

Abstract
Host-gut microbial interactions contribute to human health and disease states and an important manifestation resulting from this co-metabolism is a vast diversity of bile acids (BAs). There is increasing interest in using BAs as biomarkers to assess the health status of individuals, and therefore, an increased need for their accurate separation and identification. In this study, the negative ion fragmentation behaviors of C24 BAs were investigated by UPLC-ESI-QTOF-MS. The step-by-step fragmentation analysis revealed a distinct fragmentation mechanism for the unconjugated BAs containing a 12-hydroxyl group. The unconjugated BAs lacking 12-hydroxylation fragmented via dehydration and dehydrogenation. In contrast, the 12-hydroxylated ones, such as deoxycholic acid (DCA) and cholic acid (CA), employed dissociation routes including dehydration, loss of carbon monoxide or carbon dioxide, and dehydrogenation. All fragmentations of the 12-hydroxylated unconjugated BAs, characterized by means of stable isotope labeled standards, were associated with the rotation of the carboxylate side-chain and the subsequent rearrangements accompanied with proton transfer between 12-hydroxyl and 24-carboxyl groups. Compared to DCA, CA underwent further cleavages of the steroid skeleton. Accordingly, the effects of stereochemistry on the fragmentation pattern of CA were investigated using its stereoisomers. Based on the knowledge gained from the fragmentation analysis, a novel BA, 3β, 7β, 12α-trihydroxy-5β-cholanic acid, was identified in the postprandial urine samples of patients with nonalcoholic steatohepatitis. The analyses used in this study may contribute to a better understanding of the chemical diversity of BAs and the molecular basis of human liver diseases that involve BA synthesis, transport, and metabolism.
Graphical abstract

Bile acids (BAs) are the amphipathic products of cholesterol metabolism in vertebrates1. They play irreplaceable roles in solubilizing lipids and fat-soluble vitamins2, eliminating catabolites such as bilirubin, and aiding in the reduction of the bacteria populations in the biliary tract and small intestine3. The physiological role of BAs as important metabolic signaling molecules that modulate lipid, glucose, and energy metabolism has also gained a lot of recognition4. Because of an enterohepatic circulation driven by a series of membrane transporters5, composition of the human BA pool is entangled with a complex host-gut microbial co-metabolism6. In humans, the primary BAs, cholic acid (CA), chenodeoxycholic acid (CDCA) and their taurine and glycine conjugates, are synthesized via cytochrome P450-mediated oxidation of cholesterol through a multi-step process7. They are secreted into the gallbladder where they are stored until postprandial release into the intestine. The secondary BAs are formed in the intestine by a variety of reactions carried out by the gut microbiota, including deconjugation, 7-dehydroxylation, dehydrogenation and epimerization of hydroxyl groups. The majority (>95%) of both the primary and secondary BAs is reabsorbed by the intestine and returned to the liver where some of them undergo sulfation and glucuronidation8,9.
Human BAs are C24 molecules comprised of a C19 cyclopentanophenanthrene (steroid) nucleus and a carboxylate side-chain (Scheme 1)1,10. The structural diversities of human BAs come from several factors: (1) A/B ring fusion stereochemistry, cis/5β-H or trans/5α-H; (2) sites of hydroxylation which can occur at C3, C6, C7 and C12; (3) conjugation of glycine or taurine at the C24-carboxyl group; and (4) dehydrogenation and epimerization of hydroxyl groups. The first three factors are mainly derived from host metabolism and the last one is attributed to gut microbial biotransformation. Considering only the host synthesis and metabolism of BAs, 48 possible BA species with oxidation sites at C3, C6, C7 and/or C12 can be produced. This number increases to 384 if we consider the entire host-gut microbial co-metabolism. Therefore, it is extremely challenging to detect and differentiate the whole spectrum of BAs including various isomers and stereoisomers, many of which are present in trace amounts in biological samples. The sulfation and glucuronidation of BAs were therefore not considered in this study due to the vastly increased BA speciation, as demonstrated in the most recent analytical work of BA sulfates11.
Scheme 1.

The 2D and 3D structure of C24 bile acids highlighting the stereochemistry and common hydroxylation sites.
Bioanalysis of BAs used to be conducted by GC-MS and currently relies on LC-MS12. Excluding BA sulfates and glucuronides, the most advanced LC-MS method has detected about 50 BAs with reference to standards11,13. Characterization of unknown BAs in biological samples relies heavily on the understanding of their fragmentation mechanisms. As Murphy summarized recently14, little attention has been paid to probing the mechanistic details probably because their reported fragmentation behaviors are rather straightforward15. Particularly for the unconjugated BAs, it was generally supposed that they do not readily fragment after collisional activation of their [MH]− anion. As far as we know, there was only one report that demonstrated the characteristic fragmentation behaviors of some unconjugated BA isomers (CDCA, UDCA, HDCA and DCA), and the fragmentation mechanism was not clear16. As a result, the pseudo-multiple reaction monitoring (MRM) method has been commonly employed for the determination of BAs13,17–19. Using this kind of technique, the differentiation of unconjugated BA isomers and stereoisomers depends solely on the resolution and robustness of chromatographic methods and the characterization of novel BAs is challenging.
