

Abstract
Purpose
MUC16, a tumor biomarker and cell surface associated mucin, is overexpressed in various cancers; however, its role in lung cancer pathogenesis is unknown. Here, we have explored the mechanistic role of MUC16 in lung cancer.
Experimental Design
To identify the functional role of MUC16, stable knockdown was carried in lung cancer cells with two different shRNAs. Clinical significance of MUC16 was evaluated in lung cancer patient’s tissues using IHC. We have generated genetically engineered mouse model (KrasG12D; AdCre) to evaluate the preclinical significance of MUC16.
Results
MUC16 was overexpressed (P=0.03) in lung cancer as compared to normal. MUC16 knockdown (KD) in various lung cancer cell lines decreased the in vitro growth rate (P<0.05), migration (P<0.001), and in vivo tumor growth (P=0.007), while overexpression of MUC16-carboxyl terminal (MUC16-Cter) resulted in increased growth rate (P<0.001). Transcriptome analysis of MUC16 KD showed a downregulation (P=0.005) of TSPYL5 gene, which encodes for a testis-specific Y-like protein. Rescue studies via over-expression of MUC16-Cter in MUC16 KD cells showed activation of signaling proteins such as JAK2 (Y1007/1008), STAT3 (Y705) and glucocorticoid receptor (GR), which constitutes an important axis for the regulation of TSPYL5 for oncogenic process. Further, inhibition of STAT3 (Y705) led to decreased GR and TSPYL5 suggesting that MUC16 regulates TSPYL5 through the JAK2/STAT3/GR axis. Also, MUC16 overexpression induced cisplatin and gemcitabine resistance by down regulation of p53.
Conclusions
Our findings indicate a significant role of MUC16 in tumorigenesis and metastasis of lung cancer cells possibly via regulation of TSPYL5 through JAK2/STAT3/GR axis.
Keywords: Lung cancer, lung cancer mouse model, MUC16, TSPYL5, p53, chemoresistance and Glucocorticoid receptor
Introduction
MUC16 mucin is a large molecular weight (20 to 25 mD) glycoprotein with 22,152 amino acid residues in its protein sequence (1–3). MUC16 is a type I transmembrane protein that has three major domains: highly O-glycosylated N-terminal domain, repetitive sea urchin sperm, enterokinase and agrin (SEA) containing tandem repeat domain and a cytoplasmic domain with potential phosphorylation sites (3,4). The N-terminal portion of MUC16 interacts with mesothelin that facilitates the peritoneal metastasis of ovarian cancer cells (5,6). The SEA domain of the tandem repeat region is responsible for the cleavage process (7), whereas carboxyl-terminal region contains 32 amino acids (aa) comprising of three tyrosine, two threonine, and one serine residues, which serve as potential phosphorylation sites for intra cellular signaling (8,9).
MUC16 is overexpressed and associated with poor prognosis in ovarian (10), breast (11) and pancreatic cancer (12). MUC16 is elevated in patients with multiple brain metastases from non-small cell lung cancer (NSCLC) and it’s associated with poor prognosis (13). Another study have shown that MUC16 is elevated in stage I NSCLC patients serum samples and demonstrated that MUC16 could be good biomarker for lung cancer patients (14). Recent studies have reported that MUC16 is an extremely highly mutated gene, in various cancers including lung cancer (15). MUC16 has been shown to be associated with enhanced cancer cell growth and metastasis (4,8). During this process, MUC16 interacts with various proteins such as mesothelin (5), JAK2 (11) and Src (8) and their association facilitates cancer cell growth and metastasis.
Due to its large size, several studies have been conducted with a small portion of the carboxyl terminal (Cter) of MUC16 (344 aa and 114 aa) (4,8,16). It has been reported that MUC16-Cter has strong oncogenic role in ovarian (8) and pancreatic cancers (4). However, the mechanistic and functional role of MUC16 in lung cancer is not well understood.
Testis-specific Y-like protein 5 (TSPYL5) gene is located at chromosome 8q22 (17); it has been frequently amplified in breast cancer and is associated with a poor prognosis (18). Epping et al have demonstrated that TSPYL5 interacts with ubiquitin-specific protease 7 (USP7) that facilitates p53 degradation to suppress the tumor suppressor activity of p53 (17). TSPYL5 has been shown to be involved in cancer cell growth by activating Akt signaling and was found to be involved in radioresistance in lung cancer cells (19). The nuclear hormone receptor family protein, glucocorticoid receptor (GR) is overexpressed in lung cancer and promotes cancer cell proliferation (20). Ligand (glucocorticoid) binding to GR leads to translocation of GR from cytoplasm to nucleus, where it directly binds to DNA and is involved in gene regulation (21). In this study, we evaluated the role of MUC16 in the growth, proliferation, spread and chemosensitivity of lung cancer cells.
METHODS
Cell culture and transfection
H292, H1975 and A549 lung cancer cells were cultured in RPMI medium supplemented with 10% fetal bovine serum and antibiotics. The cell lines used in this study were recently obtained from the ATCC and revived from early-passage −140 freezer stocks. Cells were routinely inspected for phenotypic variation and mycoplasma contamination. Similarly, mouse tumor cell line K1418 were also cultured in DMEM medium with above mentioned supplements. The cells were incubated in a humidified atmosphere at 37°C with 5% CO2. Human specific MUC16-shRNA (pSUPER-Retro-shMUC16 seq1 and pSUPER-Retro-shMUC16 seq2) and mouse specific pSUPER-Retro-shmuc16 constructs were used for stable transfection of MUC16 in H292, H1975 and K1418 with respective control shRNA (4,22).
