

Abstract
Purpose
To explore whether a crosstalk exists between PARP inhibition and PD-L1/ PD-1 immune checkpoint axis, and determine if blockade of PD-L1/ PD-1 potentiates PARP inhibitor (PARPi) in tumor suppression.
Experimental Design
Breast cancer cell lines, xenograft tumors and syngeneic tumors treated with PARPi were assessed for PD-L1 expression by immunoblotting, immunohistochemistry and FACS analyses. The phospho-kinase antibody array screen was used to explore the underlying mechanism of PARPi-induced PD-L1 upregulation. The therapeutic efficacy of PARPi alone, PD-L1 blockade alone, or their combination was tested in syngeneic tumor model. The tumor-infiltrating lymphocytes and tumor cells isolated from syngeneic tumors were analyzed by CytoF and FACS to evaluate the activity of anti-tumor immunity in the tumor microenvironment.
Results
PARPis upregulated PD-L1 expression in breast cancer cell lines and animal models. Mechanistically, PARPi inactivated GSK3β, which in turn enhanced PARPi-mediated PD-L1 upregulation. PARPi attenuated anticancer immunity via upregulation of PD-L1, and blockade of PD-L1 re-sensitized PARPi-treated cancer cells to T cell killing. The combination of PARPi and anti-PD-L1 therapy compared with each agent alone significantly increased the therapeutic efficacy in vivo.
Conclusion
Our study demonstrates a crosstalk between PARPi and tumor-associated immunosuppression, and provides evidence to support the combination of PARPi and PD-L1 or PD-1 immune checkpoint blockade as a potential therapeutic approach to treat breast cancer.
Introduction
Poly (ADP-ribose) polymerase (PARP) engages in DNA base excision repair by inducing poly (ADP-ribosy)lation of itself and other target proteins [1]. PARP inhibition has been shown to be an effective therapeutic strategy against tumors associated with germline mutation in double-strand DNA repair genes by inducing synthetic lethality [1]. One PARP inhibitor (PARPi), olaparib, was approved by the U.S. Food and Drug Administration (FDA) in 2014 for the treatment of germline BRCA-mutated (gBRCAm) advanced ovarian cancer [2]. More recently, another PARPi, niraparib, which was shown to significantly prolong the progression-free survival in ovarian cancer patients, received a fast track designation from the FDA for the treatment of patients with recurrent platinum-sensitive ovarian cancer [3].
In addition to ovarian cancer, PARPi has demonstrated tremendous potential in breast cancer, and there are currently several active clinical trials evaluating PARPi-containing combination therapies for advanced breast cancer. For instance, several combinations of PARPi and targeted anticancer agents, such as inhibitors against phosphatidylinositol 3-kinase [4, 5], Wee1 kinase [6], DNA topoisomerase I [7], and DNA methyltransferase [8], have been proposed to enhance the cytotoxic effect of PARPi. In addition, c-Met-mediated phosphorylation of PARP was reported to contribute to PARPi resistance, suggesting that the combined inhibition of c-Met and PARP may benefit patients who do not respond to PARPi and whose tumors are associated with c-Met activation [9]. Thus, developing a rational combination therapy with PARPi may lead to effective anticancer strategy.
Over the last few years, there have been major breakthroughs in our understanding of tumor-associated immunosuppression. A key mechanism underlying cancer immune evasion is the expression of multiple inhibitory ligands, notably PD-L1, on the surface of cancer cells. Engagement of the PD-1 receptor on T cells by PD-L1 leads to the suppression of T cell proliferation, cytokine release, and cytolytic activity whereas blockade of co-inhibitory ligation with monoclonal antibodies, such as PD-L1 or PD1 antibodies, restores T cell function and increases therapeutic efficacy [10, 11]. The impressive and durable clinical response of checkpoint blockade immunotherapy resulted in the FDA approval of ipilimumab, nivolumab, pembrolizumab, and more recently atezolizumab for the treatment of multiple types of cancer, such as melanoma, Hodgkin’s lymphoma, and lung and bladder cancers ([12–15]). Notably, the PD-1 antibody pembrolizumab was approved as first-line treatment for patients with advanced non-small cell lung cancer and high PD-L1 expression [16].
There is accumulating evidence indicating that conventional and targeted anticancer therapies also affect tumor-targeting immune responses [17]. Thus, delineating the crosstalk between cytotoxic anticancer agents and cancer-associated immunity may lead to more efficient combinatorial regimens. Although the effects of PARPi, a targeted anticancer agent, have shown promising results in multiple cancer types, how and whether PARPi plays a role in cancer-associated immunity is still unknown. In the current study, we investigate the crosstalk between PARP inhibition and immune checkpoint, in particular, the PD-L1/PD-1 axis, which is a dominant immune checkpoint pathway in the tumor microenvironment, and further explore a mechanism-driven combination strategy to potentiate PARPi.
Methods and Materials
Cell lines
All cell lines were obtained from ATCC (Manassas, VA) and have been independently validated by STR DNA fingerprinting at MD Anderson Cancer Center. PARP1 (#sc-400046), PD-L1 (#sc-401140), and GSK3β (#sc-425249) knockout cells were established using CRISPR/Cas9 KO plasmids from Santa Cruz Biotechnology (Dallas, TX). PARP1 knockdown MDA-MB-231 cells were established as described previously [9].
