

Abstract
Chronic kidney disease (CKD) and related inflammatory responses stimulate protein-energy wasting, a complication causing loss of muscle mass. Primarily, muscle wasting results from accelerated protein degradation via autophagic/lysosomal and proteasomal pathways, but mechanisms regulating these proteolysis pathways remain unclear. Since dephosphorylation of FoxOs regulates ubiquitin/proteasome protein metabolism, we tested whether a novel nuclear phosphatase, the small C-terminal domain phosphatase (SCP) 4, regulates FoxOs signaling and, in turn, muscle wasting. In cultured mouse myoblast cells, SCP4 overexpression stimulated proteolysis while knockdown of SCP4 prevented the proteolysis stimulated by inflammatory cytokines. SCP4 overexpression led to nuclear accumulation of FoxO1/3a followed by increased expression of catabolic factors including myostatin, Atrogin-1 and MuRF-1, and induction of lysosomal-mediated proteolysis. Treatment of C2C12 myotubes with proinflammatory cytokines stimulated SCP4 expression in an NF-κB-dependent manner. In skeletal muscle of mice with CKD, SCP4 expression was up-regulated. Similarly, in skeletal muscle of patients with CKD, SCP4 expression was significantly increased. Knockdown of SCP4 significantly suppressed FoxO1/3a-mediated expression of Atrogin-1 and MuRF-1 and prevented muscle wasting in mice with CKD. Thus, SCP4 is a novel regulator of FoxO transcription factors and promotes cellular proteolysis. Hence, targeting SCP4 may prevent muscle wasting in CKD and possibly other catabolic conditions.
Keywords: Chronic kidney disease, skeletal muscle atrophy, nuclear phosphatase, FoxOs transcription factor
Introduction
Loss of lean body mass increases the risks of morbidity and mortality in patients with chronic kidney disease (CKD). Although the mechanisms leading to muscle wasting are complicated, significant progress has been made towards understanding this catabolic situation. For example, in mice with CKD, we found acidosis or cytokines impaired IGF-1/insulin signaling that triggers a caspase-3/ROCK1/PTEN negative feedback loop to suppress Akt sustainably [1–3]. A decreased Akt activity leads to decreased phosphorylation of downstream effectors, such as forkhead transcription factors (FoxOs) [4;5]. Dephosphorylation of FoxO1 at Thr24 or FoxO3a at Thr32 triggers the nuclear import and subsequently, the stimulation of the UPS proteolytic activity by increasing the expression of E3 ubiquitin ligases, Atrogin-1/ MAFbx and MuRF1 [6;7]. In addition, FoxO1 controls the expression of myostatin, a myokine which limits muscle growth and contribute to CKD-induced muscle wasting [8–10]. However, the FoxOs’ transcription activity is not only controlled by Akt signaling; phosphatases or nuclear kinases, by changing nuclear export or import of FoxOs, also influence their activities [11;12]. For example, CK1 can phosphorylate FoxOs and facilitate FoxOs’ nuclear export to decrease their activities [13].
Other inflammatory responses induced by transcription factors, such as NF-κB and signal transducer and activator of transcription 3 (STAT3) stimulate the expression of atrophy genes and contribute to muscle atrophy [14]. Several pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα), interleukin-6 (IL-6), IL-1, and interferon-γ(IFN-γ), are elevated in CKD, cancer, and other catabolic conditions, and may together contribute to muscle wasting [15;16]. NF-κB is known to mediate the cytokine-induced muscle proteolysis by activating the transcription of MuRF1 [17;18]. STAT3 is also involved in inflammatory responses that are related to muscle wasting. For example, CKD stimulates the expression of STAT3 in the muscles, which in turn increases the expression of myostatin, a growth factor belonging to the transforming growth factor (TGF) superfamily, which induces muscle atrophy [19;20]. Recent reports have enlarged the scope of power for the TGFβ superfamily. The bone morphogenetic protein (BMP) pathway reportedly acts as the fundamental hypertrophic signal and dominates myostatin signaling. Moreover, BMP-Smad1/5/8 negatively regulates a novel ubiquitin ligase MUSA1, which is required for muscle loss [21;22].
The small C-terminal domain phosphatases (SCPs) were discovered as nuclear phosphatase to dephosphorylate the C-terminal domain (CTD) of RNA polymerase II (Pol II) and hence control transcription cycle and gene expression [23;24]. There is also evidence that SCPs can modify the phosphorylation state of transcription factors and affect the transcriptional activities [25]. Our interest in SCP4 (also called CTDSPL2) was stimulated by the finding that SCP4 specifically dephosphorylates transcription factor Smad1/5/8; these reactions attenuate BMP-induced transcriptional activities [26;27]. We identified that SCP4 not only dephosphorylated Smad1/5, but also influenced FoxOs’ phosphorylation status. Furthermore, we examined how SCP4 influences muscle metabolism. Specifically, we studied whether SCP4 regulates muscle protein synthesis and degradation, and investigated whether inhibition of SCP4 would blunt muscle wasting in mice with CKD.