Generation of spontaneous lung cancer mouse model
Genetically engineered mouse models LSL-KrasG12D (B6.129-Krastm4Tyj (01XJ6)) were developed by the Tuveson lab (23). Animals that were positive for KrasG12D were infected with AdCre-Luciferase retroviral vector intra-nasally (University of Iowa, Gene and vector core, Iowa, USA). Eight weeks post-infection, the animals were injected with luciferin intra-peritoneally to monitor the tumor growth (22). Mice were fed with food and water ad libitum and subjected to 12 hrs light/dark cycle. The mice studies were performed in accordance with the U.S. Public Health Service “Guidelines for the Care and Use of Laboratory Animals” under an approved protocol by the Institutional Animal Care and Use Committee (IACUC) of the UNMC. The mouse tumor tissues were utilized for immunostaining as described previously (24).
TMA and immunohistochemistry
The clinical specimen for immunohistochemistry was a commercial Tissues Micro Array (TMA) (LC121 and LC 814, US Biomax, Rockville, MD, USA). The LC121 included 120 cases of various histological types of lung carcinoma (squamous cell carcinoma (n=20), large cell carcinoma (n=37) and adenocarcinoma (n=44) and normal lung tissues (n=10). Similarly, LC814 included 40 cases of lung carcinoma (n=40) and metastatic lymph node carcinoma (n=40). The TMA was analyzed for MUC16 expression by IHC as described previously (24).
Immunoblot analysis
Western blot assay was performed as described previously (24). The blots were incubated with following primary antibodies with respective dilutions: MUC16 (mouse, 1:1000), MUC16 (mouse, 1:1000), pJAK2 #8082, JAK2 #3230, pSTAT3 #9145, STAT3 #12640, GR #12041, pSrc #2101 (Rabbit, 1:2000, Cell signaling technology), E-cadherin (mouse, 1:1500) and N-cadherin (mouse, 1:1500) antibodies were a kind gift from Dr Keith R Johnson, UNMC, Omaha, NE, USA, CK-18 (mouse, 1:1500, Abcam #668), TSPYL5 (rabbit 1:500, Santa Cruz Technology, #sc-98185), p53 (mouse 1:500, Santa Cruz Technology, #sc-126) and anti-β-actin (mouse 1:5000, Sigma #A1978)) (diluted in 2% BSA in PBS). Similarly, immunoprecipitation assay was performed as described previously (22). The signals were detected with the ECL chemiluminescence kit (Amersham Bioscience, UK).
Quantitative real-time PCR, Growth kinetics, transwell migration, and wound healing assay
Quantitative real-time PCR (QPCR), growth kinetics, transwell migration and wound healing assays were performed as described previously (11,24).
Phosphorylation-specific JAK and STAT3 inhibition
Ruxolitinib (1 μM and 5 μM) and phospho-specific STAT3 (Y705) Inhibitor XIII, C188-9 (5 μM and 10 μM) were used to confirm the MUC16/JAK2/STAT3 downstream signaling pathway in lung cancer cells. MUC16 knockdown, MUC16-Cter overexpressed cells were treated with different concentration of ruxolitinib and C188-9 for 24 h, for control 0.01% DMSO was used.
MTT assay
The cell viability of cisplatin and gemcitabine treated lung cancer cells was determined using MTT assay as described previously (24).
Long term cisplatin treatment of lung cancer cells
We have generated the cisplatin resistant cell line H292 by continuous incubation of lung cancer cells with cisplatin as described previously with slight modification (25). H292 cells were continuously treated with increasing dose of cisplatin (100 nM, 200 nM, 400 nM, 800 nM,1600 nM and 3600 nM) for five days/week for twelve weeks and leaving two days off for recovery. After twelve weeks of cisplatin treatment, the H292 cells were used for further experiments.
Data analysis
Statistical significance was evaluated with the student t-test using sigmaPlot 11.0 software. P-values 00.05 were considered to be significant. Densitometry analyses were performed using ImageJ software for wound healing experiments. All experiments were performed in triplicates.
Results
Expression of MUC16 in lung carcinoma
To investigate the clinical significance of MUC16 in lung cancer patients, we examined the expression of MUC16 in the normal lung (n=10) and human lung cancer tissues (n=101). MUC16 was significantly overexpressed (P=0.03) in human lung carcinoma (Fig. 1A) compared to normal lung. Among the NSCLC sub-types, MUC16 was detected in a higher proportion of adenocarcinoma (19/44, 43%) as compared to the squamous (1/20, 5%) and large cell carcinoma 11/37, 29.7%) (P<0.0001 for the comparison between adenocarcinoma and squamous cell carcinoma) (Supplementary Fig. S1A). These are similar to the findings of The Cancer Genome Atlas (TCGA) database wherein MUC16 is overexpressed in a greater proportion of lung adenocarcinoma tissues compared to squamous cell carcinoma (Supplementary Fig. S1B). Furthermore, patients with high MUC16 expressing lung tumors had worse survival compared to patients who had low MUC16 expression (Fig. 1B) as demonstrated by Kaplan Meier-plotter (26). We also analyzed a commercial tissue array containing primary lung carcinoma (n=40) and corresponding metastatic lymph node patient tissues (n=40). In addition to detection of MUC16 in primary lung carcinoma, we also observed in lymph node metastatic tissues (Fig. 1C). We have generated a heat map to compare intensity of MUC16 in primary lung carcinoma and metastatic lymph node tissues using composite score (Fig. 1D). Muc16 was strongly overexpressed in mouse lung adenocarcinoma (KrasG12D; AdCre) as compared to normal bronchial tissues (Fig. 1E).
Figure 1.

MUC16 expression in lung carcinoma and association of MUC16 with lung cancer patient’s survival. A, MUC16 was observed in a smaller proportion of normal bronchial tissues (1/10) but in a higher proportion of lung carcinoma (31/101, 30.69%; P=0.03). B, MUC16 expression is associated with worse outcomes in lung cancer patients. C, MUC16 expression was retained in both primary and metastatic lymph node tissues. D, Heat map of composite score represents that MUC16 expression in both primary lung carcinoma and matched lymph node tissues. E, Immunohistochemical results show that Muc16 is strongly overexpressed in mouse lung adenocarcinoma tissues (KrasG12D; AdCre) as compared to littermate control lung tissues (KrasG12D). *P<0.05. A, C & E, Figure magnification 20X and A, lower right higher magnification 40X.