Antibodies and chemicals
PD-L1 (#13684), PARP1 (#9532), phospho-GSK3β (Ser9, #9336), GSK3β (#9315), Ki-67 (#9449) antibodies were purchased from Cell Signaling Technology, BRCA1 (sc-8326) and BRCA2 (sc-642) were from Santa Cruz, and α-Tubulin (#B-5-1-2) and β-Actin (#A2228) were obtained from Sigma-Aldrich (St. Louis, MO). Olaparib, rucaparib, and talazoparib were purchased from Selleckchem (Houston, TX), ChemieTek (Indianapolis, IN), and Selleckchem, respectively. Human CD274 (B7-H1, PD-L1) antibody for T-cell killing assay was from BioLegend (#329709). eSiRNA human BRCA1 (EHU096311), eSiRNA human BRCA2 (EHU031451) and siRNA universal negative control (SIC001) were from Sigma-Aldrich.
Human phospho-kinase antibody array
The Human Phospho-Kinase Array Kit (#ARY003B) was purchased from R&D Systems (Minneapolis, MN). Array screening was performed following the manufacturer’s protocol. Briefly, cell lysates were incubated with the array membranes. After washing, the membranes were incubated with biotinylated antibody cocktail. The amounts of phospho-kinase were assessed with streptavidin conjugated to horseradish peroxidase, followed by chemiluminescence detection. A GS-800 Calibrated Densitometer (Bio-Rad Laboratories, Hercules, CA) was used to quantify the density of each dot against the average of the internal controls on the membrane as indicated in the protocol.
Detection of cell surface PD-L1
For detection of cell surface PD-L1, cells were suspended in 100 μl of cell staining buffer (#420201, BioLegend) and incubated with APC conjugated anti-human PD-L1 antibody (#329708, BioLegend) at room temperature for 30 min. After washing in the staining buffer, stained cells were analyzed by fluorescence-activated cell sorting (FACS; BD Biosciences).
PD-L1/PD-1 binding assay
Cells (1 × 106) were incubated with 5 μg/ml recombinant human PD-1 FC chimera protein (#1086-PD-050, R&D Systems) at room temperature for 30 min. After washing in staining buffer, cells were incubated with anti-human Alexa Fluor 488 dye conjugated antibody (ThermoFisher Scientific) at room temperature for 30 min. Cells were analyzed by FACS after wash in the staining buffer. The FACS data were analyzed using Flowjo (Tree Star), and the cutoff line for relative positive percentage was set at the median of the maximum signal.
T-cell killing assay
NucLight RFP MDA-MB-231 cells (#4457, Essen Bioscience, Ann Arbor, MI) were seeded in a 96-well plate with or without olaparib. Human peripheral blood mononuclear cells (PBMCs; #70025, STEMCELL, Vancouver, BC, Canada) were activated with 100 ng/ml CD3 antibody, 100 ng/ml CD28 antibody and 10 ng/ml IL-2 (#317303; #302913; #589102, BioLegend, San Diego, CA) and then co-cultured with MDA-MB-231 cells at 10:1 ratio in the presence of fluorescence caspase 3/7 substrate (#4440, Essen Bioscience).
PD-L1 detection in xenograft tumors
All animal procedures were conducted under the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at UT MD Anderson Cancer Center. MDA-MB-231 (0.5 × 106), BT549 (1 × 106) or SUM149 (2 × 106) cells in Matrixgel (BD Biosciences, San Jose, CA) were injected into the mammary fat pads of female nude mice of 6–8 weeks of age. When tumor volume reached ~50 mm3, mice were administered olaparib (25 mg/kg) or rucaparib (5 mg/kg) orally 5 days per week for three weeks. Tumors were collected after final treatment and analyzed by immunoblotting and immunohistochemistry (IHC). IHC staining was as described previously [18]. Briefly, frozen tissue sections were fixed with 4% paraformaldehyde for 1 hour and then hydrated in PBS for 5 min at room temperature (RT). Sections were permeabilized with 0.5% triton X-100 for 15 min at RT. The endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol for 15 min at RT. After serum blocking, the slides were incubated overnight at 4 °C with human PD-L1 antibody (#13684, Cell Signaling Technology, 1:50 dilution). Slides were then incubated with biotinylated secondary antibodies for 1 hour at RT, followed by incubation with avidin-biotin-horseradish peroxidase complex. Visualization was performed using 0.125% amino-ethylcarbazole chromogen. After counterstaining with Mayer’s hematoxylin, slides were mounted.
Syngeneic tumor model treatment protocol
BALB/c mice (6–8 week female, Jackson Laboratories, Bar Harbor, Maine) were inoculated in the mammary fat pads with EMT6 (1 × 105) cells in Matrixgel. On days 3 after the inoculation, mice were injected intraperitoneally with 50 mg/kg olaparib or vehicle daily. After days 4, mice were injected intraperitoneally every 4 days with 75 μg anti-mouse PD-L1 antibody (10F.9G2, Bio X cell) or control rat IgG2b (LTF-2, Bio X cell). Tumor volumes were measured every 3 days with a digital caliper, and were calculated using the formula: π/6 × length × width2. Body weight was measured every 5 days.
Tumor cell PD-L1 expression and tumor-infiltrating lymphocyte (TIL) analysis
EMT6 tumors were excised and digested in collagenase/hyaluronidase and DNase I, and dissociated by gentleMACS Dissociator as described by the protocol of tumor dissociation kit (Miltenyi Biotec, Auburn, CA). Tumor cells and TIL were enriched and harvested separately by Percoll gradient (Sigma). Cell Surface PD-L1 of Tumor cell were stained with Brilliant Violet 421-conjugated anti-mouse PD-L1 antibody (#124315, BioLegend) and analyzed by FACS, and TIL were stained and analyzed by mass cytometry (CyTOF). The antibodies used to stain TIL were listed as followed: CD45-147Sm; CD3e-152Sm; CD4-172Yb; CD8a-168Er; IFNγ-165Ho (Fluidigm, CA).