Results
SCP4 is a nuclear phosphatase expressed in skeletal muscle cells
SCP4 consists of a regulatory domain (amino acids 1–262), which contains two nuclear locations signaling (NLS), and a catalytic phosphatase domain (amino acids 263–466) that reportedly dephosphorylates Smad1/5 (Fig. 1a). We detected that SCP4 mRNA is widely expressed in the heart, skeletal muscles, liver, kidney, and spleen (Fig. 1b) and SCP4 protein is mainly located in the nuclei of C2C12 myotubes and myofibers in normal mice and healthy individuals (Fig. 1c). Using a Phospho Explorer antibody microarray (Supplemental Table 1), we found that knockdown of SCP4 promoted the phosphorylation of more than 25% proteins (Figure 1d). KEGG enrichment pathway analysis of significantly changed proteins revealed that Cancer pathway, MAPK signaling pathway, ErbB signaling pathway and Insulin signaling pathway as the top four enriched pathways (Fig. 1e). Notably, SCP4 knockdown stimulated the phosphorylation of FoxOs and Smads (Fig. 1f), suggesting SCP4 may influence the activities of these factors by regulating their nuclear export.
Figure 1. SCP4 may regulate FoxOs and Smads signaling in skeletal muscle cells.

a: Structural diagram of small c-terminal phosphatase 4 (SCP4); note that there are two nuclear location signaling (NLS) in its regulatory domain.
b: SCP4 mRNA expression in different tissues in mice (mean±SEM, n = 3).
c: Immunostaining revealed that SCP4 is mainly located in the nuclei of C2C12 myotubes (immunoflorescent staining, red: SCP4, blue: nuclear, scale bars: 20 μm) and myofibers from mice and human (DAB staining, brown: SCP4, Differential Interference Contrast (DIC) microscopy). Scale bars: 50 μm.
d: Phosphoproteome array in C2C12 myotubes with knockdown of SCP4. Data showing the fold change of induced phosphoproteins (yellow) and suppressed phosphoproteins (blue). The inserted immunoblots show siRNA knockdown efficiency.
e: KEGG pathway analysis of induced phosphoprotein genes (fold change > 1.5) indicated that insulin (in red) and TGF-β ( in green) signaling pathways were influenced by SCP4 knockdown.
f: The fold-change of phosphorylation sites in transcription factors FoxOs and Smads after SCP4 knockdown in C2C12 myotubes.
In C2C12 myotubes, SCP4/CTDSPL2 causes atrophy by stimulating protein degradation
To ascertain the function of SCP4 in muscle protein metabolism, we measured rates of the protein synthesis and degradation in C2C12 myotubes, which had been transfected with a plasmid encoding SCP4 tagged green fluorescent protein (SCP4-GFP). As shown in Fig. 2a, overexpression of SCP4-GFP resulted in the accumulation of SCP4 in C2C12 myotubes. This induction was associated with reduction in the diameter of the myotubes (Fig. 2b). We also found that the rate of the protein synthesis was not significantly changed in myotubes with the overexpression of SCP4 (Fig. 2c); while the rate of protein degradation in these myotubes was significantly increased compared to the results in control myotubes with forced-expression of GFP only (Fig. 2d). The increase in overall protein degradation was a result of activation of both lysosomal and proteasomal proteolysis because lactacystin (a proteasomal inhibitor, 8 uM) or concanamycin A (a lysosomal inhibitor, 0.1 uM) significantly suppressed the rate of protein degradation stimulated by SCP4 (Fig. 2e). Interestingly, SCP4-induced protein degradation was partially blocked by BMP2, but largely suppressed by IGF-1 (Fig. 2f). Taken together, these results indicate that SCP4 regulates muscle degradation by being involved in at least two signaling pathways, namely BMP-Smad1/5 and Akt/FoxO1/3a signaling.
Figure 2. Overexpression of SCP4 stimulates proteolysis in C2C12 myotubes.

a: C2C12 myoblasts were transfected with control (CTL, GFP only) or SCP4-GFP plasmids. After cells being differentiated into myotubes, SCP4 overexpression was confirmed by immunoblot.
b: Overexpression of SCP4 caused a decrease in the diameter of C2C12 myotubes. Data were represented as mean±SEM and calculated from three independent experiments. *p < 0.01 vs. CTL. Scale bars: 50 μm.
c: Protein synthesis was examined in C2C12 myotubes with or without SCP4 expression. The rate of protein synthesis was measured as the incorporation of [3H]-tyrosine; measurements were done in triplicate and independently repeated six times (mean±SEM, n =6).
d: Total proteolysis was assessed in C2C12 myotubes with or without SCP4 expression. Cells were incubated with [3H] tyrosine overnight after chasing with tyrosine and washing; the released radioactivity was plotted as a percentage of total [3H] tyrosine incorporated into cell proteins. The rates of proteolysis were calculated from the linear slopes between 16 and 24 hr (mean±SEM, n = 6, **p < 0.01 vs. CTL).
e: SCP4 overexpression stimulates both lysosomal and proteasomal proteolysis in C2C12 myotubes. Total proteolysis in control (Vehicle), proteasome related proteolysis (with lysosomal inhibitor concanamycin A) and lysosome related proteolysis (with proteosomal inhibitor Lactacystin) were shown (mean±SEM, n = 6 / group, **p < 0.01 vs. Vehicle+ovSCP4).
f: SCP4 overexpression-stimulated proteolysis in C2C12 myotubes was suppressed by IGF-1 (10 ng/ml) or BMP-2 (20 ng/ml). The differences in rates of proteolysis were calculated at 12 h after adding IGF-1 or BMP-2 (mean±SEM, n = 6/group, **p < 0.01 vs. Vehicle+ovSCP4, *p < 0.05 vs. Vehicle+ovSCP4. #p<0.05 vs. IGF-1+ovSCP4).