Stable knockdown of MUC16 in human lung cancer cells (H292 and H1975) and overexpression of MUC16-Cter in A549 lung cancer cells
In order to identify the functional role of MUC16 in lung cancer, we performed stable knockdown of MUC16 (two different shRNA targets) in two human lung cancer cell lines H292 and H1975 (Fig. 2A & B). In order to examine the function of the cytoplasmic tail region of MUC16 (MUC16-Cter) on lung cancer cells, we ectopically overexpressed MUC16-Cter (F114HA) in the MUC16 negative cell line A549 (Fig. 2C).
Figure 2.
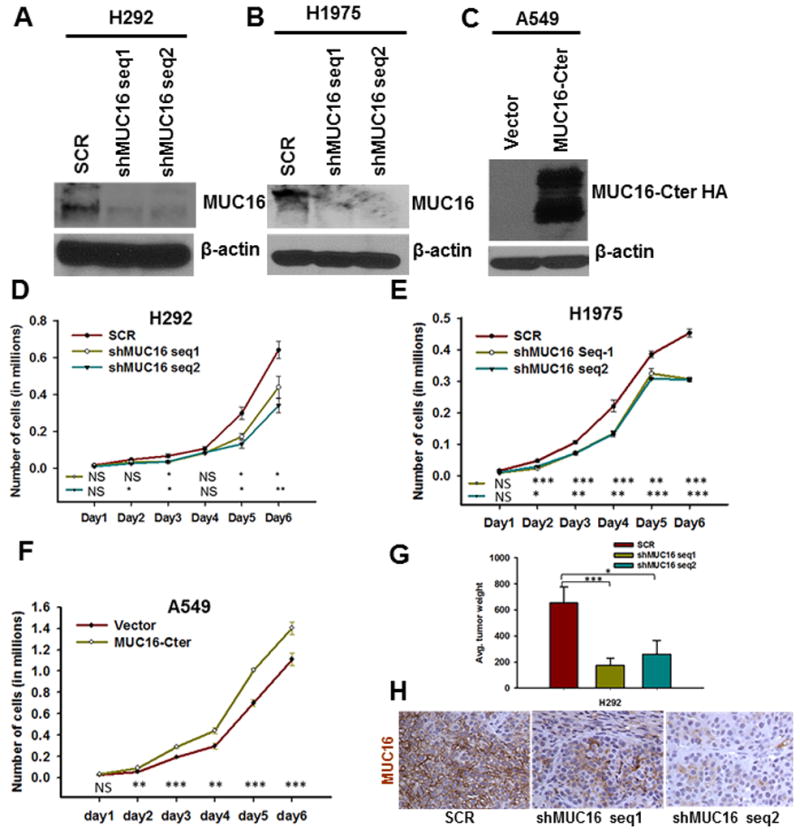
Stable of knockdown of MUC16 and ectopic overexpression of MUC16-Cter in lung cancer cells and its role in lung cancer cell growth and tumorigenicity. A & B, MUC16 is endogenously present in both H292 and H1975 lung cancer cells and its expression was silenced using pSUPER-Retro shRNA method with two different targets (shMUC16 seq1 and shMUC16 seq2). D & E, The growth of MUC16 knockdown cells (shMUC16 seq1 and shMUC16 seq2) was significantly (P<0.05) reduced. C, We ectopically overexpressed MUC16-Cter (F114HA) in MUC16 negative lung cancer cell A549. F, MUC16-Cter overexpressed lung cancer cells (A549-F114HA) had a higher growth rate (P<0.05) than vector-transfected (A549-CMV9) cells. G, We performed tumorigenic assay by sub-cutaneously injecting MUC16 knockdown and scramble in athymic mice. MUC16 knockdown cells (H292-shMUC16 seq1 (P=0.007) and seq2 (P=0.04) had significantly less tumorigenic capacity than scramble (H292-SCR) cells. H, MUC16 expression was low in tumors induced by MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells as compared to scramble (H292-SCR) cells. β-actin was used as loading control. *P<0.05, **P<0.01 and ***P<0.001, and NS non-significant. H, Figure magnification 20X.
Effect of MUC16 on lung cancer cell growth
The growth rate of MUC16 knockdown cells (H292-shMUC16 seq1 and seq2 and H1975-shMUC16 seq1 and seq2) was significantly decreased (P<0.05) compared to scramble (H292-SCR and H1975-SCR) cells in growth kinetics assays (Fig. 2D & E). Similarly, growth kinetics assays showed that MUC16-Cter overexpressed A549 (A549-F114HA) cells had significantly higher growth rate compared to vector cells (A549-CMV9) (P<0.05) (Fig. 2F). This result indicates that MUC16 might play a crucial role in lung cancer cell growth.
Role of MUC16 on tumorigenicity of lung cancer cells
MUC16 knockdown (H292-shMUC16 seq1 and seq2) and scramble (H292-SCR) cells were subcutaneously implanted in the right flank region of the athymic nude mice. After 4 weeks, mice were sacrificed and tumor weight was analyzed. MUC16 knockdown [H292-shMUC16 seq1 and seq2] cells had a significantly smaller tumor volume than scramble (H292-SCR) cells [(p=0.007) and (p=0.04) respectively] (Fig. 2G). Analysis of MUC16 expression in xenograft tissues by immunohistochemistry confirmed that MUC16 expression was decreased in tumors from knockdown (H292-shMUC16 seq1 and seq2) cells as compared to scramble (H292-SCR) cells (Fig. 2H).