Immunohistochemical staining (IHC) of human breast tumor tissue samples
IHC staining was performed as described previously [19]. Human breast tumor tissue specimens were obtained following the guidelines approved by the Institutional Review Board at MD Anderson, and written informed consent was obtained from patients in all cases. Briefly, tissue specimens were incubated with antibodies against PAR (Enzo Life Science, Clone 10H, 1:200 dilution) and PD-L1 (Abcam, Clone 288, #ab205921, 1:100 dilution) and a biotin-conjugated secondary antibody and then incubated with an avidin–biotin–peroxidase complex. Visualization was performed using amino-ethylcarbazole chromogen. According to the histological scores, the intensity of staining was ranked into four groups: high (+++), medium (++), low (+) and negative (−).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (GraphPad, San Diego, CA). Student’s t-test or one-way analysis of variance (ANOVA) was used to compare experimental data. The Pearson Chi-square test was used to analyze IHC data. A P value < 0.05 (*) was considered statistically significant.
Results
PARPi enhances PD-L1 expression in vitro and in vivo
Elevated PD-L1 expression in cancer cells has been shown to enhance PD-L1/PD-1 axis-mediated anticancer immunosuppression [11, 16]. To determine whether inhibition of PARP affects the PD-L1 protein level, we treated MDA-MB-231 and BT549 cells with two different PARP inhibitors (PARPi), olaparib and talazoparib, and determined PD-L1 expression by immunoblotting. PARPi treatment increased the total level of PD-L1 protein in both cell lines (Fig. 1A). To validate whether PARPi-induced PD-L1 upregulation is through PARP1 inhibition, we knocked down (K/D) or knocked out (K/O) PARP1 in MDA-MB-231 cells. Consistent with the results shown in Figure 1A, PD-L1 expression in the PARP1 knockdown and knockout cells was substantially higher compared with the parental cells (Fig. 1B).
Figure 1. PARPi upregulates PD-L1 protein expression in breast cancer cells.
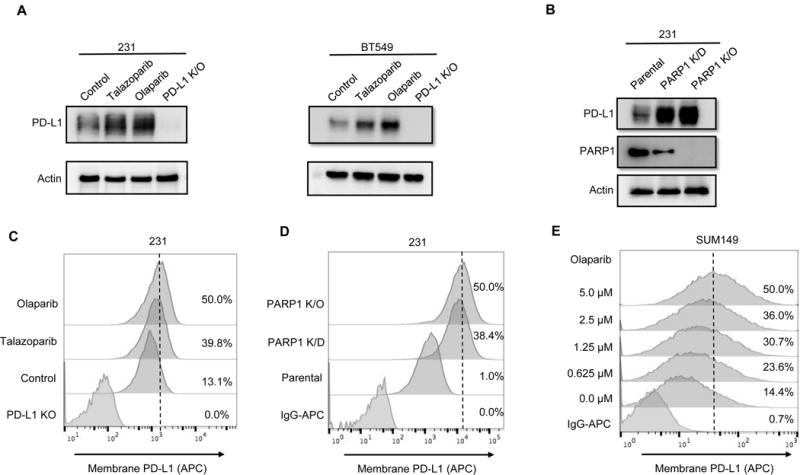
(A) MDA-MDA-MB-231 and BT549 cells were treated with 10 μM olaparib or 10 nM talazoparib for 24 hours, and subjected to immunoblotting with the indicated antibodies. PD-L1 knockout (K/O) cells were included as a negative control. (B) PD-L1 expression in PARP1 knockdown (K/D), PARP1 knockout (K/O), and MDA-MB-231 parental cells by immunoblotting. (C and D) The indicated MDA-MB-231 cells were subjected to FACS analysis for cell surface PD-L1 expression. (E) SUM149 cells were treated with the indicated concentrations of olaparib for 10 days, and cell surface PD-L1 expression was determined by FACS.
PD-L1 expressed on cell surface of cancer cells exerts immunosuppressive effects by binding to PD-1 receptor on activated T cells [20]. To determine whether the level of cell surface PD-L1 increases after PARPi treatment, MDA-MB-231 cells were treated with or without PARPi and subjected to FACS using fluorescence-labeled PD-L1 antibody. Cell surface PD-L1 levels have significantly increased after olaparib and talazoparib treatment (Fig. 1C) and the increase occurred in a dose-dependent manner (Supplementary Fig. S1). Likewise, cell surface PD-L1 levels were higher in PARP1 knockdown and knockout MDA-MB-231 cells than in parental cells (Fig. 1D). Because PARPi is commonly used to treat BRCA-deficient cancers [21], we also investigated the effects of olaparib on BRCA-mutant SUM149 cells. SUM149 cells were treated with the different concentrations of olaparib for 10 days to mimic chronic PARPi exposure in clinic, and subjected to FACS to determine PD-L1 expression. Consistent with the results in MDA-MB-231 cells, cell surface PD-L1 was significantly increased following olaparib treatment in a dose-dependent manner (Fig. 1E). To further validate the role of BRCA deficiency in PARPi-induced PD-L1 upregulation, we knocked down BRCA1 or BRCA2 in MDA-MB-231 cells and exposed cells to olaparib. Downregulation of BRCA1 or BRCA2 had virtually no effect on PARPi-induced PD-L1 expression (Supplementary, Fig. S2). These results together suggested that PARPi can upregulate cell surface PD-L1 level in both BRCA-proficient and BRCA-deficient cells.
