

Abstract
Purpose
To elucidate any differences in the exposure-response of alvocidib (flavopiridol) given by 1 hour bolus or a hybrid schedule (30 minute bolus followed by a 4 hour infusion) using a flavopiridol/cytosine arabinoside/mitoxantrone sequential protocol (FLAM) in acute leukemia patients. The hybrid schedule was devised to be pharmacologically superior in chronic leukemia based on unbound exposure.
Experimental Design
Data from 129 patients in 3 FLAM studies were used for pharmacokinetic/pharmacodynamic modeling. Newly diagnosed (62%) or relapsed/refractory (38%) patients were treated by bolus (43%) or hybrid schedule (57%). Total and unbound flavopiridol concentrations were fit using non-linear mixed-effect population pharmacokinetic methodologies. Exposure-response relationships using unbound flavopiridol AUC were explored using recursive partitioning.
Results
Flavopiridol pharmacokinetic parameters were estimated using a two-compartment model. No pharmacokinetic covariates were identified. Flavopiridol fraction unbound was 10.9% and not different between schedules. Partitioning found no association between dosing schedule and clinical response. Clinical response was associated with AUC ≥ 780 h*ng/mL for newly diagnosed patients and AUC ≥ 1690 h*ng/mL for relapsed/refractory patients. Higher exposures were not associated with increases in severe adverse events (≥ grade 3).
Conclusions
Pharmacokinetic modeling showed no difference in flavopiridol plasma protein binding for bolus versus hybrid dosing. Further trials in newly diagnosed acute leukemia patients should utilize the bolus FLAM regimen at the maximum tolerated dose (MTD) of 50 mg/m2/day. Trials in relapsed/refractory patients should use the hybrid dosing schedule at the MTD (30/60 mg/m2/day) in order to achieve the higher exposures required for maximal efficacy in this population.
Keywords: Alvocidib, Flavopiridol, Pharmacometric, Exposure-response
Introduction
Flavopiridol is a synthetic flavone that was derived from a naturally occurring compound found in the plant Dysoxylum binectariferum (1). Flavopiridol prevents cell cycle progression via cyclin dependent kinase inhibition, inhibition of transcription elongation, and induction of apoptosis (2–4). Clinical-laboratory studies were used to derive a schedule of flavopiridol followed in a timed sequential manner by ara-C and mitoxantrone (FLAM) (4–8). The sequential addition of flavopiridol to the combination regimen is intended to induce cell cycle arrest, kill non-cycling cells, and then restart the cell cycle in the remaining cells, leaving them susceptible to the effects of ara-C and mitoxantrone. Recent clinical studies have provided evidence to support the efficacy of the FLAM for poor-risk, newly diagnosed acute myelogenous leukemias (AML) as well as relapsed/refractory AML or acute lymphocytic leukemias (ALL) (9).
Flavopiridol was initially evaluated clinically using extended continuous IV infusion of 24 to 72 hours (10–13) or short bolus IV infusion (8,13,14) both of which resulted in limited efficacy. For relapsed and primary refractory AML patients treated with FLAM, the 1 hour bolus infusion MTD for flavopiridol was established at 50 mg/m2/day by dose-limiting toxicities (DLT) of profound and prolonged neutropenia at 60 mg/m2/day (8). Byrd and colleagues noted that there was differential protein binding between fetal bovine and human serum which may account for the lack of translation of in vitro activity to the clinic in chronic lymphocytic leukemia (CLL) (15). To overcome the protein binding differential, Byrd and colleagues developed a hybrid administration consisting of a 30 minute IV bolus followed immediately by a 4 hour IV infusion which has been successfully applied to refractory CLL (16). Given the improved clinical activity, the hybrid schedule was incorporated into the FLAM regimen for relapsed and refractory AML or ALL with an overall complete remission (CR) rate of 39% (17). In this study, the hybrid MTD was established at 30/60 mg/m2/day with DLTs of tumor lysis, hyperbilirubinemia, and mucositis at 30/70 mg/m2/day (17). A 3-arm randomized trial comparing FLAM and 2 other regimens for patients with relapsed and primary refractory AML demonstrated relative superiority of FLAM over the other regimens with 28% CR rate overall and 40% CR rate in patients age 60 or less, the age difference being associated with heightened mortality in the older cohort (9,18). In newly diagnosed AML patients there was no difference between the hybrid schedule and short bolus IV infusion flavopiridol as part of FLAM with regards to efficacy and toxicity with the bolus schedule resulting in higher maximum total or unbound concentrations (18). Based on the equivalency between the two dosing schedules and the less complicated administration of bolus vs. hybrid schedule, the bolus schedule was used for a randomized Phase II trial comparing FLAM to the standard of care regimen of cytarabine/daunorubicin (7+3) in newly diagnosed AML with non-favorable features (NCT01349972) (20). In this trial, FLAM induced CR in 70% vs. 46% with 7+3 (p=0.0003) or 7+3 followed by 5+2 for residual AML on day 14 of therapy (CR 57%, p=0.08). Because of the reproducibly salutary effects of FLAM in diverse adult AML populations, follow-up studies are planned for both newly diagnosed and relapsed/refractory cohorts. In this context, it is important to determine the optimal schedule for flavopiridol delivery for future clinical trials.
This report describes the results of a population pharmacokinetic analysis of total and unbound flavopiridol concentrations delivered using bolus or hybrid administration to acute leukemia patients with either a newly-diagnosed or relapsed/refractory AML. The effect of administration scheme on flavopiridol pharmacokinetic parameters and flavopiridol plasma protein binding was assessed. Exposure-response relationships between unbound flavopiridol exposure measures and clinical response or toxicities were explored along with the effect of disease history on these relationships.