MUC16 induces lung cancer cell migration through epithelial to mesenchymal transition
Transwell migration assay showed that MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells have a decreased migratory capacity (P<0.001) (Fig. 3A). On the other hand, MUC16-Cter overexpressed (A549-F114HA) cells had increased migratory capacity than vector (A549-CMV9) cells (Fig. 3B). Wound healing assays demonstrated that the migration of MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells was significantly reduced as compared to scramble (H292-SCR) cells (Fig. 3C & D).
Figure 3.

Effect of MUC16 on the migration of lung cancer cells. A, Transwell migration assay demonstrates that migration of MUC16 knockdown (H292-shMUC16 seq1 (P=0.001) and seq2 (P=0.0006)) cells was significantly decreased. B, Similarly, MUC16-Cter overexpressed cells have more migratory (P=0.02) capacity as compared to vector cells. C, Further, MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells have less migratory capacity than scramble (H292-SCR) cells as demonstrated by a wound healing assay. The wound area was stained with crystal violet for better visualization. D, The area was quantitatively calculated and normalized with wound area at 0 hrs of respective controls. E, The phosphorylation of Src (Y416) was decreased in MUC16 knockdown (H292-shMUC16 seq1 and seq2). E, Expression of mesenchymal marker N-cadherin was decreased in MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells, whereas epithelial marker CK-18 was increased. F, Increased phosphorylation of Src (Y416), increased expression of N-cadherin and downregulation of CK18 was observed in MUC16-Cter overexpressed (A549-F114HA). β-actin was used as loading control. *P<0.05, **P<0.01 and ***P<0.001. A, B & C Figure magnification 10X.
Phosphorylation of Src (Y416) was decreased in MUC16 knockdown cells (Fig. 3E). Similarly, the mesenchymal marker N-cadherin was decreased upon MUC16 knockdown, whereas epithelial marker CK18 was increased (Fig. 3E). MUC16-Cter overexpressed cells showed increased phosphorylation of Src (Y416), increased expression of N-cadherin and decreased CK-18 (Fig. 3F). This results suggest that MUC16 may have a role in the migration of lung cancer cells, possibly through Src signaling.
MUC16 downregulates TSPYL5 in lung cancer
A microarray analysis performed to analyze the MUC16 associated and regulated genes in lung cancer, revealed that TSPYL5 was significantly downregulated in MUC16 knockdown cells (P=0.005). To validate the microarray data, we confirmed the TSPYL5 downregulation in MUC16 knockdown cells by real-time PCR analysis (Fig. 4A and Supplementary Fig. S1C). Similarly, MUC16-Cter overexpressed cells have increased expression of TSPYL5 than vector cells. (Fig. 4B).
Figure 4.

MUC16 mediated downstream oncogenic signaling and inhibition of JAK2/STAT3 pathway for TSPYL5 gene expression. A, Upon MUC16 knockdown, phosphorylation of JAK2 (Y1007/1008) and STAT3 (Y705) was decreased. B, Similarly, MUC16-Cter overexpressed cells also had increased phosphorylation of JAK2 (Y1007/1008) and STAT3 (Y705). A & B, Similarly, GR and TSPYL5 were decreased in MUC16 knockdown and or increased in MUC16-Cter overexpressed lung cancer cells respectively. C, We inhibited STAT3 (Y705) using specific Inhibitor XIII, C188-9 in lung cancer. Phosphorylation of STAT3 (Y705) was decreased following pharmacological inhibition of STAT3 (Y705) using two different concentrations (5 μM and 10 μM) of C188-9. C, Total STAT3 expression remained the same. Further, as a result of STAT3 (Y705) inhibition, GR and TSPYL5 were decreased as compared to untreated cells. D, Inhibition of phospho-STAT3 inhibition in MUC16-Cter overexpressed cells confirmed the above findings. β-actin was used as loading control.
MUC16 regulates TSPYL5 through JAK/STAT3/GR pathways
Phosphorylation of JAK2 (Y1007/1008) was decreased in MUC16 knockdown cells (H292-shMUC16 seq1 and seq2) (Fig. 4A). Similarly, phosphorylation of STAT3 (Y705) was also decreased in MUC16 knockdown cells (Fig. 4A). On the other hand, MUC16-Cter overexpressed lung cancer cells (A549-F114HA) showed increased phosphorylation of JAK2 (Y1007/1008) and STAT3 (Y705) as compared to vector cells (A549-CMV9) (Fig. 4B). These results suggest that MUC16 stimulates JAK2/STAT3 signaling pathways for lung cancer cell growth. In addition, expression of the glucocorticoid receptor (GR) was decreased in MUC16 knockdown cells (Fig. 4A) as compared to scramble cells. Similarly, MUC16-Cter overexpressed (A549-F114HA) cells had a higher level of GR than vector cells (Fig. 4B).
TSPYL5 promoter studies show that promoter region of TSPYL5 has GR binding sites (Biobase software). Furthermore, STAT3 and GR act synergistically for regulation of a various subset of genes including Fox, CREB, and AP-1 (20,27,28). Here, we observed an interaction between STAT3 and GR in lung cancer by immunoprecipitation assay (Supplementary Fig. S1D). Based on this finding, we suggest that MUC16 regulates the transcription factors STAT3 and GR eventually affecting TSPYL5 gene expression.
Inhibition of JAK2 and STAT3 in lung cancer cells
We inhibited JAK1/2 (ruxolitinib) (29,30) in H292 cells. Following JAK1/2 inhibition, we analyzed the tyrosine phosphorylation of STAT3 (Y705), which is the downstream signaling target of JAK2 (31). Tyrosine phosphorylation of STAT3 (Y705) was decreased in JAK1/2 inhibitor (1 μM and 5 μM) treated cells (Supplementary Fig. S2A). Similarly, we also inhibited STAT3 (Y705) phosphorylation by STAT3 (Y705) specific Inhibitor XIII, C188-9 (32) at two different concentrations; 5 μM and 10 μM (32). Following STAT3 inhibition, we observed the decreased phosphorylation of STAT3 (Y705) (Fig. 4C). Upon STAT3 (Y705) inhibition, we observed a decreased expression of GR and TSPYL5 (Fig. 4C).