Next we asked whether PARPi may affects PD-L1 expression in tumors, we inoculated BT549 and SUM149 cells into the mammary fad pads of nude mice, and after tumor formed, administered olaparib to mice 5 days a week for 3 weeks. Tumor tissues from xenografts were isolated and subjected to immunoblotting with PD-L1 antibody. As shown, PD-L1 expression was substantially higher in the xenograft tumors from mice treated with olaparib compared with those from the untreated mice (Fig. 2A and 2B). We also assessed PD-L1 expression by IHC staining in MDA-MB-231 xenograft tumor tissues from mice that had been treated with another PARPi, rucaparib, and harvested from our previous study [9]. Mice treated with rucaparib for 3 weeks had higher PD-L1 expression in their tumors compared with control mice (Fig. 2C). Together, these results indicated that PARPi upregulates PD-L1 expression in TNBC in vitro and in vivo.
Figure 2. PARPi upregulates PD-L1 expression in xenograft tumors.
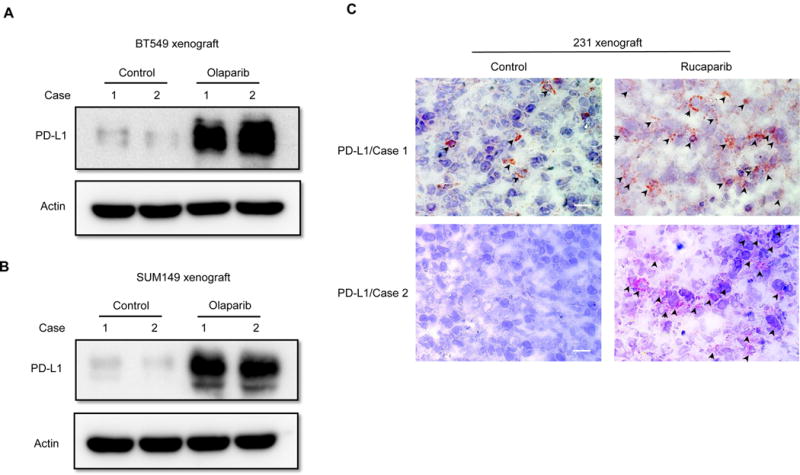
(A) BT549, (B) SUM149 and (C) MDA-MB-231 cells were inoculated into mammary fad pad of nude mice, and the mice with established tumors were treated with olaparib or rucaparib. Tumors were then isolated to evaluate PD-L1 expression by immunoblotting (A and B) or IHC staining (C). Black arrowheads indicate the detected PD-L1 signals. Scale bar, 50 μm.
GSK3β inactivation is required for PARPi-induced PD-L1 upregulation
Next, we explored the mechanism underlying PARPi-enhanced PD-L1 protein expression. Multiple signaling pathways, such as STAT, NF-κB and mTOR, have been reported to regulate PD-L1 expression level [19, 22, 23]. In an attempt to identify the potential signaling pathways that regulate PD-L1, we performed a phospho-kinase antibody array screen to identify kinases that are activated or inactivated following PARPi treatment. In the presence of PARPi treatment, the phosphorylation signal of GSK3α/β at Ser21 and Ser9, which represents its inactivated form [24], scored the second highest after the CHK2-p53 DNA repair pathway (Fig. 3A). CHK2 kinase is known to respond to DNA damage, and PARPi-induced CHK2 phosphorylation has previously been reported [25, 26], further lending support to the results of our screen. The finding that PARPi inactivates GSK3α/β is also in line with our recent report demonstrating inactivation of GSK3β (p-Ser9) stabilizes PD-L1 [27]. Those findings together prompted us to investigate whether PARPi upregulates the expression of PD-L1 via inactivation of GSK3β. To this end, we examined the status of GSK3β phosphorylation at Ser9 in response to PARPi in SUM149 cells, BT549, and MDA-MB-231 cells by immunoblotting. The results indicated that PARPi treatment induced high GSK3β Ser9 phosphorylation that was associated with PD-L1 upregulation (Fig. 3B, 3C and Supplementary, Figure S3). Knocking out GSK3β significantly increased PD-L1 expression (Fig. 3C, lane 3 vs. 1). However, PD-L1 expression level in GSK3β-knockout cells was no longer enhanced by olaparib treatment (Fig. 3C, lane 4 vs. 3). These results suggested that inactivation of GSK3β is required for the PARPi-induced PD-L1 upregulation.
Figure 3. GSK3β inactivation is required for PARPi-mediated PD-L1 upregulation.
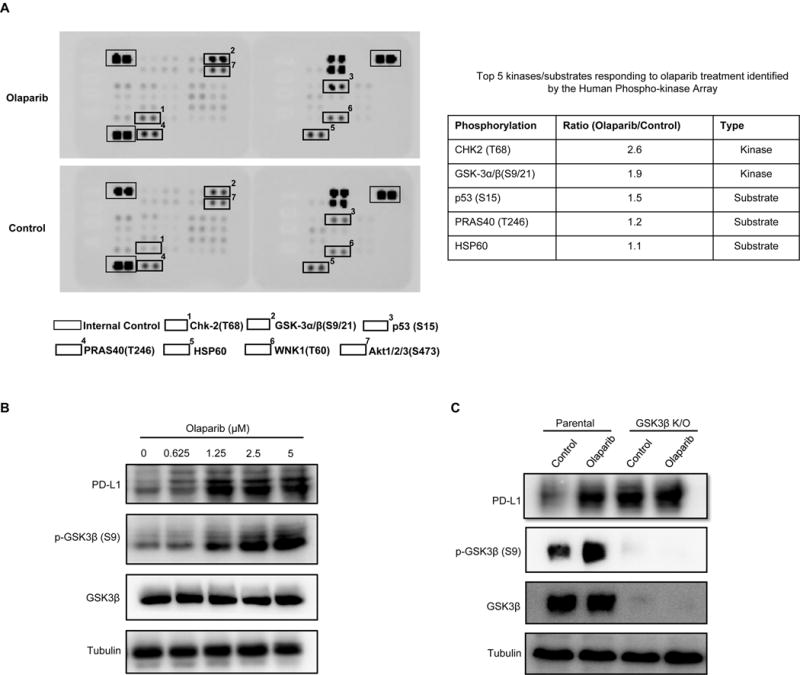
(A) SUM149 cells were treated with olaparib, and subjected to human phospho-Kinase Array. The top two responding kinases were Chk2 and GSK3α/β. (B) SUM149 cells were treated with the indicated concentrations of olaparib for 10 days, and subjected to immunoblotting with the indicated antibodies. (C) BT549 parental or GSK3β knockout cells were treated with 10 μM olaparib for 24 hours. PD-L1 expression was evaluated by immunoblotting.