In this study, our overall objective was to investigate the fragmentation behaviors of BAs. We had two specific objectives, in particular, for the unconjugated ones. The first was to understand how changes in hydroxylation sites on the steroid skeleton affected the fragmentation behaviors. The second was to determine if the stereochemistry made a difference to the fragmentation patterns. A major finding was that the presence of 12-hydroxylation committed the fragmentation down a pathway that involved the rotation of the carboxylate side-chain and the subsequent rearrangements accompanied with proton transfer between the 12-hydroxyl and 24-carboxyl groups. The elucidation of this fragmentation mechanism allowed us to identify a previously unknown BA in patient urine samples, 3β, 7β, 12α-trihydroxy-5β-cholanic acid. We believe that the knowledge gained in this work could be of great importance in the bioanalysis of BAs.
EXPERIMENTAL SECTION
Materials
Reference standards of 32 unconjugated BAs, 8 glycine-conjugated BAs and 11 taurine-conjugated BAs were purchased from Steraloids (Newport, RI), TRC (Toronto, Canada), or Sigma-Aldrich (St. Louis, MO). Their names and structures are summarized in Table S1. The stable isotope labelled BAs, including lithocholic acid-2,2,4,4-D4 (LCA-D4), ursodeoxycholic acid-2,2,4,4-D4 (UDCA-D4), deoxycholic acid-2,2,4,4-D4 (DCA-D4), cholic acid-2,2,4,4-D4 (CA-D4), glycochenodeoxycholic acid-2,2,4,4-D4 (GCDCA-D4), glycodeoxycholic acid-2,2,4,4-D4 (GDCA-D4), glycocholic acid-2,2,4,4-D4 (GCA-D4), deoxycholic acid-24-13C (DCA-24-13C) were purchased from Sigma-Aldrich or IsoSciences (King of Prussia, PA). 18O-water (97% isotopic purity) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Trifluoroacetic acid (TFA) was purchased from Thermo Fisher Scientific (Waltham, MA). The LC-MS grade methanol, acetonitrile and formic acid were obtained from Sigma-Aldrich (St. Louis, MO). Ultra-pure water was obtained by using a Milli-Q system (Millipore, Bedford).
Preparation of standard samples
The 5 mM stock solutions of each BA standard were prepared in methanol. The 5 μM or 50 μM standard samples were individually prepared by diluting the 5 mM stock solution with water-acetonitrile-formic acid (50:50:0.1, v/v). In order to be equivalent to the analytical conditions for biological samples, each BA was individually injected into the chromatographic column to obtain its chromatographic retention and fragmentation data.
Preparation of DCA-24-18O2
18O labelled DCA was prepared by acid-catalyzed oxygen exchanging method. In brief, aliquot of DCA stock solution (2 μL) was incubated at 90 °C for 1 h with 198 μL 18O-water and 2 μL TFA. In the resulting 50 μM sample, both carboxylate oxygen atoms of DCA were replaced by 18O with an isotopic purity more than 90%.
Preparation of human urine
The urine samples were collected and pooled over the 2-hour postprandial period from 7 patients with nonalcoholic steatohepatitis (NASH)20. The common urinary BAs in these samples have been quantitatively profiled in our previous work20. Using the same sample preparation method, we reanalyzed these samples by UPLC-QTOF-MS. Briefly, a 100 μL sample aliquot was evaporated to dryness. The residue was reconstituted with 50 μL each of mobile phases A and B. After centrifugation, the supernatant was analyzed.
UPLC-QTOF-MS Analysis
The analysis was performed on a Waters Acquity UPLC system coupled with a Xevo G2S Q-TOFMS (Waters, Milford, MA, USA) via electrospray ionization (ESI) interface. The chromatographic method was optimized according to our previously reported protocol19. The mobile phases consisted of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile-methanol (95:5, mobile phase B). 5 μL of each sample was injected onto an ACQUITY BEH C18 column (1.7 μm, 100 mm × 2.1 mm) (Waters, Milford, MA) maintained at 35 °C. The flow rate was 0.45 mL/min with the following mobile phase gradient: 0.0–0.5 min (5% B), 0.5–1.0 min (5–20% B), 1.0–2.0 min (20–25% B), 2.0–5.5 min (25% B), 5.5–6.0 min (25–30% B), 6.0–8.0 min (30% B), 8.0–9.0 min (30–35% B), 9.0–17.0 min (35–65% B), 17.0–18.0 min (65–100% B), 18.0–19.0 min (100% B) and 19.0–20.0 (5% B). The mass spectrometer was operated in the negative mode with a 3.0 kV capillary voltage. The source and desolvation temperatures were 150 and 550 °C, respectively. Nitrogen and argon were used as cone and collision gases, respectively. The cone gas flow and desolvation gas flow were set at 50 and 950 L/h, respectively.