Methods
Patients and Treatment
Data from patients enrolled in three clinical trials were used to study the pharmacokinetics and pharmacodynamics of flavopiridol as the initial component in polytherapy comprising flavopiridol followed sequentially by ara-C and mitoxantrone (FLAM) (17–19). Two of the studies enrolled patients with newly diagnosed, poor-risk acute myelogenous leukemias (AML; Study 1 (19) and 3 (18)) while the third study enrolled patients with relapsed and refractory acute leukemias (Study 2 (17)). All patients were adults with pathologically confirmed disease associated with ECOG performance status from 0 to 2. All studies were approved by an appropriately constituted institutional review board. All subjects were enrolled only after giving informed consent to their participation.
A 9-day cycle of FLAM treatment consisted of flavopiridol dosing for 3 days, followed by a 72 hour intravenous infusion of cytosine arabinoside at 2 gm/m2 beginning at Day 6, and a 60 to 120 minute intravenous infusion of mitoxantrone at 40 mg/m2 on Day 9. Flavopiridol bolus dosing consisted of daily intravenous administration over a 1 hour period. Flavopiridol hybrid dosing comprised daily intravenous dosing beginning with a bolus dose consisting of 30% to 43% of the total daily dose administered over a 30 minute period followed by the remainder of the daily dose administered over a 4 hour period.
Pharmacokinetic Samples
The pharmacokinetic sampling schemes were largely standardized across the 3 studies used in the analyses. However, the schemes differed slightly to accommodate the particular administration scheme used. Samples for pharmacokinetic assessment were collected pre-dose on Days 1, 2, and 3 regardless of the drug administration schedule. For bolus dosing (Studies 1 and 3), samples were collected on Day 1 at the end of the 60 minute bolus administration and again 4 hours later (5 hr post-dose). For hybrid dosing (Studies 1 and 2), samples were collected on Day 1 at the end of the 30 minute bolus administration and again after the 4 hour infusion (4.5 hr post-dose). Additional samples specific to Study 1 were drawn and included Day 3 post-dose samples at 30 min and 4.5 hr for hybrid and 60 min and 5 hr for bolus (18). All patients in Study 1 had samples drawn 24 hours and 48 hours after the last dose. A total of 780 total and unbound flavopiridol concentrations were available across the three studies. In nearly all cases there was an unbound flavopiridol concentration corresponding with a total flavopiridol concentration except for 2 cases, one with a missing total concentration and one with a missing unbound concentration.
Bioanalytical Methods
The bioanalytical methods used for the measurement of total flavopiridol in plasma were consistent across the three studies and were previously described (8,11,17). Total flavopiridol was separated using reverse-phase high performance liquid chromatography and detected using tandem mass spectroscopy. The lower limit of quantification (LLOQ) for total flavopiridol was 0.1 ng/mL. Unbound flavopiridol concentrations were measured using a micro-equilibrium dialysis method (17). All methods were appropriately validated and showed acceptable levels of performance.
Population Pharmacokinetic Analysis
Population pharmacokinetic analysis was performed using standard techniques after exploratory data analysis that included concentration versus time profiles and regression of total and unbound flavopiridol concentrations. Structural models with 2 or 3 compartments were fit to the total flavopiridol concentration versus time data. Additive, proportional, and additive/proportional residual variability models were assessed. Individual patient concentration differences from the population mean (ε) were described in the model using a normal distribution with a mean of zero and a variance of sigma (Σ). Model and data support for the estimation of between subject variability (BSV) was systematically tested. Individual patient parameter differences from the population mean (η) were described in the model using a log-normal distribution with a mean of zero and variance of omega (Ω). Covariate data (body surface area, gender, and age) were tested for the ability to explain variability in the flavopiridol pharmacokinetics. Given the availability of data from daily dosing across three days in Study 1 (bolus dosing), the ability to estimate inter-occasion variability was assessed. Model development was guided by minimization of the −2LL objective function and likelihood ratio testing, reasonableness of parameter estimates, estimate precision, as well as standard goodness of fit plots including observed concentrations (DV) versus population predicted (PRED) or individual predicted (IPRED) concentrations. Bootstrapping using 200 replicates after stratification by dosing schedule was used to obtain final precision estimates for the model. A visual predictive check (VPC) using 1000 replicates was performed to provide an overall assessment of the agreement between observed and predicted data. Non-linear mixed effects modeling used Phoenix NLME v 1.4 with its Quasi-Random Parametric Expectation Maximization (QRPEM) estimation algorithm (Certara, L.P., 210 North Tucker Boulevard Suite 350, St. Louis, MO 63101 USA).
Dosing Scheme and Plasma Protein Binding
A goal of the analysis was to determine whether there were any differences in pharmacokinetic parameters associated with use of bolus versus hybrid IV dosing. In particular, there was interest in assessing whether the fraction unbound, Fu, differed between the dose administration schemes. Three approaches were used to explore this: 1) a linear regression comparison of the bound versus unbound flavopiridol concentrations stratified by dosing scheme, 2) testing dosing scheme as a covariate on the Fu parameter, and 3) comparing the re-estimated model parameters after stratifying the dataset by dosing scheme.
Newly Diagnosed versus Relapsed/Refractory Patients
In order to understand whether there were any differences in PK for newly diagnosed patients versus relapsed/refractory patients, the dataset was stratified by disease history and then model parameters for each patient group were estimated and compared.
Pharmacokinetic/pharmacodynamic (PK/PD) Analysis
Samples for pharmacokinetic characterization were only collected in cycle 1 of treatment. Accordingly, for the current analyses, clinical response and adverse event (AE) data were compiled for the three studies using data from the first treatment cycle. A cycle was defined as completion of all doses of flavopiridol, ara-C, and mitoxantrone. Some patients underwent a subsequent cycles of treatment but these data were excluded. The intent was to explore potential relationships between the pharmacokinetics and the pharmacodynamics of the same cycle.
Complete remission (CR) was defined as follows: bone marrow showing less than 5% myeloblasts with normal maturation of all cell lines, an absolute neutrophil count (ANC) of at least 1,000/μL, and a platelet count of 100,000/μL, absence of blasts in peripheral blood, absence of identifiable leukemic cells in the bone marrow, clearance of disease-associated cytogenetic abnormalities, and clearance of any previously existing extramedullary disease (20). Complete remission with incomplete blood count recovery (CRi) was defined the same as for CR but without achievement of ANC of at least 1,000/uL and/or platelet count of 100,000/uL (20). The clinical response was converted to a binary variable where a CR or CRi was coded as 1 and any lesser response was coded as 0. There were a total of 115 responses available from the 129 patients in the 3 studies.