PARPi attenuates T-cell killing through PD-L1 induction
To understand the functional significance of PD-L1 upregulation by PARPi, we first asked whether PARPi-induced PD-L1 increases PD-1 binding on cells. Exposure of MDA-MB-231 cells to olaparib induced more PD-1 binding to PD-L1 on the cell surface (Fig. 4A). Similar results were observed in MDA-MB-231 PARP1 K/D and K/O cells (Fig. 4B), and in SUM149 cells treated with different concentrations of olaparib (Fig. 4C). Next, to determine whether PARPi-mediated PD-L1 upregulation which resulted in increased PD-1 binding affects T-cell function, we performed a T-cell-mediated killing assay by co-culturing activated human peripheral blood mononuclear cells (PBMCs) with MDA-MB-231 cells labeled with nuclear red fluorescence protein (RFP; NucLight Red MDA-MB-231) in the presence or absence of olaparib. As expected, olaparib efficiently inhibited cancer cell proliferation (black vs. blue in Fig. 4D, left panel; Supplementary, Fig. S4A; Supplementary, Movie S1 vs. S2) but did not inhibit activated PBMCs proliferation (Supplementary, Fig. S4B). Interestingly, although MDA-MB-231 cells were sensitive to the T-cell killing (black vs. red in Fig. 4D, left panel; Supplementary, Movie S3), those that were treated with olaparib were strongly resistant to activated T-cell killing (blue vs. yellow in Fig. 4D, left panel; Fig. 4D, right panel; Supplementary, Movie S4), supporting the notion that upregulation of PD-L1 by PARPi may render the PARPi-treated cancer cells more resistant to T-cell killing. On the basis of the above results, we tested the T-cell killing effects of the combination of PD-L1 antibody and olaparib. The results showed that blockage of PD-L1 re-sensitized PARPi-treated MDA-MB-231 cells to activated T-cell killing, and that the PARPi-PD-L1 antibody combination was more effective than each agent alone (Fig. 4E).
Figure 4. Olaparib attenuates PD-1 binding and T-cell-mediated cell death in TNBC cells.
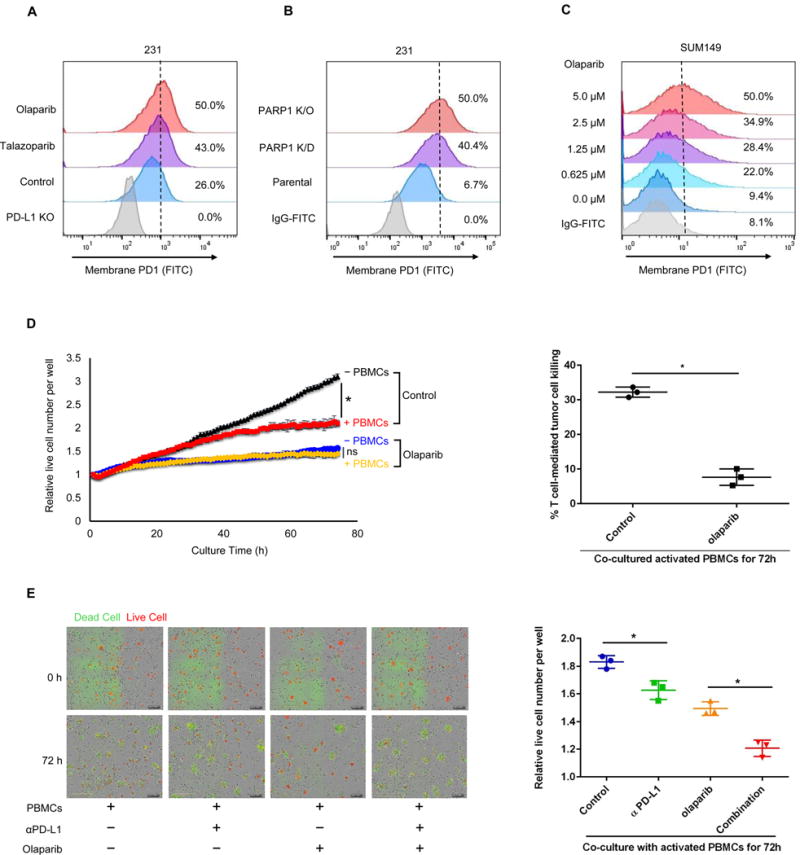
(A) FACS analysis of cell surface PD-1 binding of MDA-MB-231 cells treated with 10 μM olaparib or 10 nM talazoparib for 24 hours. (B) FACS analysis of PARP1 knockdown (K/D), PARP1 knockout (K/O), and parental MDA-MB-231 cells. (C) SUM149 cells were treated with the indicated concentrations of olaparib for 10 days. (D) MDA-MB-231 cells expressing nuclear RFP protein were first treated with or without olaparib (10 μM) for 3 hours and then co-cultured with or without activated peripheral blood mononuclear cells (PBMCs). Left, quantitation showing the number of live cells per well, counting the number of red fluorescent objects, normalized to that at the zero time point. Right, the percent of T cell-meditated tumor cell killing observed at 72 hours in activated PBMC co-culture with control or olaparib-treated cells (normalized to co-culture without PBMCs). (E) Left, representative merged images showing red fluorescent (nuclear restricted RFP), and green fluorescent (Caspase 3/7 substrate) objects in MDA-MB-231 cells co-cultured with activated PBMCs at 0 and 72 hours. Images were taken using the IncuCyte Zoom microscope. Right, quantitation showing the number of live cells following treatment with olaparib (10 μM), PD-L1 antibody (PD-L1 Ab; 10 μg/ml), or the combination co-cultured with activated PBMCs for 72 hours. The number of live cells (red fluorescent objects) were counted and normalized to that at the zero time point. * P < 0.05; ns, not significant.