SCP4 blocks FoxO1/3a nuclear export and enhances Atrogin-1 and MuRF1 expression
To investigate the mechanism by which SCP4 influences muscle protein metabolism, we examined how the overexpression of SCP4 changes the gene expression profiles in C2C12 myotubers. By using the “Myogenesis & Myopathy PCR Array”, we analyzed the expression of 84 key genes involved in skeletal muscle protein metabolism, function, and disease-related processes (Fig. 3a). As shown in Fig. 3b, SCP4 overexpression upregulated ~20% of the tested genes, including myostatin, Atrogin-1 and MuRF1, three important factors mediating muscle proteolysis. By using qPCR, we confirmed the induction of myostatin (Fig. 3c) and Atrogin-1 and MuRF1 by SCP4 overexpression (Fig. 3d). In addition, we found that the expression of MUSA1 (muscle ubiquitin ligase of the SCF complex in atrophy-1, a newly identified muscle ubiquitin E3 ligase regulated by BMP signaling [22]), was upregulated by SCP4 overexpression (Fig. 3d, right panel). Myostatin, Atrogin-1 and MURF1 expression were controlled by Akt/FoxOs signaling, but we did not detect any change in p-Akt level due to SCP4 overexpression in C2C12 myotubes (Fig. 3e). Instead, we found that the overexpression of SCP4 significantly suppressed p-FoxO1/3a (Fig. 3f). To confirm that SCP4 targets FoxO1/3a to stimulate atrogene expressions, we studied the nuclear trafficking of FoxO1 in C2C12 cells overexpressing SCP4-GFP and FoxO1 tagged with red fluorescent protein. Using live cell confocal microscopy, we detected that there were remarkable amounts of FoxO1-RFP located in the nuclei of C2C12 cells cultured in serum-free media (Fig. 3g, 0 min). FoxO1-RFP started to traffic out the nuclei which had not been transfected with SCP4-GFP 15 min after insulin (2 μg/ml) was added; within 60 min after the addition of insulin, most of the FoxO1-RFP had moved into the cytoplasm (Fig. 3g, middle panel, arrow, and Fig. 3h). In contrast, in the cells that overexpressed SCP4-GFP and FoxO1-RFP, addition of insulin did not drive FoxO1-RFP out of the nucleus (Fig. 3e, middle panel, asterisk). These results indicate that SCP4 retains FoxO1 in the nucleus even though phosphor-Akt remains unchanged.
Figure 3. SCP4 increases atrogenes expression via regulation of FoxO1 nuclear export.

a: Heat map of Myogenesis & Myopathy PCR Array analysis in C2C12 myotubes with or without overexpression of SCP4. Arrows indicate the genes’ relevance to muscle proteolysis (n=3/group).
b: Statistical analysis of Myogenesis & Myopathy PCR Array as volcano plots. The fold-changes in gene expression between control (CTL) and SCP4 overexpression (ovSCP4) is plotted on the x-axis, and false discovery rate (FDR)–adjusted significance is plotted on the y axis (–log10 scale). Upregulated and downregulated genes in each SCP4 group are indicated in yellow and blue respectively. The names of some relevant antibodies are shown. The horizontal line represents a p = 0.05.
c and d: The increase in Myostatin, Atrogin-1 and MuRF1 stimulated by SCP4 as confirmed by real-time quantitative PCR. The expression of MUSA1, an E3 ligase regulated by Smad1 signaling, also increased (mean±SEM; n = 3).
e: Immunoblots showed that SCP4 overexpression minimally influences the level of p-Akt in C2C12 myotubes
f: Overexpression of SCP4 in C2C12 myotubes significantly suppresses the levels of p-FoxO1 and p-FoxO3a (mean±SEM, n = 3/group, **p < 0.01).
g: Confocal images of a representative experiment showing that SCP4 (green) sequestrates FoxO1 (red) in nucleus even upon insulin stimulation. C2C12 myoblasts were co-transfected with SCP4-GFP and FoxO1-RFP; after 24 h of serum-free incubation, the cells were treated with insulin (2 ug/ml), and the changes in the subcellular location of the FoxO1 was monitored with a confocal microscope. Asterisk indicated a cell expressing both SCP4-GFP and FoxO1-RFP. Arrow showed a cell that only expresses FoxO1-RFP.
h: FoxO1-RFP nuclear export was quantified as the attenuation of red fluorescent intensity in nuclei (** p<0.01 vs. Cells with ovSCP4. n=12). Scale bars: 25 μm.