Similar experiments were also performed in MUC16-Cter overexpressed in A549 cells. Following JAK1/2 inhibition, we observed decreased phosphorylation of STAT3 (Y705) (Supplementary Fig. S2B). In addition, STAT3 (Y705) inhibition in MUC16-Cter overexpressed cells resulted in decreased GR and TSPYL5 as compared to untreated cells (Fig. 4D). These results indicate that MUC16 mediated JAK2/STAT3/GR signaling leads to TSPYL5 gene regulation, which in turn may cause lung cancer cell growth and metastasis.
MUC16 contributes to cisplatin and gemcitabine resistance
MUC16 knockdown cells (H292-shMUC16 seq1 and seq 2, H1975-shMUC16 seq1 and seq 2) were more sensitive to cisplatin (Fig. 5A & supplementary Fig. S3A) and gemcitabine (Fig. 5B & supplementary Fig. S3B) as demonstrated by MTT assay. Upon cisplatin treatment, MUC16 knockdown cells had higher apoptosis (Supplementary Fig. S3C). In contrast, no significant change was observed in the untreated scramble (H292-SCR) and MUC16 knockdown (H292-shMUC16 seq1 and shMUC16 seq2) cells (Supplementary Fig. S3C). Similarly, MUC16-Cter overexpressed lung cancer cells (A549-F114HA) were more resistant to the cytotoxic effects of cisplatin (Fig. 5C) and gemcitabine (Fig. 5D).
Figure 5.

Role of MUC16 in cisplatin and gemcitabine resistance. A & B, We treated MUC16 knockdown (H292-shMUC16 seq1 and seq2) and scramble (H292-SCR) cells with various concentrations of cisplatin and gemcitabine. MTT assay results show that MUC16 knockdown (P<0.05) cells were more responsive to cisplatin (A) and gemcitabine (B). C & D, Similarly, MUC16-Cter overexpressed cells (P<0.05) were more resistant to cisplatin (C) and gemcitabine (D). E & F, Muc16 knockdown (K1418-shMuc16) mouse GEMM tumor cells (P<0.05) were more sensitive to cisplatin (E) and gemcitabine (F) than scramble cells. Mechanism of MUC16 mediated chemoresistance. G, Upon MUC16 knockdown, expression of p53 was increased as compared to scramble. H, The expression of p53 was high in tumors derived by subcutaneous injection of MUC16 knockdown cells as compared to tumors derived by injection of scramble cells. I, Similarly, MUC16-Cter overexpressed cell had a lower p53 expression. β-actin was used as loading control. *P<0.05, **P<0.01, ***P<0.001, and NS non-significant. I, Figure magnification 20X.
In order to find out the therapeutic role of Muc16 on chemoresistance, we generated a mouse tumor cell line from genetically engineered mouse lung cancer (KrasG12D; AdCre) tissues. The cell line K1418 has endogenous Muc16 and it was stably knocked down by mouse Muc16 specific shRNA (Supplementary Fig. S3D). MTT assays on these cell lines showed that Muc16 knockdown (K1418-shMuc16) cells were more sensitive to cisplatin (Fig. 5E) and gemcitabine (Fig. 5F). These results indicate that MUC16 confers cisplatin and gemcitabine resistance in lung cancer cells.
Mechanism of MUC16 mediated chemoresistance
Upon MUC16 silencing, expression of p53 (wild type) was increased compared to scramble cells (Fig. 5G). Similarly, the p53 target gene p21 was also upregulated in MUC16 knockdown cells (Supplementary Fig. S3E). We also observed increased expression of p53 in MUC16 knockdown (H292-shMUC16 seq1 and seq2) cells from implanted xenograft tumor tissues (Fig. 5H). Similarly, p53 was downregulated in MUC16-Cter overexpressed cells (Fig. 5I). Overall, our results indicate that MUC16 regulates TSPYL5 expression, which downregulates p53 and its associated genes thereby leading to chemoresistance.
Upregulation of MUC16 in cisplatin resistant lung cancer cells
We developed cisplatin resistant cell lines by exposing various concentrations (100nm – 3.6 μM) of cisplatin. MUC16 transcript was upregulated in cisplatin resistant cell lines (P=0.02) as compared to parental cells (Supplementary Fig. S4a). These results suggest that MUC16 contributes to chemoresistance in lung cancer.
Stable knockdown of TSPYL5 in H292 lung cancer
In order to find out the role of TSPYL5 in lung cancer chemoresistance, we performed stable knockdown of TSPYL5 in H292 cells (Supplementary Fig. S4B and S4C). The p53 expression was increased in TSPYL5 knockdown cells compared to scramble cells (Supplementary Fig. 4B).
Overexpression of MUC16-Cter in MUC16 knockdown H292 cells
To confirm the MUC16 mediated JAK2/STAT3/GR/TSPYL5 signaling in lung cancer, we performed rescue experiments by overexpressing MUC16-Cter in MUC16 knockdown H292 (MUC16-Cter/H292-shMUC16) cells. We observed a restoration of GR and TSPYL5 expression in MUC16-Cter transfected MUC16 knockdown cells (Fig. 6A). Similarly, p53 expression was downregulated in the presence of MUC16-Cter as compared to vector transfected MUC16 knockdown (CMV9/H292-shMUC16) cells (Fig. 6A). These results suggest that MUC16 mediates JAK2/STAT3/GR signaling for TSPYL5 gene regulation in lung cancer.
Figure 6.