Combination with PD-L1 blockade sensitizes PARPi therapy
Next, we sought to determine whether PD-L1 blockade could further potentiate PARPi anti-tumor efficacy in vivo. We first treated a murine breast cancer cell lines, EMT6, with olaparib with results showing significant induction of PD-L1 by olaparib (Fig. 5A). Consequently, we evaluated PARPi and anti-PD-L1 treatment alone or in combination in the EMT6 syngeneic mouse model. Consistent with our observations in vitro, both olaparib and anti-PD-L1 restricted tumor growth but the combined treatment demonstrated better therapeutic benefit than each treatment alone (Fig. 5B and 5C). There were significantly fewer Ki-67 positive tumor cells in the combined treatment compared with each treatment alone (Fig. 5D). Mice that received the combination treatment did not show any significant changes in body weight or elevation in liver enzyme (ALT and AST), and kidney toxicity marker, blood urea nitrogen (Supplementary, Fig. S5). At the end of treatment, tumors resected from mice were subjected to dissociation, and tumor cells and tumor-infiltrating lymphocytes (TIL) were separately harvested. Consistent with the observation in xenograft mice model (Fig. 2), PARPi significantly upregulated PD-L1 expression on tumor cell surface as determined by FACS in EMT6 syngeneic mice (Fig. 5E). Analysis of TILs by CytoF showed that tumor-infiltrated cytotoxic CD8+ T-cell population, as measured by the level of IFNγ, decreased after PARPi treatment. Meanwhile, the addition of anti-PD-L1 restored the cytotoxic CD8+ T-cell population (Fig. 5F). These results suggested that PARPi enhances cancer-associated immunosuppression through upregulation of PD-L1 and that PD-L1 blockade potentiates PARPi therapy.
Figure 5. PARPi-induced PD-L1 upregulation suppresses anticancer immunity and blockade of PD-L1 potentiates PARPi.
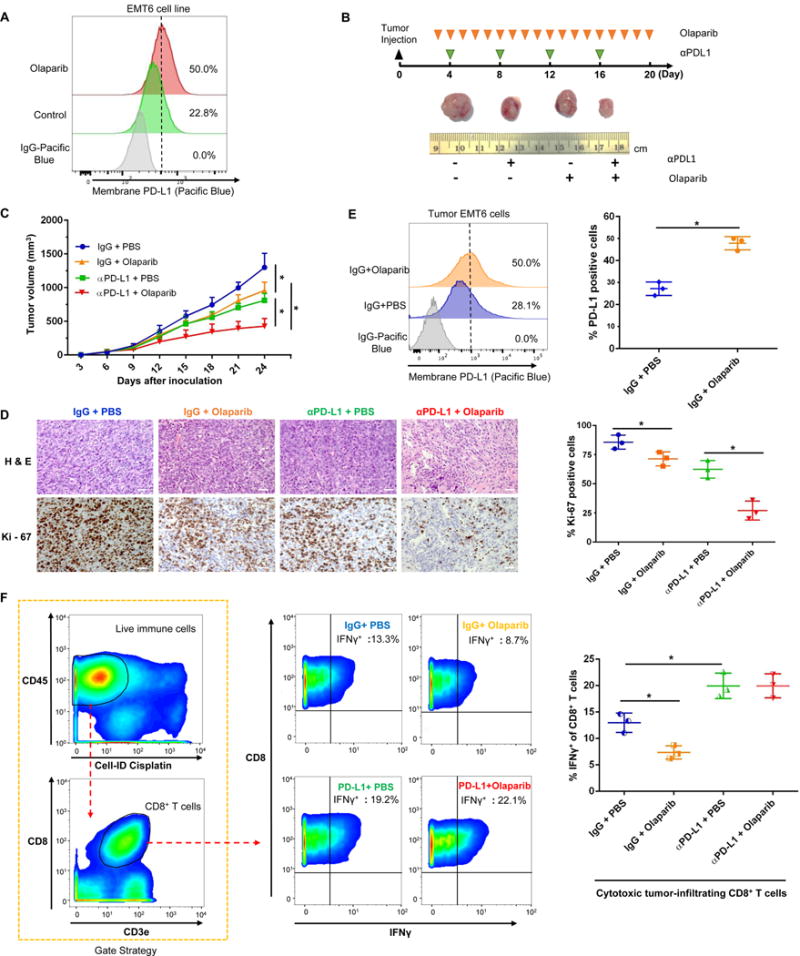
(A) EMT6 cells were treated with 10 μM olaparib for 24 h. Cell surface PD-L1 were analyzed by FACS. (B) Representative images of tumors after olaparib and/or anti-PD-L1 antibody treatment at the indicated time points in the EMT6 syngeneic mouse model. (C) Effects of olaparib and/or anti-PD-L1 antibody treatment on tumor growth in EMT6 syngeneic mouse model treated (n = 8). Tumors were measured at the indicated time points and dissected for tumor cell PD-L1 expression analysis, TIL analysis, and pathological analysis at endpoint. (D) Hematoxylin and eosin (H&E) and Ki-67 staining of EMT6 tumors. Scale bar, 50 μm. (E) Cell surface PD-L1 expression in EMT6 cells derived from EMT6 mouse tumors. (F) Cytotoxic CD8+ T cells population (IFNγ+ CD8+ CD3+ CD45+) in TILs isolated from EMT6 tumors by CyTOF analysis. * P < 0.05