The fragments data were collected by MS/MS scans (0.036 s/scan) of the [M-H]− anion under various collision energies (CE) from m/z 50–600 in centroid mode. For analysis of 5 μM standard samples and urine samples, the LM resolution was set at 4.7 and the HM resolution was set at 15.0. The resulting MS/MS spectra incorporated isotopic contributions. In order to narrow down the quadrupole transmission window and exclude isotopic contributors, the LM resolution was set at 25.0, where the 50 μM standard samples were analyzed to compensate the sacrificed sensitivity. Leucine enkephalin was infused via the reference probe as lockspray to ensure m/z accuracy.
Data processing
The UPLC-QTOF-MS raw data was processed by MassLynx (V4.1, Waters, Milford, MA, USA). The ion chromatogram of each BA was acquired at the pseudo-MRM transition ([M-H]− > [M-H]−) at CE of 20 V for the unconjugated and glycine-conjugated BAs and 40 V for the taurine-conjugated BAs. The retention data were recorded after smoothing (mean method, twice at the window size of ±1 scan). The MS/MS spectrum of each BA was obtained by combining the scans within the whole peak width (about 10–20 scans) and subtracting the baseline.
RESULTS AND DISCUSSION
Chromatographic behaviors associated with stereochemistry
In this work, the high-definition fragmentation data of each BA standard was individually captured by product ion scans after chromatographic separation. The technique was consistently used for the subsequent analysis of unknown BAs in urine samples. Figure 1 summarizes the acquired ion chromatograms of all BA standards with each pseudo-MRM transition color-coded. The retention time (tR) of each BA is listed in Table S1. Some isomers that are detected in the same MRM transition were a challenge to separate, such as 3,7,12-trihydroxy-cholanic acid and 3,6,7-trihydroxy-cholanic acid, which each have a theoretical total of sixteen stereoisomers. The resolution of seven of them, CA, ACA, HCA, βMCA, αMCA, ωMCA and UCA, has been reported13. It was found in this study that βCA and ωMCA were co-eluted. We thought that the LC-MS/MS profiling of BAs should consider the possibility of such co-elution. Therefore the differentiation of them is in need for unique MRM transitions based on fragmentation studies.
Figure 1.

The ion chromatograms of 32 unconjugated bile acids (U01–U32), 8 glycine-conjugated bile acids (G01–G08) and 11 taurine-conjugated bile acids (T01–T11).
In order to gain a preliminary insight into the effects of stereochemistry on the retention time of BAs, the retention data of 15 pairs of stereoisomers were comparatively analyzed. As shown in Figure S1A, the shorter retention time was observed for the trans/5α-H A/B ring fusion relative to the cis/5β-H A/B ring fusion. Such a trend required further characterization with more standards. Figure S1B illustrates the effect of hydroxyl orientation. The BAs containing a β-hydroxyl group generally had longer retention time than the corresponding isomer with an α-hydroxyl group. Two exceptions to this trend were αMCA and βMCA and their taurine-conjugates. Visually associated with the 3D structure illustrated in Scheme 1, the decreased chromatographic retention by β-hydroxylation appeared to be associated with the structural variations that potentially disturb the amphipathic conformation. The general trend that β-hydroxylation reduces the retention time may aid in the characterization of unknown BA stereoisomers in biological samples.
Optimization of collision energy
In order to capture the complete fragmentation data for various kinds of BAs, the dissociations of the unconjugated, glycine-conjugated and taurine-conjugated BAs were comparatively investigated under different collision energies. Our data was consistent with the results of CDCA, GCDCA and TCDCA recently acquired by an API 5000 triple quadrupole mass spectrometer15. The unconjugated and glycine-conjugated BAs were dissociated at the collision voltage from 20 V to 40 V, while the collision voltage range for dissociation of the taurine-conjugated BAs was from 40 to 60 V (Figure S2). About 90–95% of the unconjugated, glycine-conjugated and taurine-conjugated BAs were respectively dissociated at CEs of 40, 30 and 60 V, at which the abundance of fragment ions also evolved the most. The MS/MS spectra of analytes were thus associated with the above CE levels unless specified otherwise. The fragmentation studies were carried out step-by-step according to the complexity of structures.