Restoration of MUC16 mediated pathways in lung cancer. A, To determine the recue effect of MUC16, we transfected MUC16-Cter (F114HA) in MUC16 knockdown (H292-shMUC16) cells. Restoration of phospho STAT3 (Y705), GR and TSPYL5 was observed in MUC16-Cter overexpressed in MUC16 knockdown cells as compared to vector transfected MUC16 knockdown cells. As expected, p53 expression was low in MUC16-Cter overexpressed in MUC16 knockdown cells. Schematic representation for MUC16 signaling in lung cancer cell growth and mechanistic role of MUC16 in chemoresistance in lung cancer. B, MUC16 phosphorylates JAK2 (Y1007/1008) and STAT3 (Y705) leading to translocation of STAT3 into the nucleus, where it recruits the glucocorticoid receptor (GR). The GR regulates TSPYL5 gene for lung cancer cell growth and metastasis. Inhibition of STAT3 phosphorylation by C188-9 leads to decreased expression of its target gene TSPYL5. In summary, MUC16 promotes JAK2/STAT3/GR signaling axis for TSPYL5 gene expression. This in turn promotes lung cancer cell growth and metastasis. MUC16/TSPYL5 downregulates p53 (wild-type) leading to chemoresistance of lung cancer cells.
Discussion
We observed that MUC16 is overexpressed in human lung adenocarcinoma and overexpressed in genetically engineered mouse lung cancer tissues (KrasG12D; AdCre), which suggests that MUC16 may have a crucial role in lung cancer pathogenesis.
MUC16 promotes cancer cell growth in breast and pancreas (4,11,16). Here, we observed that MUC16 knockdown cells had less growth and tumorigenic properties than control cells. Similarly, MUC16-Cter induced lung cancer cell growth in relative to control cells, suggesting that MUC16 might play a critical role in lung cancer cell growth. In addition, MUC16 was overexpressed in both human primary lung cancer and corresponding lymph node metastases. MUC16 knockdown cells showed significantly reduced migration relative to scramble cells, which suggests that MUC16 may be involved in lung cancer metastasis. Phosphorylation of Src (Y416) was high in MUC16 expressing cells, suggesting that Src phosphorylation is important for MUC16 mediated lung cancer cell migration. Akita et al have reported that the tyrosine phosphorylation of MUC16-Cter is important in ovarian cancer cell migration and it has been shown that MUC16-Cter interacts with Src family kinases that mediate ovarian cancer cell migration (8). Further, EMT markers were significantly altered based on MUC16 expression. The epithelial marker CK-18 was decreased and the mesenchymal marker N-cadherin was increased in MUC16 expressing cells where migration was high, thereby suggesting that MUC16 may be involved in the epithelial to mesenchymal transition during lung cancer cell metastasis.
Decreased phosphorylation of JAK2 (Y1007/1008) and STAT3 (Y705) was observed in MUC16 knockdown cells. Similarly, increased phosphorylation of JAK2 (Y1007/1008) and STAT3 (Y705) was seen in MUC16-Cter overexpressed lung cancer, which indicates that MUC16 may mediate JAK2/STAT3 downstream signaling in lung cancer cells. The role of JAK2/STAT3 has been well established in the past, with several studies demonstrating that JAK2/STAT3 signaling is necessary for lung cancer cell growth (33–35).
TSPYL5 has been shown to be involved in cancer cell growth and metastasis in various cancers (17–19). Our microarray data have demonstrated that TSPYL5 was significantly decreased in MUC16 knockdown cells. Further, glucocorticoid receptor, a regulator for TSPYL5 (by promoter analysis, Biobase software) was also decreased in MUC16 knockdown cells and increased in the MUC16-Cter overexpressed cells. We have also observed an interaction between STAT3 and GR in lung cancer cells, suggesting that STAT3 binds with GR and regulates TSPYL5 gene expression. Previous studies have demonstrated that the transcription factor STAT3 and GR synergistically regulate various genes (27,28,36). Upon STAT3 inhibition, we observed decreased GR and TSPYL5 expression, which suggested that MUC16 regulates glucocorticoid receptor for TSPYL5 gene expression through its action on STAT3. STAT3 has been shown to recruit the GR and regulate gene expression (20,27,28). In support of our findings, the GeneCards database shows that TSPYL5 promoter has GR binding sites, which suggests that GR regulate TSPYL5 gene expression. Overall, our findings suggest that MUC16 regulates TSPYL5 via JAK2/STAT3/GR signaling axis for lung cancer cell growth and metastasis.
MUC16 has been shown to be involved in chemoresistance of ovarian cancer cells (37); however, the mechanism behind the MUC16 mediated chemoresistance is not well understood. Cisplatin, a platinum analog, is a DNA damaging agent, widely used in treatment of lung cancer (38,39). Similarly, gemcitabine is a nucleoside analog that is commonly utilized for the treatment of lung cancer patients (40). We observed that MUC16 knockdown (both human and mouse tumor) cells were highly sensitive to cisplatin and gemcitabine, while MUC16-Cter overexpressed cells were more resistant. These results suggest that MUC16 might have a role in chemoresistance in lung cancer cells. TSPYL5 has been implicated in radio- and chemo-resistance in various cancers including lung and breast cancer (17,19). Overexpression of TSPYL5 suppresses p53 function and its target genes by regulating ubiquitin-specific protease 7 (USP7) that cause p53 degradation (17). In the present study, TSPYL5 was significantly downregulated in MUC16 knockdown cells. Similarly, expression of p53 and its target gene p21 was increased in MUC16 knockdown cells. In addition, p53 expression was drastically downregulated in MUC16-Cter overexpressed cells compared to vector cells. Furthermore, increased expression of p53 was observed in MUC16 knockdown cell xenografts, where less tumor growth was seen.