Correlations of PARylation and PD-L1 expression in human tumor tissues
PARP exerts its biological function through its PARylation enzyme activity, and PARP inhibitors are designed to inhibit its enzyme activity. Thus, the extent of protein PARylation was utilized to assess the efficacy of PARP inhibition [7]. To further validate our findings in human cancer patient samples, we analyzed the correlations between PARylation level and PD-L1 expression in human breast tumor specimens using IHC. High level of protein PARylation was detected in 87 (75.0%) of the 116 specimens, of which 65 (74.7%) cases showed low PD-L1 expression (Figure 6, top). The Pearson Chi-Square test further showed the inverse correlation between PARylation level and PD-L1 expression exists in human cancer patient specimens (Figure 6, bottom). These results supported the notion that high PARP enzyme activity suppresses PD-L1 expression.
Figure 6. Inverse correlation between PAR and PD-L1 in surgical specimens of breast cancer.
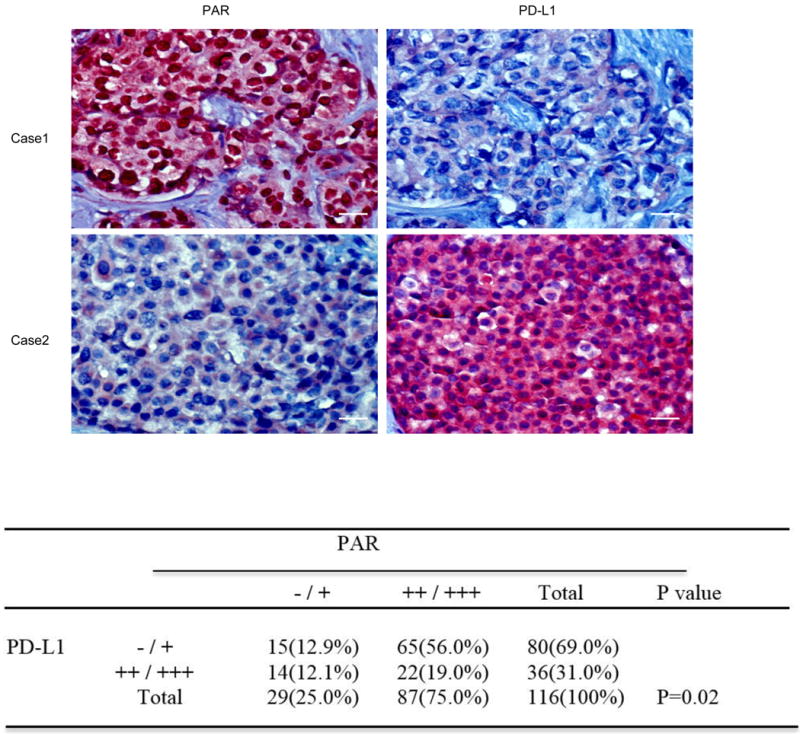
Top, representative images of IHC staining of PAR and PD-L1 in human breast cancer tissues (n = 116). Scale bar, 50 μm. Bottom, correlation analysis between PAR and PDL-1 was analyzed using the Pearson Chi-Square test (P = 0.02). A P value of less than 0.05 was considered to be statistically significant.
Discussion
Although the cytotoxic effects of PARPi have been well studied, the role of PARP inhibition in cancer-associated immunity is still largely unknown. In this study, we demonstrated that PARPi upregulates PD-L1 expression primarily through GSK3β inactivation. PARPi renders cancer cells more resistant to T-cell-mediated cell death, and PD-L1 blockade potentiates PARPi in vitro and in vivo. These data strongly suggested that PD-L1 upregulation by PARPi treatment attenuates PARPi therapeutic efficacy via tumor-associated immunosuppression, and simultaneous inhibition of PARP and PD-L1 may benefit breast cancer patients. There are currently three clinical trials testing the combination of PARPi (olaparib, niraparib, and BGB-290) and PD-L1 or PD-1 antibody in multiple cancer types (NCT02484404; NCT02657889; NCT02660034). The results of the current study provided scientific basis for these clinical trials.