A decisive role for 12-hydroxylation in the fragmentation pattern of unconjugated BAs
The MS/MS spectra of the representative unconjugated BAs are illustrated in Figure 2. It was evident from the fragmentation patterns of BAs with 12-hydroxylation, that this functional group played a special role in their dissociation mechanism. The product ions were few in number in the spectra of those BAs without 12-hydroxylation, such as LCA, CDCA, HDCA and HCA. The major detected fragments in their spectra were the dehydrated product ions. In contrast, a variety of fragments were detected in the spectra of those BAs possessing a 12-hydroxyl group, such as DCA, isoDCA and CA. The product ion, [M-H-CH2O2]− was the major fragment of DCA and isoDCA. For CA, [M-H-H2O-CH2O2]− was one of the major fragments. This characteristic fragmentation was briefly mentioned in a recent review12, and it has been proposed that it allows the differentiation of CDCA, UDCA, HDCA and DCA16. The product ion [M-H-CH2O2]− of DCA was proposed to be derived from the loss of a formic acid (HCOOH)15 but no explanation was given as to why loss of formic acid was not observed in the spectra of those BAs without 12-hydroxylation. In the experiments described below we confirmed that the dissociation mechanisms involve the rotation of the carboxylate side-chain and the subsequent rearrangements accompanied with proton transfer between 12-hydroxyl and 24-carboxyl groups. Characterizations of major dissociation mechanisms are discussed in the below sections using representative examples.
Figure 2.
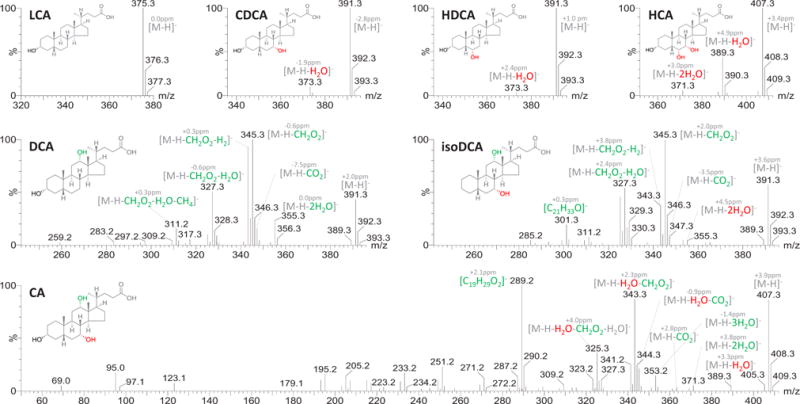
MS/MS spectra of the representative unconjugated BAs (5 μM).
Dehydration and dehydrogenation of the unconjugated BAs without 12-hydroxylation
Dehydration was almost the exclusive fragmentation pathway detected in the spectra of the unconjugated BAs without 12-hydroxylation. As shown in Figures S3, the dehydrated product ions were not detected in LCA, LCA-D4, isoLCA and alloLCA, but became detectable in those with additional hydroxyl groups at C7 or/and C6 (CDCA, UDCA, βUDCA, HDCA, βHDCA, muroCA and HCA). It was therefore concluded that the 6-hydroxyl group and 7-hydroxyl group were more inclined to be dehydrated than the 3-hydroxyl group regardless of stereochemistry. This conclusion was validated by the spectrum of UDCA-D4, in which the dehydrated product ion of the 7β-hydroxyl group ([M-H-H2O]−) rather than that of 3α-hydroxyl group ([M-H-DHO]−) was detected.
Besides dehydration, dehydrogenation was another fragmentation pathway detected in the spectra of HCA and its stereoisomers including αMCA, βMCA and ωMCA (Figure S4). It was observed that the hydroxyl orientations at C6 and C7 have a significant impact on the dehydrogenation. Both ωMCA and αMCA have a trans-conformation of the two hydroxyl groups at C6 and C7 (6α, 7β or 6β, 7α) and underwent more dehydrogenation. Following the dehydrogenation, another product ion [M-H-H2-CH2O]− (ωMCA for example, m/z 375.2553, bias = 3.2ppm) evolved in the spectra of ωMCA and αMCA. The fragmentation mechanisms of αMCA and ωMCA require further confirmatory studies, however, their dehydrogenation behavior is especially useful in their discrimination from a variety of trihydroxyl BAs.