Knockdown of SCP4 blunts proteolysis stimulated by serum-depletion in C2C12 cells
Since SCP4 overexpression causes atrophy of C2C12 myotubes, inhibition of SCP4 could prevent proteolysis induced by serum depletion. To examine this possibility, we used a siRNA approach and measured the rate of degradation stimulated by serum depletion in C2C12 myotubes. C2C12 myotubes were transfected with siRNAs for 48 h, followed by an incubation in serum-free media for 36 h. The knockdown efficiency was confirmed with western blot, the protein level of SCP4 was 90% lower compared to the myotubes transfected with control siRNA (Fig. 4a). As expected, we found that the knockdown of SCP4 significantly suppressed the rate of protein degradation that was stimulated by serum depletion (Fig. 4b). The response was associated also with the suppression of Myostatin, Atrogin-1 and MuRF1 expressions (Fig. 4c). Silencing SCP4 did not affect the p-Akt level but increased the levels of p-FoxO1 and p-FoxO3a (Fig. 4d). These results demonstrate that silencing SCP4 suppresses FoxO1/3a mediated muscle proteolysis induced by catabolic stimuli.
Figure 4. Knockdown of SCP4 blunts proteolysis stimulated by serum-depletion in C2C12 cells.

a: C2C12 myotubes were transfected with scramble control siRNA (siCTL) or SCP4 specific siRNA (siSCP4) for 48 h in DMEM containing 2% horse serum. The knockdown efficiency was confirmed by immunoblots.
b: At 48 h after transfection, C2C12 myotubes with siCTL or siSCP4 were switched to serum-free media (SF) for 36 h. The rates of proteolysis were calculated from the linear slopes between 24 and 36 h (mean±SEM, n = 3/group, *p < 0.05).
c: The mRNA level of Myostatin, Atrogin-1 and MuRF1 were assessed by RT-PCR in siCTL and siSCP4 myotubes, which were treated with 2% horse serum or serum-free media for 36 h (mean±SEM, n = 3/group, **p < 0.01).
d: The levels of p-Akt, p-FoxO1, p-FoxO3a were assessed by immunoblots in siCTL and siSCP4 myotubes that had been treated with 2% horse or serum-free media for 36 h. Note that serum depletion decreases p-Akt in both siCTL and siSCP4 cells, but this response did not affect the p-FoxOs in myotubes with SCP4 knockdown
Overexpression of SCP4 in muscle results in myofiber atrophy in mice
Since overexpression of SCP4 stimulates protein degradation in C2C12 myotubes, we next determined whether forced expression of SCP4 is sufficient to cause muscle fiber atrophy. The tibialis anterior (TA) muscles of normal C57B6J mice were transfected with control GFP or GFP-SCP4 plasmid using electroporation. We first validated the efficiency of transfection by examining the fluorescence intensity in muscle fibers transfected with the control GFP plasmid. As shown in the Supplemental Fig. 1a, more than 70% of myofibers expressed GFP at 14 days after transfection. TA muscle sections were then stained with anti-dystrophin antibody to outline myofibers. In muscles that were transfected with SCP4-GFP, we observed a remarkable SCP4-GFP expression in the nuclear fractions (Fig. 5a). We then examined the cross-sectional area (CSA) of myofibers expressing either GFP or SCP4-GFP. Forced SCP4 expression caused a ~36% decrease in fiber CSAs compared to myofibers transfected with GFP (Supplemental Fig 1b). This result was confirmed by finding a leftward shift in the distribution of the CSAs of myofibers that had forced expression of SCP4-GFP (Fig. 5b). We also detected that there was a decrease in both p-FoxO1 and p-FoxO3a compared to results in muscles transfected with the control GFP (Fig. 5c). Consistent with these results, the expression of Myostatin, Atrogin-1, MuRF1 and MUSA1 were increased in muscles electroporated with SCP4-GFP (Fig. 5d). Thus, forced expression of SCP4 sufficiently causes myofibers atrophy even in normal mice, highlighting its catabolic effects in muscle protein metabolism.
Figure 5. Forced expression of SCP4 in muscle results in myofiber atrophy in mice.

a: TA muscles of normal C57B6J mice were transfected with GFP control (GFP) and SCP4-GFP plasmids using electroporation. After 2 weeks, the total nuclear SCP4 (endogenous SCP4 plus SCP4-GFP) was assessed using immunoblots (n=5, **p<0.01); cryosections (6 μm) were immunostained with anti-dystrophin antibody to outline myofibers (red fluorescence). Scale bars: 50 μm.
b: The distribution of myofiber sizes in TA muscles transfected with SCP4-GFP was shifted leftward compared with the result in TA muscles electroporated with CTL-GFP. Data were obtained from 5 animals in each group.