A list of 11 adverse events of interest was compiled: elevated alanine aminotransferase (ALT), elevated aspartate aminotransferase (AST), hyperbilirubinemia, diarrhea, nausea, vomiting, mucositis, rash, elevated creatinine, tumor lysis syndrome (TLS), and cardiac-related adverse events. In order to focus the analyses on only the most clinically relevant adverse events, the data sets from each study were systematically processed to retain only those adverse events 1) from cycle 1 of treatment, 2) with an attribution of possibly, probably, or definitely related to the investigational agents, 3) with a severity grade of 3 or higher, and 4) from patients in the PK population. Adverse events (AE) were graded by NCI Common Toxicity Criteria under the version active during the conduct of the trial.
Pharmacokinetic/pharmacodynamic analyses were performed with unbound flavopiridol exposures based on a mechanistic assumption (i.e. it is only the unbound flavopiridol that is able to interact with receptors and cause downstream effects). Post-hoc unbound flavopiridol pharmacokinetic profiles from 0 to 96 hours using the final pharmacokinetic model were summarized as AUC for use in exposure-response analyses.
Data for clinical response and adverse event incidence were stratified by quartiles of flavopiridol unbound AUC, disease history, or dosing schedule. Differences across stratification factors were tested by Fisher’s exact test for categorical variables and ANOVA for continuous variables, except for unbound AUC and Cmax which were tested with the non-parametric Kruskal-Wallis test. Graphical analyses included plots of clinical response probability (with 95% confidence interval) versus quartiles of flavopiridol unbound AUC (h*ng/mL), first for all patients and then stratified by disease history.
Recursive partitioning was used to explore flavopiridol unbound AUC, disease history, and dosing scheme as predictor variables for clinical response. This technique iteratively selected the most important predictor variable (and threshold level for continuous variables) and used it to make binary splits of the data into branches. The iterative splits continued until there were too few values for additional splitting (i.e. 20 in a node) or an additional split would not improve the overall fit (21). A pruning procedure was subsequently used on the raw partitioning tree in an attempt to cut the tree back to the point where overall predictive accuracy was maximized, thus preventing overfitting of the data (22).
PK/PD Simulations
Using the flavopiridol PK model, stochastic simulations were performed (N=500 subjects per dosing scenario; without residual variability) to generate 96-hour unbound flavopiridol PK profiles for bolus 50 mg/m2/day and hybrid 20/30, 25/35, 30/40, 30/50, 30/60, and 30/70 mg/m2/day dosing. For each PK profile, the unbound flavopiridol AUC from 0 to 96 hours was calculated. The clinical response probabilities obtained from the recursive partitioning results at the specific decision nodes were used to generate an overall probability of clinical response for each dosing scenario by summarizing the mean of assigned individual probabilities from the 500 simulations.
Statistical and graphical analyses used the R software package (23).
Results
Patient Demographics
Baseline characteristics for all patients are presented in Supplemental Table 1. Bolus flavopiridol was administered to 56 patients (43% of the overall population) who received the previously determined maximum tolerated dose (MTD) of 50 mg/m2/day (8). Hybrid flavopiridol was administered to 73 patients (57% of the overall population) who received total daily doses ranging from 50 to 100 mg/m2/day with the initial bolus component of the schedule comprising from 30 to 43% of the total daily dose. The overall population of 129 patients was equally represented with respect to gender and had a median age of 58 years. Nearly two thirds of the patients had poor-risk, newly-diagnosed AML while the remaining third of patients had relapsed or refractory acute leukemia.
Population Pharmacokinetic Analysis
Observed total flavopiridol concentration versus time profiles for bolus and hybrid administrations are shown in Supplemental Figure 1. The relationship between total and unbound flavopiridol using all samples is presented in Figure 1. The relationship appeared linear across the range of observed total flavopiridol concentrations, from just above the LLOQ to nearly 5000 ng/mL, with values clustered relatively tightly around the regression line.
Figure 1. Relationship between Total and Unbound Flavopiridol Concentrations, Stratified by Dosing Scheme.
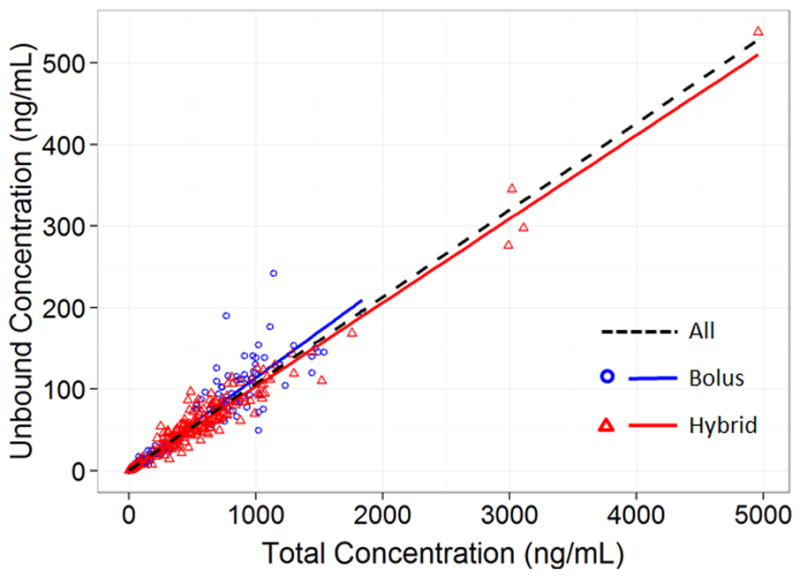
Linear regression was used to assess the relationship between paired total and unbound concentrations for samples from all subjects (N=779 samples), bolus-treated subjects (N=340 samples), or hybrid-treated subjects (N=439 samples).