The detail fragmentation mechanisms of DCA involving interaction between 12-hydroxyl group and 24-carboxyl group
As the simplest case of a naturally-occurring, unconjugated, 12-hydroxylated BA, DCA served as an ideal model to demonstrate why the 12-hydroxyl group plays such a determining role in the fragmentation behavior. The studies of DCA were done using a comparative analysis of its analogues including NorDCA, DCA-24-13C, DCA-24-18O2 and DCA-D4 (Figure 3). Deciphering of the dissociation mechanisms started with the analysis of a twice dehydrated ion, [M-H-2H2O]−, which was generally hypothesized from the dehydration of both the 3α- and 12α-hydroxyl groups. In the study of DCA-D4, however, it was observed that the 3α-hydroxyl group did not contribute to either of the two dehydrations. We then proposed that the 24-carboxyl group was somehow involved in the gas-phase decomposition of DCA by interaction with the 12-hydroxyl group.
Figure 3.

MS/MS spectra of NorDCA, DCA, DCA-24-13C, DCA-24-18O2 and DCA-D4 (50 μM).
As shown in Scheme 2, the involvement of 24-carboxyl group in the dissociation of DCA was believed to be initialized by the rotation of carboxylate side-chain that presumably happens during ESI. The resulting hydrogen-bonded structure stabilizes the negative charge and facilitates the proton transfer between the carboxylate and the 12-hydroxyl group. There were at least three possible mechanisms for the major fragment [M-H-CH2O2]−. The first mechanism might be associated with the formation of an unstable octalactone intermediate via dehydration (Scheme 2A). However, it was not clear how the lactone intermediate underwent the second dehydration. The other two mechanisms were associated with ion-dipole complexes that have been well established for gas-phase decomposition21. Two kinds of ion-neutral complexes were proposed according to the proton transfer among 12-hydroxyl group, 24-carboxyl group and its α-hydrogen. The first one was a formate complex that rearranges from a HO-C=O anion complex (Scheme 2B) and the other was a hydroxide complex (Scheme 2C). Both mechanisms explained the loss of 24-carbon in the MS/MS spectrum of DCA-24-13C where the mass loss corresponding to [M-H-13CH2O2]− was detected (Figure 3). Compared to the formate complex, the hydroxide complex explained well the neutral loss of carbon monoxide (CO) and the whole series of fragment ions including [M-H-H2O]−, [M-H-2H2O]−, [M-H-H2O-CO]− and [M-H-2H2O-CO]− detected in the spectra of NorDCA, DCA, DCA-D4 and DCA-24-13C. The concerted losses of oxygen atoms at specific sites were additionally confirmed by the MS/MS spectrum of DCA-24-18O2 where mass losses corresponding to [M-H-H218O]−, [M-H-H218O-H2O]− and [M-H-H218O-C18O]− were detected (Figure 3). Some other dissociation mechanisms including loss of 24-carboxylate (CO2), demethylation and dehydrogenation were also observed in the spectra of DCA and its analogues. Since the loss of CO2 has never been detected in the unconjugated BAs lacking 12-hydroxylation, the way that CO2 is dissociated from DCA is worthy of discussion. We hypothesized that loss of CO2 was also initiated by the rotation of the carboxylate side-chain. The intramolecular hydrogen bond formed between 12-hydroxyl and 24-carboxyl groups could potentially facilitate the transfer of negative charge to the 12-hydroxyl group. This mechanism could explain the presence of the fragment ions [M-H-13CO2]− and [M-H-C18O2]− in the spectra of DCA-24-13C and DCA-24-18O2, respectively. Following the loss of CO2 or CO, the dehydration of 3-hydroxyl group and the subsquent demethylation were observed. Dehydrogenation was observed along the whole fragmentation pathway, especially from the product ion [M-H-H2O-CO]−. The proposed CID fragmentation pathways for DCA are illustrated in Scheme 2. The high-definition m/z bias was less than ±5 ppm for major fragment ions except for some unstable intermediates detected with a low intensity. Our hypothesis at this point is that 12-hydroxylation provides a common fragmentation pathway for BAs that have this group.
Scheme 2.
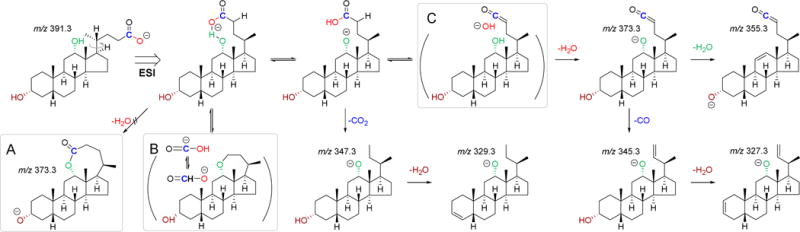
The proposed negative ion CID fragmentation pathways of DCA. The mechanism via an octalactone intermediate (A), the mechanism via a formate complex (B) and the mechanism via a hydroxide complex (C).