Higuchi et al. recently investigated the combination of PARPi and CTLA4 antibody in the BR5-AKT ovarian cancer syngeneic mouse model and claimed to have observed a synergistic therapeutic effect [28]; however, they indicated they did not observe such synergistic effect using the anti-PD-1 and PARPi combination in the same animal model. It is worthwhile to mention that PD-1 blockade in the BR5-AKT syngeneic mice did not affect T-cell activation or cytokine induction in the peritoneal tumor environment in their study [28], and therefore, synergistic effects may not be observed in combination with PARPi under their experimental condition. Moreover, because BR5-AKT tumor display high AKT activities that already inhibit GSK3β [29], it is possible that PARPi cannot further inhibit GSK3β and upregulate PD-L1 in the BR5-AKT tumors, and thus the synergistic effects were not observed. In contrast, the results from our study indicated that PARPi upregulates PD-L1 in EMT6 tumors and PD-L1 blockade attenuated immunosuppression activity (Fig. 5F), which allowed us to observe an anti-PD-L1 therapy-potentiated antitumor activity of PARPi. Meanwhile, other studies have reported that chemotherapeutic agents, gemcitabine and paclitaxel, can induce PD-L1 in ovarian cancer cells [30, 31]. The combination of paclitaxel and PD-L1/PD-1 blockade enhanced antitumor efficacy in an ID8 ovarian syngeneic mouse model [30]. Therefore, whether the combination of PD-L1 blockade and PARPi induces synergistic effect in ovarian cancer warrants further investigation in a suitable animal model. Nonetheless, the mechanism of interaction between PARP and PD-L1/PD-1 as shown in the current study is timely and provides scientific basis to develop more effective combination therapies consisting of two powerful anti-cancer agents.
Supplementary Material
Statement of Translational Relevance.
With the recent approval of therapeutic antibodies that block CTLA4, PD-1, and PD-L1, immune checkpoints have emerged as new targets in cancer therapy. In addition, there is accumulating evidence highlighting the role of cancer-associated immunity in patient response to cytotoxic anticancer agents. Inhibitors of poly (ADP-ribose) polymerase (PARP) have shown substantial cytotoxic effects against tumors with defects in DNA damage response. However, whether a crosstalk between PARP inhibition and immune checkpoints exists remains unclear. Here, we show that PARP inhibitor (PARPi) treatment upregulates tumor cell PD-L1 expression, which attenuates PARPi efficacy via cancer-associated immunosuppression. The blockade of PD-L1 can restore the attenuated anti-tumor immunity and potentiate PARPi in tumor suppression. This study provides a scientific rationale for the evaluation of PD-L1 / PD-1 blockade with PARPi in clinical trials.
Acknowledgments
This work was partially supported by the following: the National Institutes of Health (CCSG CA016672); Cancer Prevention & Research Institutes of Texas (DP150052 and RP160710); Breast Cancer Research Foundation grant (to M.-C.H. and G.N.H.); Patel Memorial Breast Cancer Endowment Fund; National Breast Cancer Foundation, Inc.; The University of Texas MD Anderson Cancer Center-China Medical University and Hospital Sister Institution Fund (to M.-C.H.); Ministry of Science and Technology, International Research-intensive Centers of Excellence in Taiwan (I-RiCE; MOST 105-2911-I-002-302); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW105-TDU-B-212-134003).
Footnotes
Conflicts of Interest
The authors have declared that no conflict of interest exists.
References
- 1.Sonnenblick A, et al. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- 2.Kim G, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015;21(19):4257–61. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 3.Mirza MR, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016 doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 4.Ibrahim YH, et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discovery. 2012;2(11):1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juvekar A, et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discovery. 2012;2(11):1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karnak D, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085–96. doi: 10.1158/1078-0432.CCR-14-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kummar S, et al. Phase I Study of PARP Inhibitor ABT-888 in Combination with Topotecan in Adults with Refractory Solid Tumors and Lymphomas. Cancer Research. 2011;71(17):5626–5634. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muvarak NE, et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer Cell. 2016;30(4):637–650. doi: 10.1016/j.ccell.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Y, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22(2):194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 12.Hodi FS. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma (vol 363, pg 711, 2010) New England Journal of Medicine. 2010;363(13):1290–1290. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robert C, et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. New England Journal of Medicine. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 14.Garon EB, et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. New England Journal of Medicine. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg JE, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reck M, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016 doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 17.Galluzzi L, et al. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28(6):690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Xia W, et al. Phosphorylation/cytoplasmic localization of p21Cip1/WAF1 is associated with HER2/neu overexpression and provides a novel combination predictor for poor prognosis in breast cancer patients. Clin Cancer Res. 2004;10(11):3815–24. doi: 10.1158/1078-0432.CCR-03-0527. [DOI] [PubMed] [Google Scholar]
- 19.Lim SO, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016 doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaufman B, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellucci R, et al. JAK1 and JAK2 Modulate Tumor Cell Susceptibility To Natural Killer (NK) Cells Through Regulation Of PDL1 Expression. Blood. 2013;122(21) [Google Scholar]
- 23.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 24.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2(10):769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 25.Hirao A, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287(5459):1824–7. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 26.Anderson VE, et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011;71(2):463–72. doi: 10.1158/0008-5472.CAN-10-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li CW, Lim SO, Xia W, Lee HH, Li CC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Higuchi T, et al. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res. 2015;3(11):1257–68. doi: 10.1158/2326-6066.CIR-15-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing DY, Orsulic S. A mouse model for the molecular characterization of Brca1-associated ovarian carcinoma. Cancer Research. 2006;66(18):8949–8953. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng J, et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015;75(23):5034–45. doi: 10.1158/0008-5472.CAN-14-3098. [DOI] [PubMed] [Google Scholar]
- 31.Ding ZC, et al. Immunosuppressive Myeloid Cells Induced by Chemotherapy Attenuate Antitumor CD4(+) T-Cell Responses through the PD-1-PD-L1 Axis. Cancer Research. 2014;74(13):3441–3453. doi: 10.1158/0008-5472.CAN-13-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.