c: Overexpression of SCP4 in TA muscles resulted in a decrease in p-FoxO1 and p-FoxO3a. A statistical analysis of immunoblots was shown on the right (mean±SEM, n = 5/group, **p<0.01,
d: The mRNA level of Myostatin, Atrogin-1, MuRF1 and MUSA1 were examined using RT-PCR in in TA muscles expressed GFP-CTL or GFP-SCP4 (mean±SEM, n = 5/group, **p < 0.01, *p<0.05)
CKD stimulates SCP4 expression in skeletal muscles
To investigate the pathophysiological relevance of SCP in muscle catabolism, we measured SCP4 mRNA and protein level in muscles from mice with CKD. There was significant increase in mRNA and protein in the muscles of mice with CKD (Fig. 6a, b). To determine whether SCP4 is induced by CKD in patients, we examined its expression in samples of rectus abdominis muscle from CKD patients and age/gender match controls [28]. There were significant increases in SCP4 protein level in muscles of CKD patients compared to healthy subjects (Fig. 6c, d). Next, we ascertained the mechanism by which CKD stimulates SCP4 expression in C2C12 myotubes. By analyzing the promoter region of SCP4, we found that there were many binding sites for inflammatory transcription factors, including NF-κB, E2Fs, Stat1/3, and C/EBPs (supplemental Fig. 2a). This stimulated us to investigate if CKD-related cytokines increase SCP4 expression in muscles. As shown in Fig. 6e, SCP4 expression (both mRNA and protein) was significantly increased by IL-6, IFN-γ or LPS treatments. The combination of IL-6, INF-γand LPS also significantly stimulated the expression of SCP4. Because the combination of the four cytokines is a potent stimulator for NF-κB activation [29], these results indicated that NF-κB signaling might be a major regulator mediating SCP4 expression. To confirm this possibility, we performed a ChIP assay to test whether NF-κB (p65) associates with the putative NF-κB binding sites in the promoter regions of the SCP4 gene (Supplemental Fig. 2b). Treatment of mixed cytokines enriched DNA fragments that cover the sequences in the promoter of SCP4 in the immunoprecipitates using anti-NF-κB (p65) antibody (Fig. 6f). We also found that addition of QNZ, an NF-κB inhibitor, completely blocked SCP4 expression stimulated by the cytokine mixture (Fig. 6g, h). These results suggest that CKD-stimulated NF-κB activation mediates the expression of SCP4 in muscle.
Figure 6. CKD stimulates SCP4 expression via NF-κB signaling in muscles.

a: The expression of SCP4 in the muscle of CKD mice was assessed by RT-PCR (left panel) and immunoblots (right panel). Data were presented as the mean±SEM (*p < 0.05, n = 5).
b: Representative differential interference contrast (DIC) image of SCP4 immunostaining in muscle of control (CTL) and CKD mice (DAB staining, brown: SCP4, Original magnification ×400).
c: The expression of SCP4 in muscles of controls and CKD patients was examined using immunoblotting (mean±SEM, n = 5, *p < 0.05).
d: Representative DIC image of SCP4 immunostaining in muscles of control subjects and patients with CKD (DAB staining, brown: SCP4. Scale bars: 50 μm).
e: In C2C12 myotubes, the expression of SCP4 was examined by RT-PCR (upper panel) and immunoblots (lower panel) treated with different kinds of cytokines. The mixture (Mix) is a combination of Il-6, LPS and IFN-γ, which was reportedly as a potent NF-κB signaling activator (mean±SEM, n = 3/group, **p < 0.01).
f: The ChIP assay showed that the cytokine mix significantly stimulated NF-κB (p65) binding to SCP4 promoter; normal rabbit IgG was used as the mock control (mean±SEM; n = 3, *p < 0.05).
g and h: immunoblots showed that QNZ blocked SCP4 expression stimulated by the cytokine mix in C2C12 myotubes.
Inhibition of SCP4 protects against muscle wasting in mice with CKD
Because our in vitro experiments showed that the knockdown of SCP4 suppresses muscle protein degradation by increasing FoxO1/3a nuclear export, we hypothesized that inhibition of SCP4 would block muscle wasting in CKD mice. We tested this possibility using siRNA-based gene knockdown in the mice model of CKD. We electroporated siRNAs into TA muscles of mice with subtotal nephrectomy and pair-fed them for 2 weeks. After electroporation, SCP4 protein was reduced more than 70% compared with the results from the mice electroporated with a scrambled, control siRNA (Fig. 7a). In CKD mice, there was a significant decrease in TA muscle weight after transfected with control siRNA. In contrast, when TA muscles of CKD mice were transfected with SCP4 siRNA, the reduction of TA muscle weight was significantly improved (Fig. 7b). The improvement also was confirmed by an increase in average cross-sectional area of the myofibers (Fig. 7c); the distribution of myofibers in TA muscles shifted rightward when compared to results in CKD mice that had been electroporated with control siRNA (Fig. 7d). Consistent with the findings, the expression of Myostatin, Atrogin-1 and MuRF1 significantly decreased in TA muscles of CKD mice that had been electroporated with siSCP4 (Fig. 7e). In CKD mice, we detected that both p-FoxO1 and p-FoxO3a were increased in TA muscles with SCP4 knockdown compared to results in muscles of CKD mice electroporated with control siRNA (Fig. 7f). Thus, the improvement in muscle mass in CKD mice following SCP4 inhibition was likely due to the suppression of FoxO1 and FoxO3a transcriptional activity. We summarized that SCP4 inhibition prevents muscle wasting in mice with CKD via the increase of FoxO1/3a nuclear export (Fig. 8).
Figure 7. SCP4 knockdown protects CKD-induced loss of muscle mass.