TSPYL5 knockdown in lung cancer cells resulted in an increased expression of p53. These results suggest that MUC16 suppresses p53 via TSPYL5 in lung cancer cells. Furthermore, in cisplatin resistant lung cancer cells, there was an increased expression of MUC16, which strongly implicates MUC16 in chemoresistance in lung cancer. To confirm the role of MUC16 mediated signaling pathways in lung cancer, we overexpressed MUC16-Cter in MUC16 knockdown down cells and showed the restoration of JAK2/STAT3/GR/TSPYL5 oncogenic signaling pathways. Overall, these results demonstrate that MUC16 regulates TSPYL5, leading to decreased tumor suppressor activity of p53 (41,42), promoting lung cancer cell growth and chemoresistance.
Conclusion
MUC16 is overexpressed in lung cancer tissues, specifically in adenocarcinoma. MUC16 mediates JAK2/STAT3/GR downstream signaling pathways, resulting in lung cancer cell growth and migration through TSPYL5. In addition, MUC16 confers resistance to cisplatin and gemcitabine by upregulating TSPYL5, which suppresses p53 activity. In conclusion, we found that MUC16 is a key player during lung cancer progression, metastasis, and chemoresistance (Fig. 6b). Targeting MUC16 may increase the response to lung cancer tissues to cytotoxic chemotherapy.
Supplementary Material
Translational Relevance.
Although MUC16 has been shown to be involved in the growth and metastasis of several cancers, its role in lung carcinoma remains unclear. Herein, we have shown subtype-specific expression of MUC16 in lung adenocarcinoma. MUC16 expression seems to increase the aggressiveness of lung cancer cells. In addition, MUC16 appears to mediate chemoresistance via expression of TSPYL5 and consequent inactivation of p53 (wild-type) in lung cancer. Targeting the MUC16/TSPYL5 pathway may help in decreasing the aggressiveness and metastatic potential of lung cancer cells and in overcoming chemoresistance, thereby improving outcomes.
Acknowledgments
The authors acknowledge the valuable technical support from Mrs. Kavita Mallya, Microarray Core Facility for gene expression analysis, Cell Sorting Facilities for cell cycle/apoptosis analysis, and the Confocal Facility for imaging assistance.
Financial Support: The work is, in part, supported by grants from the US Department of Veterans’ Affairs; Summer Undergraduate Research Program, UNMC Department of Internal Medicine; Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727) and UO1 CA111294, P50 CA127297, U54 CA163120, RO1 CA183459, RO1 CA195586, K22 CA175260 and P20 GM103480).
Grant Support
The work is partly supported by grants from the US Department of Veterans’ Affairs, UNMC Department of Internal Medicine Summer Undergraduate Research Program and Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727) and National Institutes of Health (UO1 CA111294, P50 CA127297, U54 CA163120, RO1 CA183459, RO1 CA195586, K22 CA175260 and P20 GM103480).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors Contributions
Conception and design: Imayavaramban Lakshmanan, Surinder K Batra and Apar K Ganti
Development of methodology: Imayavaramban Lakshmanan
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Imayavaramban Lakshmanan, Moorthy P. Ponnusamy, Parthasarathy Seshacharyulu, Satyanarayana Rachagani, Shereen Salfity, Abigail Thomas, Srustidhar Das, Prabin D. Majhi, Rama Krishna Nimmakayala, and Raghupathy Vengoji
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Imayavaramban Lakshmanan, Surinder K Batra and Apar K Ganti
Writing, review, and/or revision of the manuscript: All authors
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Surinder K Batra and Apar K. Ganti
Study supervision: Surinder K Batra and Apar K Ganti
References
- 1.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 2.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9(12):874–85. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das S, Batra SK. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer Res. 2015;75(22):4669–74. doi: 10.1158/0008-5472.CAN-15-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das S, Rachagani S, Torres-Gonzalez MP, Lakshmanan I, Majhi PD, Smith LM, et al. Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget. 2015;6(8):5772–87. doi: 10.18632/oncotarget.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gubbels JA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006;5(1):50. doi: 10.1186/1476-4598-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279(10):9190–8. doi: 10.1074/jbc.M312372200. [DOI] [PubMed] [Google Scholar]
- 7.Maeda T, Inoue M, Koshiba S, Yabuki T, Aoki M, Nunokawa E, et al. Solution structure of the SEA domain from the murine homologue of ovarian cancer antigen CA125 (MUC16) J Biol Chem. 2004;279(13):13174–82. doi: 10.1074/jbc.M309417200. [DOI] [PubMed] [Google Scholar]
- 8.Akita K, Tanaka M, Tanida S, Mori Y, Toda M, Nakada H. CA125/MUC16 interacts with Src family kinases, and over-expression of its C-terminal fragment in human epithelial cancer cells reduces cell-cell adhesion. Eur J Cell Biol. 2013;92(8–9):257–63. doi: 10.1016/j.ejcb.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Yin BW, Lloyd KO. Molecular cloning of the CA125 ovarian cancer antigen: identification as a new mucin, MUC16. J Biol Chem. 2001;276(29):27371–5. doi: 10.1074/jbc.M103554200. [DOI] [PubMed] [Google Scholar]
- 10.Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi: 10.1186/1476-4598-13-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakshmanan I, Ponnusamy MP, Das S, Chakraborty S, Haridas D, Mukhopadhyay P, et al. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene. 2012;31(7):805–17. doi: 10.1038/onc.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haridas D, Chakraborty S, Ponnusamy MP, Lakshmanan I, Rachagani S, Cruz E, et al. Pathobiological implications of MUC16 expression in pancreatic cancer. PLoS One. 2011;6(10):e26839. doi: 10.1371/journal.pone.0026839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng YC, Wu R, Wang SL, Chi F, Xing R, Cai WS, et al. Serum CA125 level predicts prognosis in patients with multiple brain metastases from non-small cell lung cancer before and after treatment of whole-brain radiotherapy. Med Oncol. 2014;31(7):48. doi: 10.1007/s12032-014-0048-y. [DOI] [PubMed] [Google Scholar]