Both 2 and 3 compartment structural models were assessed for flavopiridol but poor parameter estimate precision suggested that either there was not enough information content in the dataset to support 3 compartments or alternatively that a 3 compartment model was not the best representation for flavopiridol pharmacokinetics. Of note, previous flavopiridol models reported in the literature have all used 2 compartment models even when richer sampling schemes were used (8,24–26). A 2 compartment structure was therefore adopted to describe flavopiridol pharmacokinetics.
Between subject variability (BSV) was systematically tested in the base model to assess the extent to which overall variability could be assigned to differences between patients. The best model included BSV on clearance (Cl), volume of distribution (V), inter-compartmental clearance (Q), and peripheral volume of distribution (V2). The addition of BSV on Fu was assessed but did not improve the model objective function. The BSV estimate for Fu was low (6.71 %) but because the η-shrinkage was high (71.6 %), the value was suspect. Therefore, BSV on Fu was ultimately not incorporated into the model. Inspection revealed a high correlation between etas (η), in particular between the ηCl and ηV pair (correlation 0.847) and the ηQ and ηV2 pair (correlation 0.675). In order to deal with the correlation, a full block omega structure was implemented and significantly improved the model by reducing the objective function 78 units. Post hoc-estimated concentration versus time profiles were analyzed and revealed no apparent occasion-based differences.
Body surface area, age, and sex were simultaneously added to the model to assess whether they could help explain a portion of the variability in the pharmacokinetic parameters. The covariates were only added to parameters for which there was a mechanistic rationale for a relationship. Body surface area was added to Cl, V, Q, and V2. Age and sex were added only to the Cl parameter. Body surface area and age were centered on the population median values for these covariates. Considering the 95% confidence intervals for parameter estimates, the only covariate relationship with an effect distinguishable from zero was between BSA and V. BSA ranged in the study population from 1.4 m2 to 2.7 m2 with a median of 1.9 m2. The covariate relationship suggested that a patient with the lowest BSA would have a V approximately 85% of the population mean V. A patient with the highest BSA would have a V approximately 120% of the population mean V. Although statistically significant, the relationship between BSA and V was likely not of clinical importance. None of the tested covariates were incorporated in the final model.
The parameter estimates from the final flavopiridol pharmacokinetic model are shown in Table 1. The diagnostic plots in Supplemental Figure 2 showed good agreement between observed and either population or individual predicted total and unbound flavopiridol concentrations. Local regression (loess) lines fit to the data were nearly superimposable with the lines of unity. The residual plots displayed in Supplemental Figure 3 did not reveal any untoward trends, indicating the sufficiency of the proportional residual variability models used. The visual predictive check shown in Supplemental Figure 4 revealed an overall good fit between the observed and simulated total and unbound flavopiridol concentrations. Supplemental Figure 5 presents representative individual plots of PRED and IPRED versus DV. These plots portray the relatively good fit of the model and also highlight the different pharmacokinetic profile shapes associated with bolus versus hybrid dosing.
Table 1.
Final Flavopiridol Pharmacokinetic Model Parameter Estimates
| Parameter | Estimate | %RSE | Bootstrap (200 runs) | |||
|---|---|---|---|---|---|---|
| Estimate | %RSE | 2.5% CI | 97.5% CI | |||
| tvFu (%) | 10.9 | 2.00 | 10.9 | 2.03 | 10.5 | 11.4 |
| tvV (L) | 71.0 | 4.69 | 70.8 | 4.90 | 63.8 | 77.3 |
| tvV2 (L) | 88.8 | 8.34 | 91.4 | 13.6 | 67.4 | 118 |
| tvCl (L/h) | 33.4 | 3.87 | 33.7 | 4.54 | 30.9 | 36.6 |
| tvCl2 (L/h) | 8.95 | 12.3 | 9.28 | 16.2 | 6.40 | 12.5 |
| Prop. RV (total) | 34.9% | 2.04 | 34.7% | 6.26 | 30.5% | 38.3% |
| Prop. RV (unbound) | 34.1% | 2.51 | 33.7% | 5.05 | 30.4% | 37.2% |
| Parameter | Estimate | %RSE | Bootstrap (200 runs) | η Shrinkage | ||
| Estimate | %RSE | |||||
| ηV (%) | 41.0 | 44.3 | 42.7 | 66.2 | 9.79% | |
| ηCl (%) | 48.5 | 37.0 | 47.8 | 45.2 | 4.75% | |
| ηCl2 (%) | 199 | 9.33 | 161 | 26.4 | 5.63% | |
| ηV2 (%) | 98.3 | 22.3 | 100 | 26.5 | 11.2% | |
RSE= relative standard error; CI= confidence interval; Prop. RV = proportional residual variability
Dosing Scheme and Plasma Protein Binding
Simple linear regression of the bound versus unbound flavopiridol concentrations stratified by dosing scheme showed a slope-based unbound fraction of 11.4% for bolus dosing and 10.3% for hybrid dosing (p-value <0.0001; Figure 1). However, more comprehensive analyses using the pharmacokinetic model showed a universal unbound fraction of 10.9% (Table 1) with no difference in the unbound fraction between the dosing schemes. Dosing scheme added as a covariate on the Fu parameter did not improve the model objective function, indicating no difference in Fu for bolus versus hybrid dosing. When separate pharmacokinetic analyses were performed for each dosing scheme (results not shown), all the structural parameters were similar between the two schemes, including Fu, again providing evidence for the universal Fu of 10.9%. The mean Fu (95% confidence interval) for bolus dosing was 11.0% (10.4–11.6%) and for hybrid dosing was 10.8% (10.1–11.5%).
Newly Diagnosed versus Relapsed/Refractory Patients
All the structural PK parameter estimates were similar for newly diagnosed versus relapsed/refractory patients except for the V2 parameter. Newly diagnosed patients had a V2 of 89.1 L while relapsed/refractory patients had a V2 of 139 L. The V2 estimates for the two groups had non-overlapping 95% confidence intervals. Simulations of the typical PK profiles associated with the population parameter estimates from the newly diagnosed and relapsed/refractory models showed only minimal changes (data not shown). A single PK model was therefore subsequently used to describe the pharmacokinetics of all patients regardless of disease history.