Similar fragmentation mechanisms were seen for CA relative to DCA but with additional cleavage of the steroid skeleton
Elucidation of the dissociation mechanisms for DCA greatly facilitated the fragmentation analysis of CA. The spectra of NorCA, CA and CA-D4 are comparatively shown in Figure 4. After the dehydration of the 7α-hydroxyl group, the following fragmentation of CA was similar to that of DCA as it involved the rotation of the carboxylate side-chain and the subsequent rearrangements (Scheme 3). Mass losses corresponding to [M-H-2H2O-CO]− and [M-H-H2O-CO2]− were both detected in parallel to those seen for DCA. The fragmentation of CA showed a more distinct cleavage pattern of the steroid skeleton not observed for DCA, resulting in a series of product ions within the m/z range of 180–300, such as [C19H29O2]−, [C16H27O2]−, [C15H21O2]− and [C13H23O]−. The most diagnostic ring-cleavage fragment ion of CA was [C19H29O2]− (m/z 289.2166, −2.4 ppm). Almost the same fragment pattern with a mass difference of CH2 was observed for CA and NorCA, indicating that the ring-cleavage occurred in either ring-A and/or ring-B rather than ring-C or ring-D. Confirmation of the ring cleavage positions came from the analysis of the MS/MS spectrum of CA-D4. According to the loss of deuterium on the corresponding fragments of CA-D4, [C19H29O2]− was proposed to be derived from the cleavage of ring-A form C1–C10 to C3–C4 (A1,10-3,4), and [C16H27O2]− and [C14H21O]− was proposed to be derived from the cleavages of ring-B form C5–C6 to C9–C10 (B5,6–9,10) and C6–C7 to C9–C10 (B6,7–9,10), respectively. The putative product ions derived from ring cleavages of CA are illustrated in Scheme 3.
Figure 4.

MS/MS spectra of NorCA, CA-D4, CA, βCA, UCA and ACA (50 μM).
Scheme 3.

The proposed negative CID fragmentation pathways of CA, βCA, UCA and ACA.
Effects of stereochemistry on the fragmentation patterns of 3,7,12-trihydroxy-cholanic acid
In theory, there are sixteen stereoisomers of 3,7,12-trihydroxy-cholanic acid when one considers the possible A/B ring fusion styles and hydroxyl orientations. Three stereoisomers, βCA, UCA and ACA, were selected to initially evaluate the effects of stereochemistry on the dissociation patterns (Figure 4). The 3β-hydroxyl group did not alter the fragmentation pattern, while both the 7β-hydroxyl group in UCA and the trans A/B ring juncture in ACA had a significant impact on the observed fragmentation patterns relative to CA. The major changes in the spectrum of UCA in contrast to that of CA were the significantly altered ring cleavage dissociations. The A1,10–3,4 cleavage of UCA was attenuated, resulting in much less intensity of the m/z 289.2 diagnostic ion. The B5,6–9,10 cleavage of UCA was undetectable, leading to the disappearance of the m/z 251.2 fragment. A novel fragment ion, [C17H21O]− (m/z 241.2), was indicative of the A1,10–4,5 cleavage of UCA (Scheme 3).
The differences in the fragmentation of ACA from that of CA were more complex because the trans A/B ring juncture of ACA results in a “planar” skeleton. Although C24 carboxyl side chain rotation was the initiation of fragmentation parallel to CA, the dissociation of ACA differed from that of CA in two important ways. First, the 7α-hydroxyl group of ACA did not undergo dehydration prior to the dissociations of the intermediate with an intramolecular hydrogen bond. As a result, [M-H-H2O-CO]− (m/z 361.3) instead of [M-H-2H2O-CO]− (m/z 343.3) became the major diagnostic fragment of this pathway. Secondly, there were additional ring-cleavage mechanisms of ACA, producing diagnostic fragments such as [C21H33O2]− (m/z 317.2) and [C20H31O2]− (m/z 303.2) respectively from A1,2–3,4 and A1,10–3,4 cleavage mechanisms (Scheme 3). Despite that the underlying mechanisms remain unclear, this is the first report that highlights the difference in fragmentation patterns between the stereoisomers of BAs. The observation may turn out to be extremely useful for the analysis of this group of analytes in biological samples as some of them may have very similar chromatographic behaviors.
Validation of the dissociation mechanisms for 12-hydroxylated BAs by online resources
Some of the special BAs that would have been useful to validate the proposed dissociation mechanisms for the unconjugated 12-hydroxylated BAs, for example, the stereoisomers of 12-hydroxy-cholanic acid, were not commercially available. Fortunately, there we were able to locate the MS spectra of a total of 12 stereoisomers of monohydroxy-cholanic acid at C3, C7 or C12 in MASSBANK (http://www.massbank.ip/index.html). The data was acquired by directly infusing samples into a TOFMS via ESI interface using the negative mode (JMS-T100LP, JEOL Ltd.). Although the instrument was not a tandem MS, the major fragment ions of analytes were detected to a certain extent at the highest orifice voltage (−150 V). As summarized in Table S2, the diagnostic fragment ions, [M-H-CO2]− and [M-H-H2O-CO]−, were only detected in the spectra of all four stereoisomers of 12-hydroxy-cholanic acid. The observations in these reference MS were perfectly consistent with our proposed dissociation mechanism, and confirmed that the mechanism was independent of the stereochemistry of 12-hydroxyl group.