a: TA muscles of control (CTL) and CKD mice were transfected with scramble, control siRNA (siCTL) or siRNA for SCP4 (siSCP4). The expression of SCP4 (mRNA and protein) was examined by RT-PCR (left penal) or immunoblots (right panel) at 12 days after electroporation (mean±SEM, n = 5 /group).
b: The weight of TA muscles (normalized by the tibia length) was significantly improved in CKD mice with SCP4 knockdown (mean±SEM, n = 5/group).
c and d: The cross-sectional area of myofibers in TA muscles of CTL and CKD mice following electroporation with siCTL and siSCP4 are shown. The distribution of myofiber sizes in muscles from sham-operated mice treated with siCTL and siSCP4 was comparable (upper panel, data were obtained from three animals in each group). In the muscles of CKD mice treated with siSCP4, the distribution of myofiber sizes was shifted toward the right. Data were obtained from five animals in each group. Scale bars: 50 μm.
e: The expression of myostatin, Atrogin-1/MAFbx and MuRF1 mRNAs was accessed by RT-PCR. SCP4 knockdown suppressed Atrogin-1 and MuRF1 expression in the muscle of CKD mice (mean±SEM, n = 5/group).
f: p-FoxO1 (Ser 256) and p-FoxO3a (Ser 253) were examined using immunoblotting in muscles of sham-operated and CKD mice electroporated with siCTL or siSCP4. Along with a SCP4 knockdown, p-FoxO1 and p-FoxO3a levels were significantly increased in the muscle of CKD mice (mean±SEM, n = 5/group, *p < 0.05 vs. CKD+siSCP4).
Figure 8. A summative diagram of SCP4 causes muscle wasting in CKD.
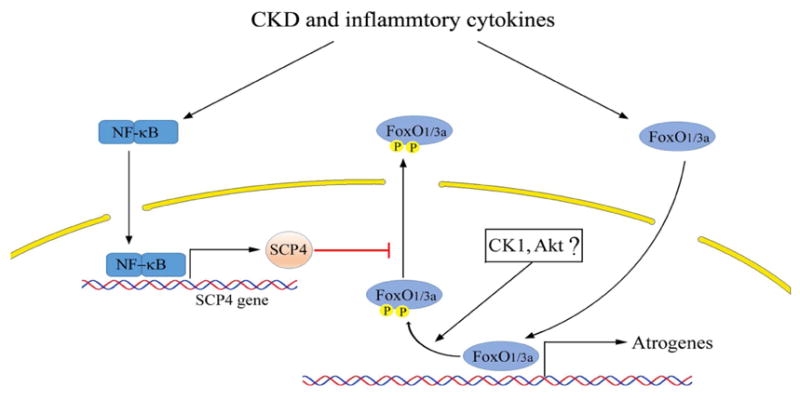
CKD and associated inflammation activates NF-kB leading to SCP4 expression. In nucleus, SCP4 dephosphorylates FoxO1/3a and prevents FoxO1/3a nuclear export resulting in atrogrnes expression and increased muscle proteolysis. Therefore, inhibition of SCP4 prevents muscle wasting via the suppression of FoxO1/3a.
Discussion
Recently, it was reported that the phosphatase SCP4, working as a Smad phosphatase, negatively regulates BMP signaling [25;27]. We observed that SCP4 not only suppresses BMP signaling but also counteracts IGF-1/insulin signaling (Fig. 2F). These distinct properties of SCP4 underscore its importance in regulating muscle protein metabolism because IGF-1 and BMP signaling are essential for maintaining skeletal muscle mass. By identifying the function of SCP4 in muscle protein metabolism, we have uncovered a novel mechanism by which CKD stimulates muscle protein wasting: CKD and inflammatory cytokines activate NF-κB pathway to stimulate SCP4 expression, and SCP4 dephosphorylates nuclear FoxO1/3a increasing their nuclear accumulation as a result of the activation of muscle proteolysis pathways, including autophagic/lysosomal and ubiquitin-proteasomal pathways [30;31]. Since the activation of FoxO1/3a is implicated in many types of muscle atrophy [32;33], we speculate that the activation of SCP4 could be a critical mechanism for muscle wasting occurring in other catabolic conditions, such as diabetes, sepsis and cancer cachexia.
What controls FoxO1/3a activity during muscle proteolysis? FoxO1/3a are quite stable proteins that continuously shuttle between the nucleus and cytoplasm and depend on cellular metabolic demand for achieving this dynamic equilibrium [34]. When responding to ligand-activated signaling (e.g. insulin signaling), FoxO1/3a are phosphorylated by Akt and this reaction produces a docking site to bind with 14-3-3 proteins, preventing FoxO1/3a nuclear import [12]. When responding to ligand-independent signaling (e.g. AMPK signaling), FoxO1/3a are also phosphorylated, but in this case, the phosphorylation (at Ser 329) facilitates their nuclear import [35]. Alternatively, dephosphorylation of FoxO1/3a dramatically influences their subcellular localization and transcription activities. For example, certain cytoplasm phosphatases, such as PP2A, reportedly dephosphorylate FoxO1/3a and promote their nuclear translocation [36]. In the nucleus, there are also protein kinases that phosphorylate FoxO1/3a and facilitate their nuclear export. For example, nuclear Akt reportedly phosphorylates FoxO1/3a to facilitate their cytoplasmic translocation; Casein kinase-1 can phosphorylate FoxO1/3a (at S318 and S321) to directly promote their nuclear export [13]. However, whether there are antagonistic phosphatases to balance nuclear protein kinase remains unexplored. Our results suggested that SCP4 might be a nuclear antagonistic phosphatase, perhaps by directly dephosphorylating FoxO1/3a. In this manner, SCP4 inhibits the nuclear export signaling (NES) of FoxO1/3a and produces the dephosphorylation-dependent nuclear sequestration and hence transcriptional activity in response to catabolic stimuli.