Pharmacokinetic/pharmacodynamic (PK/PD) Analysis
Unbound flavopiridol concentrations were used to assess pharmacokinetic/pharmacodynamic relationships on a mechanistic basis but because there was a constant relationship between unbound and bound concentrations in the pharmacokinetic model, the results would have been the same had total flavopiridol concentrations been used in the analyses.
Higher flavopiridol unbound AUC was associated with a higher probability of clinical response in an analysis of all patients. The same analysis stratified by patient disease history confirmed that newly diagnosed patients generally responded better than relapsed/refractory patients.
Recursive partitioning using unbound AUC and disease history showed that AUC ≥ 780 h*ng/mL was associated with better response for newly diagnosed patients while relapsed-refractory patients required an AUC ≥ 1690 h*ng/mL to achieve similar efficacy. When dosing scheme (bolus or hybrid) was added to the partitioning model as a potential explanatory variable, the partitioning algorithm assessed it of low importance and did not include it in the classification tree.
The incidence of 11 pre-selected adverse events (ALT, AST, bilirubin, cardiac-related, creatinine, diarrhea, mucositis, nausea, rash, TLS, and vomiting) with severity grade of ≥ 3 were relatively rare with AST elevations showing the highest incidence (8.7%). There were no trends detected for increasing incidence of any of the adverse events across increasing quartiles of flavopiridol unbound AUC exposure (Table 2).
Table 2.
Clinical Response and Adverse Event Incidence Stratified by Quartiles of Flavopiridol Unbound AUC
| AUC Quartiles | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Variable | Category/Units | 1 | 2 | 3 | 4 | p-value |
| n | 29 | 29 | 28 | 29 | ||
|
| ||||||
| Unbound AUC (median [range]) | h* ng/mL | 683 [444, 806] | 946 [808, 1061] | 1252 [1078, 1437] | 1961 [1456, 8234] | <0.001 |
|
| ||||||
| Response (%) | Non-responder | 15 (51.7) | 14 (48.3) | 7 (25.0) | 8 (27.6) | 0.077 |
| Responder | 14 (48.3) | 15 (51.7) | 21 (75.0) | 21 (72.4) | ||
|
| ||||||
| Disease History (%) | Newly diagnosed | 18 (62.1) | 18 (62.1) | 22 (78.6) | 13 (44.8) | 0.077 |
| Relapse-refractory | 11 (37.9) | 11 (37.9) | 6 (21.4) | 16 (55.2) | ||
|
| ||||||
| Dosing Scheme (%) | Bolus | 18 (62.1) | 15 (51.7) | 13 (46.4) | 4 (13.8) | 0.001 |
| Hybrid | 11 (37.9) | 14 (48.3) | 15 (53.6) | 25 (86.2) | ||
|
| ||||||
| Dose Level (%) | 20/30* | 3 (10.3) | 2 (6.9) | 0 (0.0) | 0 (0.0) | NA |
| 25/35* | 2 (6.9) | 3 (10.3) | 1 (3.6) | 0 (0.0) | ||
| 30/40* | 5 (17.2) | 3 (10.3) | 10 (35.7) | 9 (31.0) | ||
| 30/50* | 0 (0.0) | 3 (10.3) | 1 (3.6) | 0 (0.0) | ||
| 30/60* | 1 (3.4) | 3 (10.3) | 3 (10.7) | 15 (51.7) | ||
| 30/70* | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (3.4) | ||
| 50** | 18 (62.1) | 15 (51.7) | 13 (46.4) | 4 (13.8) | ||
|
| ||||||
| Daily Dose (mean (sd)) | mg/m2 | 55.5 (10.2) | 60.3 (14.5) | 62.9 (13.8) | 78.6 (15.1) | <0.001 |
|
| ||||||
| ALT (%) | 0 | 26 (89.7) | 29 (100) | 27 (96.4) | 28 (96.6) | 0.335 |
| 1 | 3 (10.3) | 0 (0.0) | 1 (3.6) | 1 (3.4) | ||
|
| ||||||
| AST (%) | 0 | 26 (89.7) | 27 (93.1) | 26 (92.9) | 26 (89.7) | 1 |
| 1 | 3 (10.3) | 2 (6.9) | 2 (7.1) | 3 (10.3) | ||
|
| ||||||
| BILI (%) | 0 | 27 (93.1) | 28 (96.6) | 27 (96.4) | 26 (89.7) | 0.834 |
| 1 | 2 (6.9) | 1 (3.4) | 1 (3.6) | 3 (10.3) | ||
|
| ||||||
| Cardiac (%) | 0 | 28 (96.6) | 29 (100) | 28 (100) | 29 (100) | 1 |
| 1 | 1 (3.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
|
| ||||||
| Creatinine (%) | 0 | 29 (100) | 29 (100) | 28 (100) | 29 (100) | NA |
| 1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
|
| ||||||
| Diarrhea (%) | 0 | 29 (100) | 27 (93.1) | 28 (100) | 28 (96.6) | 0.615 |
| 1 | 0 (0.0) | 2 (6.9) | 0 (0.0) | 1 (3.4) | ||
|
| ||||||
| Mucositis (%) | 0 | 28 (96.6) | 28 (96.6) | 28 (100) | 27 (93.1) | 0.901 |
| 1 | 1 (3.4) | 1 (3.4) | 0 (0.0) | 2 (6.9) | ||
|
| ||||||
| Nausea (%) | 0 | 27 (93.1) | 29 (100) | 26 (92.9) | 28 (96.6) | 0.569 |
| 1 | 2 (6.9) | 0 (0.0) | 2 (7.1) | 1 (3.4) | ||
|
| ||||||
| Rash (%) | 0 | 29 (100) | 29 (100) | 28 (100) | 29 (100) | NA |
| 1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
|
| ||||||
| TLS (%) | 0 | 28 (96.6) | 26 (89.7) | 28 (100) | 27 (93.1) | 0.509 |
| 1 | 1 (3.4) | 3 (10.3) | 0 (0.0) | 2 (6.9) | ||
|
| ||||||
| Vomiting (%) | 0 | 28 (96.6) | 29 (100) | 26 (92.9) | 29 (100) | 0.191 |
| 1 | 1 (3.4) | 0 (0.0) | 2 (7.1) | 0 (0.0) | ||
|
| ||||||
| Unbound Cmax (median [range]) | ng/mL | 80.9 [31.6, 111] | 86.4 [39.3, 147] | 89.0 [51.3, 152] | 120 [69.8, 375] | <0.001 |
bolus:infusion in mg/m2/day units;
in units of mg/m2/day; for binary variables, presence or absence are denoted by 1 or 0, respectively;
’newdiag’ = newly diagnosed patient; ‘rel-refr’ = relapsed/refractory patient
PK/PD Simulations
Using the recursive partitioning results, newly diagnosed patients were assigned a 38% probability of clinical response for an AUC < 780 h*ng/mL and 77% for an AUC ≥ 780 h*ng/mL. Relapsed/refractory patients were assigned a 38% probability of clinical response for an AUC < 1690 h*ng/mL and 77% for an AUC ≥ 1690 h*ng/mL. Mean clinical probabilities over 500 simulations from the assigned baseline probabilities were generated over the different dosing scenarios. The bolus MTD of 50 mg/m2/day was associated with clinical response probabilities of 42% or 63% for relapsed/refractory or newly diagnosed patients, respectively. Using the hybrid MTD of 30/60 mg/m2/day, clinical response probabilities of 56% or 74% were simulated for relapsed/refractory or newly diagnosed patients, respectively.