Fragmentation mechanisms of glycine-conjugated and taurine-conjugated BAs
The fragmentation mechanisms of conjugated BAs have been well summarized14,15. In this study, we examined the roles of the hydroxylation sites on the steroid core in the fragmentation patterns using a variety of glycine-conjugated and taurine-conjugated BAs. The MS/MS spectra of the conjugated BAs are summarized in Figure S5 and Figure S6, respectively. It was clearly observed that the effects of hydroxyl groups on the fragmentation of the steroid skeleton were submerged by the strong signals related to the dissociations of the amide side chain.
For the glycine-conjugated BAs, the cleavage of the glycineamide bond on the C24 side chain included two pathways. The first pathway began with the break of the amide bond, producing a deprotonated glycine (m/z 74.0) and the neutralized steroid skeleton. The second pathway begins with the loss of CO2 sequentially followed by the dehydrogenation of N-methyl radical. After side chain cleavage, dehydration was the major detected mechanism on the steroid nucleus. With comparisons of GLCA, GCDCA, GHDCA, GDCA, GCA with and without deuterium labeling (Figures S5), the dehydration capability of hydroxyl groups was found to decrease with 7α< 6α< 12α< 3α. For the taurine-conjugated BAs, the dissociations of the steroid nucleus were obscured by the signals derived from cleavage of the taurine-amide side chain, which produced three major fragments including SO3− (m/z 80.0), [CH2CHSO3]− (m/z 107.0), the deprotonated taurine (m/z 124.0). According to the above observations, it was proposed that the characterization of novel conjugated BAs by negative fragmentation analysis may not be achieved merely with the test of its original conjugated form. The additional test of the deconjugated form may greatly contribute to the structural characterizations.
Identification of 3β, 7β, 12α-trihydroxy-5β-cholanic acid, a novel BA in urine of NASH patients
Based on the knowledge gained from the described fragmentation analysis, we have identified a novel BA in the post-prandial urine samples of NASH patients. The ion chromatogram of the pseudo- MRM transition of trihydroxy-BAs in a representative urine sample is shown in Figure 5A. In comparison to the corresponding standards (Figure 5B), a novel trihydroxy-BA was detected with higher intensity than CA, βCA and UCA. Its retention time was shorter than UCA, βCA and CA and its MS/MS spectrum (Figure 5C) matched well with the fragmentation patterns of 3,7,12-trihydroxy-cholanic acid, indicating that it might be a stereoisomer of CA. In comparison with the spectra of CA, βCA, UCA and ACA, the fragmentation pattern of this analyte was the most similar to that of UCA. Summarizing the chromatographic and fragmentation features of this novel BA, we tentatively identified it as 3β, 7β, 12α-trihydroxy-5β-cholanic acid (βUCA). The validation of our results came from comparison with the MS/MS spectrum of a βUCA authentic standard (Figures 5D), which was synthesized by Professor Dr. Takashi Iida two decades before22. The two spectra were almost same with the high-definition m/z bias of major fragment ions less than 5 ppm. We believe that more BA stereoisomers can be identified in biological samples using the MS/MS spectral analysis strategy developed in this research. There is need for more authentic standards for the fragmentation library so that the identification of novel BAs can be facilitated.
Figure 5.
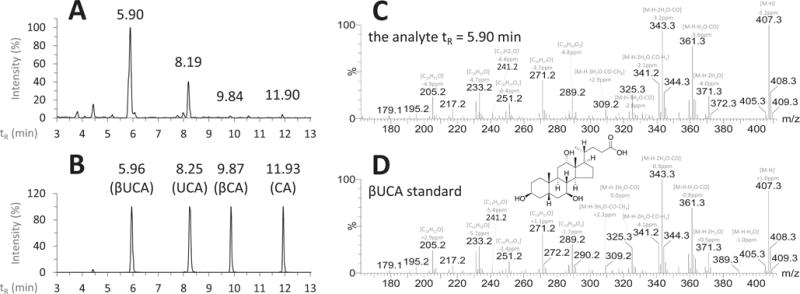
Identification of βUCA in the post-prandial urine samples of patients with nonalcoholic steatohepatitis. The ion chromatograms (pseudo-MRM transition m/z 407.3 > 407.3, CE = 30 V) of the representative urine sample (A) and the standards (B); the high-definition product ions scanned MS/MS spectra of βUCA in the urine sample (C) and the standard sample (5 μM) gifted by Prof. Dr. Takashi Iida (D).