- 14.Ma S, Shen L, Qian N, Chen K. The prognostic values of CA125, CA19. 9, NSE, AND SCC for stage I NSCLC are limited. Cancer Biomark. 2011;10(3–4):155–62. doi: 10.3233/CBM-2012-0246. [DOI] [PubMed] [Google Scholar]
- 15.Kim N, Hong Y, Kwon D, Yoon S. Somatic mutaome profile in human cancer tissues. Genomics Inform. 2013;11(4):239–44. doi: 10.5808/GI.2013.11.4.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theriault C, Pinard M, Comamala M, Migneault M, Beaudin J, Matte I, et al. MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecol Oncol. 2011;121(3):434–43. doi: 10.1016/j.ygyno.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Epping MT, Meijer LA, Krijgsman O, Bos JL, Pandolfi PP, Bernards R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat Cell Biol. 2011;13(1):102–8. doi: 10.1038/ncb2142. [DOI] [PubMed] [Google Scholar]
- 18.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 19.Kim EJ, Lee SY, Kim TR, Choi SI, Cho EW, Kim KC, et al. TSPYL5 is involved in cell growth and the resistance to radiation in A549 cells via the regulation of p21(WAF1/Cip1) and PTEN/AKT pathway. Biochem Biophys Res Commun. 2010;392(3):448–53. doi: 10.1016/j.bbrc.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 20.Matthews LC, Berry AA, Morgan DJ, Poolman TM, Bauer K, Kramer F, et al. Glucocorticoid receptor regulates accurate chromosome segregation and is associated with malignancy. Proc Natl Acad Sci U S A. 2015;112(17):5479–84. doi: 10.1073/pnas.1411356112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell. 2008;29(5):611–24. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Lakshmanan I, Rachagani S, Hauke R, Krishn SR, Paknikar S, Seshacharyulu P, et al. MUC5AC interactions with integrin beta4 enhances the migration of lung cancer cells through FAK signaling. Oncogene. 2016:10. doi: 10.1038/onc.2015.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 24.Majhi PD, Lakshmanan I, Ponnusamy MP, Jain M, Das S, Kaur S, et al. Pathobiological implications of MUC4 in non-small-cell lung cancer. J Thorac Oncol. 2013;8(4):398–407. doi: 10.1097/JTO.0b013e3182829e06. [DOI] [PubMed] [Google Scholar]
- 25.Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O’Flaherty JD, et al. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS One. 2013;8(1):e54193. doi: 10.1371/journal.pone.0054193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langlais D, Couture C, Balsalobre A, Drouin J. Regulatory network analyses reveal genome-wide potentiation of LIF signaling by glucocorticoids and define an innate cell defense response. PLoS Genet. 2008;4(10):e1000224. doi: 10.1371/journal.pgen.1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell. 2012;47(1):38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 29.Barosi G, Klersy C, Villani L, Bonetti E, Catarsi P, Poletto V, et al. JAK2 allele burden 50% is associated with response to ruxolitinib in persons with MPN-associated myelofibrosis and splenomegaly requiring therapy. Leukemia. 2016 doi: 10.1038/leu.2016.45. [DOI] [PubMed] [Google Scholar]
- 30.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123(22):e123–e33. doi: 10.1182/blood-2014-02-554634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–46. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 32.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117(21):5701–9. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One. 2012;7(2):e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Yin P, DID, Luo G, Zheng L, Wei J, et al. IL-6 regulates MMP-10 expression via JAK2/STAT3 signaling pathway in a human lung adenocarcinoma cell line. Anticancer Res. 2009;29(11):4497–501. [PubMed] [Google Scholar]
- 35.Zhao M, Gao FH, Wang JY, Liu F, Yuan HH, Zhang WY, et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73(3):366–74. doi: 10.1016/j.lungcan.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Takeda T, Kurachi H, Yamamoto T, Nishio Y, Nakatsuji Y, Morishige K, et al. Crosstalk between the interleukin-6 (IL-6)-JAK-STAT and the glucocorticoid-nuclear receptor pathway: synergistic activation of IL-6 response element by IL-6 and glucocorticoid. J Endocrinol. 1998;159(2):323–30. doi: 10.1677/joe.0.1590323. [DOI] [PubMed] [Google Scholar]
- 37.Gardner GJ, Baser RE, Brady MF, Bristow RE, Markman M, Spriggs D, et al. CA125 regression in ovarian cancer patients treated with intravenous versus intraperitoneal platinum-based chemotherapy: a gynecologic oncology group study. Gynecol Oncol. 2012;124(2):216–20. doi: 10.1016/j.ygyno.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 38.Gatzemeier U, von PJ, Vynnychenko I, Zatloukal P, de MF, Eberhardt WE, et al. First-cycle rash and survival in patients with advanced non-small-cell lung cancer receiving cetuximab in combination with first-line chemotherapy: a subgroup analysis of data from the FLEX phase 3 study. Lancet Oncol. 2011;12(1):30–7. doi: 10.1016/S1470-2045(10)70278-3. [DOI] [PubMed] [Google Scholar]
- 39.Pless M, Stupp R, Ris HB, Stahel RA, Weder W, Thierstein S, et al. Induction chemoradiation in stage IIIA/N2 non-small-cell lung cancer: a phase 3 randomised trial. Lancet. 2015;386(9998):1049–56. doi: 10.1016/S0140-6736(15)60294-X. [DOI] [PubMed] [Google Scholar]
- 40.Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015;16(7):763–74. doi: 10.1016/S1470-2045(15)00021-2. [DOI] [PubMed] [Google Scholar]
- 41.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22(47):7486–95. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 42.Lai SL, Perng RP, Hwang J. p53 gene status modulates the chemosensitivity of non-small cell lung cancer cells. J Biomed Sci. 2000;7(1):64–70. doi: 10.1007/BF02255920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.