Discussion
Several flavopiridol pharmacometric models have been published. Karp et al documented a two-compartment population pharmacokinetic model that used total flavopiridol concentrations from patients with acute leukemias (8). Efforts from Ji et al, Ni et al, and Phelps et al modeled flavopiridol and the glucuronide metabolite of flavopiridol in patients with chronic lymphocytic leukemia (24–26). These models identified relationships between the glucuronide metabolite and tumor lysis syndrome, the role of transporter polymorphisms on disposition and response, and relationships between flavopiridol and response or cytokine-release syndrome.
The analyses herein focused on the pharmacokinetics and pharmacodynamics of total and unbound flavopiridol in acute leukemias and aimed to assess the impact of bolus versus hybrid administration schedules as well as explore differences between newly diagnosed and relapsed/refractory patients. The parameter estimates for the final flavopiridol pharmacokinetic model were comparable to those in the previously published models. Although a statistically significant relationship between BSA and central volume of distribution was detected, no covariates with a clinically meaningful impact on flavopiridol pharmacokinetics were identified or added to the model.
The flavopiridol hybrid dosing schedule was designed by Byrd et al using in vitro and clinical pharmacokinetic data, presumably with the expectation of achieving flavopiridol exposures more efficacious than those achievable with either a bolus, a continuous infusion, or an infusion with a true loading bolus dose (16). This approach was hypothesized to overcome the high plasma protein binding of flavopiridol. Our results showed that there was no evidence of binding saturation over the range of exposures achieved. This is consistent with the in vitro work from Myatt et al that showed binding site saturation was not reached even at 20-fold molar excess of flavopiridol over human serum albumin (HSA) (27). Flavopiridol concentrations at the upper end of the observed range (~5,000 ng/mL) were equivalent to ~0.012 mM while HSA typically circulates at ~0.6 mM. Importantly, our results also showed that the estimated fraction unbound did not change for bolus versus hybrid dosing. Serum albumin, a prognostic biomarker in newly diagnosed and relapsed/refractory AML (28,29), was not explicitly tested as a PK covariate. However, protein binding, the most plausible mechanism for an effect of albumin on flavopiridol PK, showed low variability between subjects.
Patients with relapsed/refractory disease appeared to require greater flavopiridol exposure (AUC ≥ 1690 h*ng/mL) than newly diagnosed patients (AUC ≥ 780 h*ng/mL) to achieve the same maximal level of clinical response (77%) in the patient population. In the patients we studied, relapsed/refractory patients were, on average, treated with a higher daily dose of flavopiridol (~1.4-fold) that resulted in a greater range of flavopiridol unbound AUC (Supplemental Table 2). There were no meaningful differences in pharmacokinetic parameter estimates between newly diagnosed and relapsed/refractory patients suggesting that the differences in response between these patient groups does not have a pharmacokinetic basis.
In newly diagnosed patients, administration of flavopiridol at the maximum tolerated bolus dose of 50 mg/m2/day should be used. Simulations using a 50 mg/m2/day bolus dose showed a median unbound AUC of 927 h*ng/mL, a value in excess of the efficacy improvement threshold of 780 h*ng/mL for newly diagnosed patients and predicted to result in a 63% clinical response rate. In this population of patients, consistent with the results of the Phase II FLAM trial directly comparing bolus versus hybrid dosing, there may be little advantage in either efficacy or safety associated with use of the hybrid dosing schedule (Supplemental Table 3). Use of bolus dosing importantly avoids the added inconvenience of the hybrid schedule and the additional energy and effort it requires of the pharmacy, nursing staff, and patients.
Relapsed/refractory patients should use the maximum tolerated hybrid flavopiridol dose of 30/60 mg/m2/day. Simulations using the hybrid MTD showed a median unbound AUC of 1631 h*ng/mL, an exposure essentially at the efficacy improvement threshold of 1690 h*ng/mL for relapsed/refractory patients. In this population, the added burden of the hybrid schedule is warranted as it improves the likelihood of clinical response for these patients from 42% to 56%.
This work was associated with some limitations. Adverse event information was only available as worst grade per cycle. This prevented a strict time-matching of adverse events to each of the specific sequentially-administered drugs in the FLAM cycle.