CONCLUSION
In this work, the negative ion fragmentation mechanisms of C24 bile acids were characterized by the UPLC-ESI-QTOF-MS analysis of a total of 51 authentic BA standards. The analysis of unconjugated BAs showed a significant difference between those with and without a 12-hydroxyl group. The fragmentation mechanisms of the unconjugated BAs containing a 12-hydroxyl group were decoded using isotope-labeled standards. A series of dissociation mechanisms, including dehydration, loss of CO and CO2, dehydrogenation and cleavages of the steroid skeleton, was revealed. A common starting point for the fragmentation processes was the rotation of the carboxylate side-chain that facilitates the proton transfer between 12-hydroxyl and 24-carboxyl groups. The knowledge gained in this study has been utilized successfully to identify βUCA in the post-prandial urine samples of NASH patients. The analytical strategy proposed in this work is believed to be of great importance in identification of novel BAs in biological samples as well as in the improvement of method specificity in quantitative metabolomics studies.
Supplementary Material
Acknowledgments
We are grateful to Prof. Dr. Takashi Iida (Nihon University) for the gift of βUCA authentic standard and contributions for the MS data in the public online resources MASSBANK. This study was supported, in part, by National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM041935. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Hofmann AF, Hagey LR, Krasowski MD. J Lipid Res. 2010;51:226–246. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carey MC, Small DM. Arch Intern Med. 1972;130:506–527. [PubMed] [Google Scholar]
- 3.Hofmann AF. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 5.Halilbasic E, Claudel T, Trauner M. J Hepatol. 2013;58:155–168. doi: 10.1016/j.jhep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson PA, Karpen SJ. J Lipid Res. 2015;56:1085–1099. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell DW. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 8.Trottier J, Verreault M, Grepper S, Monte D, Belanger J, Kaeding J, Caron P, Inaba TT, Barbier O. Hepatology. 2006;44:1158–1170. doi: 10.1002/hep.21362. [DOI] [PubMed] [Google Scholar]
- 9.Alnouti Y. Toxicol Sci. 2009;108:225–246. doi: 10.1093/toxsci/kfn268. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann AF, Hagey LR. J Lipid Res. 2014;55:1553–1595. doi: 10.1194/jlr.R049437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarafian MH, Lewis MR, Pechlivanis A, Ralphs S, McPhail MJ, Patel VC, Dumas ME, Holmes E, Nicholson JK. Anal Chem. 2015;87:9662–9670. doi: 10.1021/acs.analchem.5b01556. [DOI] [PubMed] [Google Scholar]
- 12.Griffiths WJ, Sjovall J. J Lipid Res. 2010;51:23–41. doi: 10.1194/jlr.R001941-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han J, Liu Y, Wang R, Yang J, Ling V, Borchers CH. Anal Chem. 2015;87:1127–1136. doi: 10.1021/ac503816u. [DOI] [PubMed] [Google Scholar]
- 14.Murphy RC. In: New Developments in Mass Spectrometry No. 4, Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids. Gaskell SJ, editor. Royal Society of Chemistry; 2014. pp. 263–269. [Google Scholar]
- 15.Maekawa M, Shimada M, Iida T, Goto J, Mano N. Steroids. 2014;80:80–91. doi: 10.1016/j.steroids.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Qiao X, Ye M, Liu CF, Yang WZ, Miao WJ, Dong J, Guo DA. Steroids. 2012;77:204–211. doi: 10.1016/j.steroids.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Canaveras JC, Donato MT, Castell JV, Lahoz A. J Lipid Res. 2012;53:2231–2241. doi: 10.1194/jlr.D028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Canaveras JC, Donato MT, Lahoz A. Methods Mol Biol. 2014;1198:233–247. doi: 10.1007/978-1-4939-1258-2_15. [DOI] [PubMed] [Google Scholar]
- 19.Xie G, Wang Y, Wang X, Zhao A, Chen T, Ni Y, Wong L, Zhang H, Zhang J, Liu C, Liu P, Jia W. J Proteome Res. 2015;14:850–859. doi: 10.1021/pr500920q. [DOI] [PubMed] [Google Scholar]
- 20.Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, Brouwer KL, Sidney Barritt At. Dig Dis Sci. 2015;60:3318–3328. doi: 10.1007/s10620-015-3776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morton TH. In: Encyclopedia of Mass Spectrometry, Volume 1, Collisional Activation and Dissociation:Via Ionneutral Complexes. Armentrout PB, editor. Elsevier, B.V.; 2003. pp. 467–479. [Google Scholar]
- 22.Iida T, Chang FC. J Org Chem. 1982;47:2972–2978. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.