJ, Anastopoulos AD, Reid R. ADHD Rating Scale-IV: Checklists, norms, and clinical interpretation. New York: Guilford Press; 1998. [Google Scholar]
- 31.Guy W. Clinical global impression scale. ECDEU Assess Man Psychopharmacol-Revis Vol DHEW Publ No ADM. 1976;76(338):218–222. [Google Scholar]
- 32.Kooij SJ, Bejerot S, Blackwell A, Caci H, Casas-Brugue M, Carpentier PJ, et al. European consensus statement on diagnosis and treatment of adult ADHD: The European Network Adult ADHD. BMC Psychiatry. 2010;10:67. doi: 10.1186/1471-244X-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elvanse 20 mg, 30 mg, 40 mg, 50 mg, 60 mg and 70 mg capsules, hard [prescribing information]. Hampshire: Shire Pharmaceuticals Limited; 2016. https://www.medicines.org.uk/emc/medicine/27442/SPC/Elvanse+30mg,+50mg+%26+70mg+Capsules,+hard/. Accessed 12 Nov.
- 34.Adler LA, Dirks B, Deas PF, Raychaudhuri A, Dauphin MR, Lasser RA, et al. Lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder who report clinically significant impairment in executive function: results from a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(7):694–702. doi: 10.4088/JCP.12m08144. [DOI] [PubMed] [Google Scholar]
- 35.McGough JJ, Biederman J, Wigal SB, Lopez FA, McCracken JT, Spencer T, et al. Long-term tolerability and effectiveness of once-daily mixed amphetamine salts (Adderall XR) in children with ADHD. J Am Acad Child Adolesc Psychiatry. 2005;44(6):530–538. doi: 10.1097/01.chi.0000157550.94702.a2. [DOI] [PubMed] [Google Scholar]
- 36.Wilens T, McBurnett K, Stein M, Lerner M, Spencer T, Wolraich M. ADHD treatment with once-daily OROS methylphenidate: final results from a long-term open-label study. J Am Acad Child Adolesc Psychiatry. 2005;44(10):1015–1023. doi: 10.1097/01.chi.0000173291.28688.e7. [DOI] [PubMed] [Google Scholar]
- 37.Faraone SV, Spencer TJ, Kollins SH, Glatt SJ. Effects of lisdexamfetamine dimesylate treatment for ADHD on growth. J Am Acad Child Adolesc Psychiatry. 2010;49(1):24–32. doi: 10.1097/00004583-201001000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Swanson JM, Elliott GR, Greenhill LL, Wigal T, Arnold LE, Vitiello B, et al. Effects of stimulant medication on growth rates across 3 years in the MTA follow-up. J Am Acad Child Adolesc Psychiatry. 2007;46(8):1015–1027. doi: 10.1097/chi.0b013e3180686d7e. [DOI] [PubMed] [Google Scholar]
- 39.Cortese S, Faraone SV, Konofal E, Lecendreux M. Sleep in children with attention-deficit/hyperactivity disorder: meta-analysis of subjective and objective studies. J Am Acad Child Adolesc Psychiatry. 2009;48(9):894–908. doi: 10.1097/CHI.0b013e3181ac09c9. [DOI] [PubMed] [Google Scholar]
- 40.Faraone SV, Glatt SJ, Bukstein OG, Lopez FA, Arnold LE, Findling RL. Effects of once-daily oral and transdermal methylphenidate on sleep behavior of children with ADHD. J Atten Disord. 2009;12(4):308–315. doi: 10.1177/1087054708314844. [DOI] [PubMed] [Google Scholar]
- 41.Giblin JM, Strobel AL. Effect of lisdexamfetamine dimesylate on sleep in children with ADHD. J Atten Disord. 2011;15(6):491–498. doi: 10.1177/1087054710371195. [DOI] [PubMed] [Google Scholar]
- 42.Kim HW, Yoon IY, Cho SC, Kim BN, Chung S, Lee H, et al. The effect of OROS methylphenidate on the sleep of children with attention-deficit/hyperactivity disorder. Int Clin Psychopharmacol. 2010;25(2):107–115. doi: 10.1097/YIC.0b013e3283364411. [DOI] [PubMed] [Google Scholar]
- 43.Kidwell KM, Van Dyk TR, Lundahl A, Nelson TD. Stimulant medications and sleep for youth with ADHD: a meta-analysis. Pediatrics. 2015;136(6):1144–1153. doi: 10.1542/peds.2015-1708. [DOI] [PubMed] [Google Scholar]
- 44.Vyvanse [prescribing information]. Lexington, MA: Shire LLC; 2017. http://pi.shirecontent.com/pi/pdfs/vyvanse_usa_eng.pdf. Accessed Nov 12.
- 45.Martinez-Raga J, Knecht C, Szerman N, Martinez MI. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15–30. doi: 10.1007/s40263-012-0019-9. [DOI] [PubMed] [Google Scholar]
- 46.Westover AN, Halm EA. Do prescription stimulants increase the risk of adverse cardiovascular events? A systematic review. BMC Cardiovasc Disord. 2012;12(1):41. doi: 10.1186/1471-2261-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winterstein AG. Cardiovascular safety of stimulants in children: findings from recent population-based cohort studies. Curr Psychiatry Rep. 2013;15(8):379. doi: 10.1007/s11920-013-0379-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.