A population model was developed that robustly characterized flavopiridol pharmacokinetics and the constant linear relationship between total and unbound flavopiridol concentrations. Exposure-response analyses helped define the utility of bolus flavopiridol dosing for newly diagnosed patients and hybrid dosing for relapsed/refractory patients to maximize efficacy in each population. Future studies evaluating these insights are recommended. A randomized Phase 2 study of FLAM (flavopiridol hybrid 30/60 mg/m2/day) versus cytosine arabinoside/mitoxantrone (AM) treatment in relapsed/refractory AML patients is currently under development.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 2. Flavopiridol Exposure-Response: Probability of Clinical Response versus Quartiles of Flavopiridol Unbound AUC (h*ng/mL) for A) All Patients or B) Patients Stratified by Disease History.

Quartiles of flavopiridol unbound AUC were plotted as the median of the quartile. Vertical bars are the 95% confidence interval for probability of clinical response.
Figure 3. Recursive Partitioning of Clinical Response by Flavopiridol Unbound AUC (h*ng/mL) and Patient History.

Observed clinical response data from 115 patients were subjected to recursive partitioning. Rectangles represent terminal nodes. Ovals represent splits with the value of the partitioning variable (unbound AUC or patient history) labeled on the branches.
Figure 4. PK/PD Simulations of Clinical Response Probability for Bolus or Hybrid Flavopiridol FLAM Dosing Scenarios.
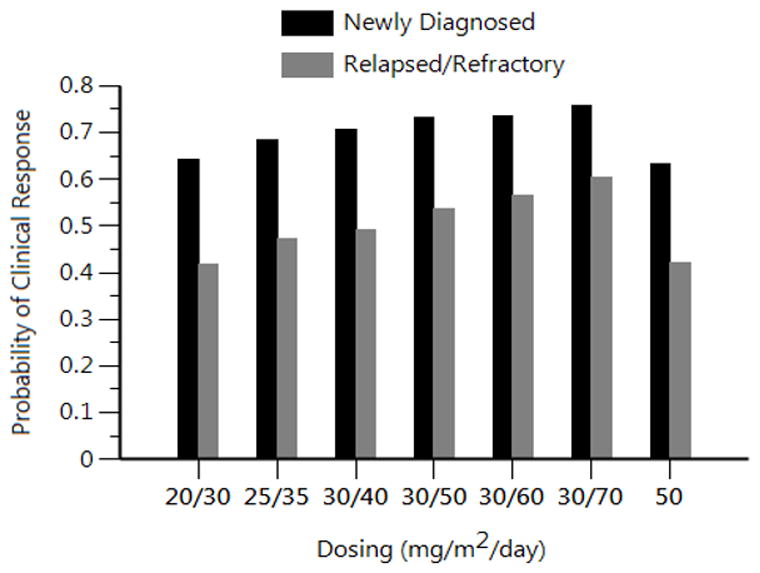
Stochastic simulations (N=500 per scenario) from the flavopiridol PK model across a range of bolus and hybrid doses were translated into clinical response probabilities using the recursive partitioning results. Single dosing values (e.g. 50) represent bolus dosing. Dual dosing values (e.g. 20/30) represent hybrid dosing with the first value indicating the dose used for the 30 min bolus and the second value indicating the dose used for the 4 hour infusion portion of the hybrid regimen.
Statement of Translational Relevance.
Pharmacokinetic and recursive partitioning models were developed to assess alvocidib (flavopiridol) exposure-response in the setting of a flavopiridol/cytosine arabinoside/mitoxantrone sequential protocol (FLAM) in patients with acute leukemias. These models were developed using sparse sampling from several studies and provided a quantitative understanding of the implications of bolus versus hybrid (short infusion followed by long infusion) flavopiridol dosing.
A Phase II trial of FLAM in adults with newly diagnosed acute myelogenous leukemia (AML) directly comparing bolus versus hybrid dosing showed no difference in efficacy or toxicity. While modeling showed no differences between the schedules in the fraction of plasma unbound flavopiridol, achievement of clinical response was associated with an area under the curve (AUC) of ≥ 780h*ng/mL for newly diagnosed and ≥1690 h*ng/mL for relapsed/refractory AML patients. Based on maximum tolerated doses and exposure thresholds for maximized efficacy, there are roles for both bolus and hybrid flavopiridol dosing in AML.
Acknowledgments
Financial support: The project described was supported in part by NCI Cooperative Agreement U01 CA070095, and UM1CA186691 (JEK and MAR) and by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973 and UL1 TR 001079). Grant Number UL1 TR 001079 is from the National Center for Advancing Translational Sciences (NCATS) a component of the NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS or NIH.
The authors thank the Johns Hopkins Sidney Kimmel Cancer Center nursing staff for superb medical care, and the patients and their families, without whose partnership we could never have conducted the trial and from whom we have learned critical information that will help us to improve the treatment of these diseases. The authors thank Dr. Gary Rosner for his scientific input.
Footnotes
Disclosure: The authors declare no potential conflicts of interest.