Another important finding is that CKD stimulates SCP4 expression in muscle. To understand the underling mechanism, we treated muscle cells with proinflammatory cytokines and identified NF-κB to be responsible for SCP4 transcription (Fig. 6). These results partially explained why proinflammatory cytokines are able to simultaneously stimulate both MuRF1 and Atrogin-1 / MAFbx expression: cytokines stimulate NF-κB to cause MuRF1expression, meanwhile, NF-κB promotes SCP4 expression to activate FoxO1/3a and hence Atrogin-1/MAFbx expression. Another mechanism by which SCP4 up-regulates Atrogin-1 and MuRF-1 is increase in myostatin, because evidence suggests that myostatin upregulates atrophy-related ubiquitin ligases in a FoxO-dependent manner [8].
There are only few reports describing the strategy of phosphatase inhibition to prevent muscle wasting in catabolic conditions. For example, we reported that the phosphatase PTEN inhibition protects muscle wasting in CKD mice [3;4]; knockdown of protein phosphatase 2C-alpha (PP2Cα) protects angiotensin II-induced muscle wasting [37]. In the present study, we demonstrated a novel nuclear phosphatase, SCP4, which regulates muscle protein degradation by preventing FoxO1/3a nuclear translocation and activity. Upregulation of SCP4 enhances FoxO1/3a transcription activities by preventing their nuclear export; conversely, SCP4 inhibition in catabolic condition suppresses FoxO1/3a activity and hence muscle proteolysis in CKD. Since FoxO1/3a mediated autophagy/lysosomal and proteasomal pathways commonly involve different kinds of catabolism, the discovery of SCP4 as a regulator of FoxO1/3a signaling could lead to a new therapeutic intervention to prevent muscle wasting induced by CKD or other catabolic conditions.
Methods
CKD Models and Electroporation
Male C57BL/6 mice (12-week-old) were housed with 12 h light/dark cycles, and all animal procedures were approved in Baylor College of Medicine’s Institutional Animal Care and Use Committee. The CKD model was created using subtotal nephrectomy as previously described [4]. Six weeks after subtotal nephrectomy, CKD mice and sham controls underwent electroporation and pair-feeding for 2 weeks. For pair-deeding, the amount of chow eaten by CKD mouse is fed to the sham-control mouse, this sequence is repeated for the duration of the experiment. For electroporation, 20 μg of plasmid (pXF-GFP-SCP4, pXF-GFP-only) or 0.01 nmol of siRNA (scramble siRNA or Smart Pool siRNA targeting SCP4, GE Dharmacon, Lafayette, CO) in 20 μl of phosphate-buffered saline were injected into TA muscles. In each mouse, the left TA muscle was received empty vector or scramble siRNA; the right TA muscle was received pXF-SCP4 or siSCP4. Electroporation was performed at 80 V for 10 pulses (100 ms each pulse, 200 ms intervals). Nuclei fractions from TA muscle were isolated as described [38].
Human Muscle Biopsies
During placement of a peritoneal dialysis catheter in CKD patients, the rectus abdominis muscle was biopsied, frozen at −80°C and stored until analyzed. Biopsy of this muscle was obtained from healthy subjects during abdominal hernia surgeries. The procedures were approved by the Ethical Committee of the Department of Internal Medicine of the University, Genoa, Italy, in accordance with the Helsinki declaration regarding ethics of human research [28].
Cell culture and Confocal living imaging
C2C12 cells were cultured in Dulbecco’s modified Eagle’s medium as described [39]. For overexpression or knockdown of SCP4, plasmid DNA (500 ng/ml) or siRNA (0.01nmol) was introduced into cells using an Invitrogen electroporation transfection system [40]. For monitoring FoxO1 nuclear export, C2C12 myoblasts were co-transfected with pXF-GFP-SCP4 and pdsRED-Mono-N1-FoxO1 plasmids. After 24 h, cells were incubated in serum-free for another 24 h. After the baseline image was taken using a Nikon A1-Rs Inverted Laser Scanning Microscope (set as 0 min at this point), insulin was added to the selected cells (2 ug/ml) and images were taken for 60 min at 15-min increments. To quantify the FoxO1-Red nuclear translocation, we measured the decrease of red fluorescent intensity in nuclear using NIH imageJ. Total of 12 cells in each group were examined. The pdsRED-Mono-N1-FoxO1 was a gift from Domenico Accili (Addgene plasmid # 34678).
Protein synthesis and degradation using isotopic
C2C12 myotubes were incubated in media containing L-[3, 5–3H] tyrosine (5 μCi/ml; PerkinElmer, USA) for 24 h and following the procedures as described [39]. The released radioactivity was plotted as a percentage of total [3H] tyrosine incorporated into total cell proteins. The rates of proteolysis were calculated from the linear slopes between 24 and 36 h.
RNA Preparation and Real-time qPCR
The procedures of RNA Preparation and Real-time qPCR were previously described [39]. All mRNA expressions were normalized to RPL39 [41;42] or beta-actin. The primer sets are listed in supplemental Figure 1c.