References
- 1.Naik RG, Kattige SL, Bhat SV, Alreja B, de Souza NJ, Rupp RH. An antiinflammatory cum immunomodulatory piperidinylbenzopyranone from dysoxylum binectariferum : isolation, structure and total synthesis. Tetrahedron. 1988;44(7):2081–6. [Google Scholar]
- 2.Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone that induces cell death in noncycling A549 human lung carcinoma cells. Cancer Res. 1996;56(21):4856–61. [PubMed] [Google Scholar]
- 3.Sedlacek HH. Mechanisms of action of flavopiridol. Crit Rev Oncol Hematol. 2001;38(2):139–70. doi: 10.1016/s1040-8428(00)00124-4. [DOI] [PubMed] [Google Scholar]
- 4.Senderowicz AM, Sausville EA. Preclinical and clinical development of cyclin-dependent kinase modulators. J Natl Cancer Inst. 2000;92(5):376–87. doi: 10.1093/jnci/92.5.376. [DOI] [PubMed] [Google Scholar]
- 5.Lee YK, Isham CR, Kaufman SH, Bible KC. Flavopiridol disrupts STAT3/DNA interactions, attenuates STAT3-directed transcription, and combines with the Jak kinase inhibitor AG490 to achieve cytotoxic synergy. Mol Cancer Ther. 2006;5(1):138–48. doi: 10.1158/1535-7163.MCT-05-0235. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24(11):1770–83. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 7.Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, et al. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res. 2003;63(8):1822–33. [PubMed] [Google Scholar]
- 8.Karp JE, Passaniti A, Gojo I, Kaufmann S, Bible K, Garimella TS, et al. Phase I and pharmacokinetic study of flavopiridol followed by 1-beta-D-arabinofuranosylcytosine and mitoxantrone in relapsed and refractory adult acute leukemias. Clin Cancer Res. 2005;11(23):8403–12. doi: 10.1158/1078-0432.CCR-05-1201. [DOI] [PubMed] [Google Scholar]
- 9.Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res. 2015;39(12):1312–8. doi: 10.1016/j.leukres.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil N, Villalba L, et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol. 1998;16(9):2986–99. doi: 10.1200/JCO.1998.16.9.2986. [DOI] [PubMed] [Google Scholar]
- 11.Rudek MA, Bauer KS, Jr, Lush RM, 3rd, Stinson SF, Senderowicz AM, Headlee DJ, et al. Clinical pharmacology of flavopiridol following a 72-hour continuous infusion. Ann Pharmacother. 2003;37(10):1369–74. doi: 10.1345/aph.1C404. [DOI] [PubMed] [Google Scholar]
- 12.Flinn IW, Byrd JC, Bartlett N, Kipps T, Gribben J, Thomas D, et al. Flavopiridol administered as a 24-hour continuous infusion in chronic lymphocytic leukemia lacks clinical activity. Leuk Res. 2005;29(11):1253–7. doi: 10.1016/j.leukres.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Byrd JC, Peterson BL, Gabrilove J, Odenike OM, Grever MR, Rai K, et al. Treatment of relapsed chronic lymphocytic leukemia by 72-hour continuous infusion or 1-hour bolus infusion of flavopiridol: results from Cancer and Leukemia Group B study 19805. Clin Cancer Res. 2005;11(11):4176–81. doi: 10.1158/1078-0432.CCR-04-2276. [DOI] [PubMed] [Google Scholar]
- 14.Tan AR, Headlee D, Messmann R, Sausville EA, Arbuck SG, Murgo AJ, et al. Phase I clinical and pharmacokinetic study of flavopiridol administered as a daily 1-hour infusion in patients with advanced neoplasms. J Clin Oncol. 2002;20(19):4074–82. doi: 10.1200/JCO.2002.01.043. [DOI] [PubMed] [Google Scholar]
- 15.Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, et al. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood. 1998;92(10):3804–16. [PubMed] [Google Scholar]
- 16.Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, Fischer B, et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood. 2007;109(2):399–404. doi: 10.1182/blood-2006-05-020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karp JE, Smith BD, Resar LS, Greer JM, Blackford A, Zhao M, et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood. 2011;117(12):3302–10. doi: 10.1182/blood-2010-09-310862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karp JE, Garrett-Mayer E, Estey EH, Rudek MA, Smith BD, Greer JM, et al. Randomized phase II study of two schedules of flavopiridol given as timed sequential therapy with cytosine arabinoside and mitoxantrone for adults with newly diagnosed, poor-risk acute myelogenous leukemia. Haematologica. 2012;97(11):1736–42. doi: 10.3324/haematol.2012.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karp JE, Blackford A, Smith BD, Alino K, Seung AH, Bolanos-Meade J, et al. Clinical activity of sequential flavopiridol, cytosine arabinoside, and mitoxantrone for adults with newly diagnosed, poor-risk acute myelogenous leukemia. Leuk Res. 2010;34(7):877–82. doi: 10.1016/j.leukres.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–74. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 21.Therneau TM, Atkinson EJ. Technical Report. Vol. 61. Mayo Clinic; 2015. An introduction to recursive partitioning using the RPART routines. http://www.mayo.edu/hsr/techrpt/61.pdf. [Google Scholar]
- 22.Strobl C, Malley J, Tutz G. An introduction to recursive partitioning: rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol Methods. 2009;14(4):323–48. doi: 10.1037/a0016973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.RCoreTeam. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. http://www.R-project.org/ [Google Scholar]
- 24.Ji J, Mould DR, Blum KA, Ruppert AS, Poi M, Zhao Y, et al. A pharmacokinetic/pharmacodynamic model of tumor lysis syndrome in chronic lymphocytic leukemia patients treated with flavopiridol. Clin Cancer Res. 2013;19(5):1269–80. doi: 10.1158/1078-0432.CCR-12-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phelps MA, Lin TS, Johnson AJ, Hurh E, Rozewski DM, Farley KL, et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood. 2009;113(12):2637–45. doi: 10.1182/blood-2008-07-168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni W, Ji J, Dai Z, Papp A, Johnson AJ, Ahn S, et al. Flavopiridol pharmacogenetics: clinical and functional evidence for the role of SLCO1B1/OATP1B1 in flavopiridol disposition. PLoS One. 2010;5(11):e13792. doi: 10.1371/journal.pone.0013792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myatt D, Johnson L, Baumli S, Siligardi G. The binding of flavopiridol to blood serum albumin. Chirality. 2010;22(Suppl 1):E40–3. doi: 10.1002/chir.20925. [DOI] [PubMed] [Google Scholar]
- 28.Khan AM, Lancet JE, Kharfan-Dabaja MA, Al Ali NH, List AF, Komrokji RS. Albumin is a prognostic factor for overall survival in newly diagnosed patients with acute myeloid leukemia (AML) Blood. 2011;118(21):4253. [Google Scholar]
- 29.Komrokji RS, Kharfan-Dabaja MA, Price SL, Wetzstein GA, List FA, Fernandez HF, et al. Albumin is a prognostic factor for response and overall survival in relapsed or refractory acute myeloid leukemia (AML) Blood. 2009;114(22):4685. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.