RT² profiler PCR array analysis
Total RNA was reverse-transcribed to cDNA using the First Strand cDNA Kits (Qiagen Sciences, Germantown, MD). RT² Profiler™ PCR Array (Myogenesis & Myopathy, PAMM-099Z) was performed following the manufactory’s instruction (Qiagen Sciences).
Western blot analysis
The method of western blotting was previously described [4]. The antibodies GAPDH, beta-tubulin, p-FoxO1 (Ser 256), p-FoxO3a (Ser 253), p-Akt and anti-SCP4 (CTDSPL2) were purchased from Cell Signaling Technology (Cambridge, MA).
Phospho-specific protein microarray analysis
Cell lysate (~100 μg) from C2C12 myotubes transfected with scramble siRNA or siSCP4 were subjected to a Phospho Explorer antibody microarray (Full Moon Biosystems, Sunnyvale, CA) according to the manufacturer’s protocols.
Immunostaining and myofibers size measurement
After being fixed with 4% formaldehyde, the frozen tibialis anterior muscles’ sections (5 μm) were subjected to immunostaining or immunohistochemistry as described [3]. For measuring myofiber diameter, C2C12 myotubes were transfected with adenovirus bearing GFP for 24 h to outline cells. Myotube images were captured using Nikon inverted microscope. Diameter was measured between two adjacent nuclei (avoiding regions of clustered nuclei) using National Institutes of Health (NIH) Image J 1.63 software. For a single myotuber, the average of 3~4 measurement was calculated as diameter of the myotube. Total 200 myotubes/well were examined and the mean of their diameters was represented as diameter of long multinucleate myotubes.
Promoter binding site and ChIP assay
The transcription factor binding site in promoter region of SCP4 (CTDSPL2) gene was obtained use online software provide by Qiangen website (Supplemental Figure 2a). We then analyzed ~2400 bp promoter region upstream of start codon (nm_6748574) using P-Match (1.0) program and identified three putative NF-κB binging sites (Supplemental Figure 2b). The chromatin immunoprecipitation (ChIP) assay was performed using a SimpleChIP Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology) following the manufacturer’s instruction.
Statistical analysis
Results are presented as mean ± SEM. For experiments comparing two groups, we analyzed results by two-tail unpaired Student’s t-tests. When more than two groups were compared, two-way ANOVA followed by Newman-Keul’s test were used to analyze the differences between the two interested groups. Differences were considered statistically significant at p < 0.05 (*).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We acknowledge the National Institutes of Health Grants (5RO1 AR063686 to Z.H) for supporting this study. S.S.T is supported by the VA Career Development Award (1IK2 BX002492) from the United States Department of Veterans Affairs, Biomedical Laboratory Research and Development Program, and by Michael E. DeBakey, VA Medical.
Footnotes
Author contributions statement: X.L, R.Y, L.S, R.L carried out experiments, study design and data analysis; G.G, Y.W, S.S.T, R.L and Z.H interpreted the data; Z.H wrote the paper. All authors had final approval of the submitted versions.
Disclosure: All the authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Franch HA, Price SR. Molecular signaling pathways regulating muscle proteolysis during atrophy. Curr Opin Clin Nutr Metab Care. 2005;8:271–275. doi: 10.1097/01.mco.0000165005.01331.45. [DOI] [PubMed] [Google Scholar]
- 2.Lee SW, Dai G, Hu Z, et al. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15:1537–1545. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 3.Peng H, Cao J, Yu R, et al. CKD Stimulates Muscle Protein Loss Via Rho-associated Protein Kinase 1 Activation. J Am Soc Nephrol. 2016;27:509–519. doi: 10.1681/ASN.2014121208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu J, Li R, Workeneh B, et al. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012;82:401–411. doi: 10.1038/ki.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milan G, Romanello V, Pescatore F, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z, Lee IH, Wang X, et al. PTEN expression contributes to the regulation of muscle protein degradation in diabetes. Diabetes. 2007;56:2449–2456. doi: 10.2337/db06-1731. [DOI] [PubMed] [Google Scholar]
- 8.McFarlane C, Plummer E, Thomas M, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J Cell Physiol. 2006;209:501–514. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- 9.Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007;292:C188–C199. doi: 10.1152/ajpcell.00542.2005. [DOI] [PubMed] [Google Scholar]
- 10.Verzola D, Procopio V, Sofia A, et al. Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int. 2011;79:773–782. doi: 10.1038/ki.2010.494. [DOI] [PubMed] [Google Scholar]
- 11.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–169. doi: 10.1016/j.tibs.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011;1813:1938–1945. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Rena G, Woods YL, Prescott AR, et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 2002;21:2263–2271. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li YP, Chen Y, John J, et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011;30:4323–4335. doi: 10.1038/emboj.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doyle A, Zhang G, Abdel Fattah EA, et al. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011;25:99–110. doi: 10.1096/fj.10-164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee D, Goldberg AL. Muscle Wasting in Fasting Requires Activation of NF-kappaB and Inhibition of AKT/Mechanistic Target of Rapamycin (mTOR) by the Protein Acetylase, GCN5. J Biol Chem. 2015;290:30269–30279. doi: 10.1074/jbc.M115.685164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hindi SM, Mishra V, Bhatnagar S, et al. Regulatory circuitry of TWEAK-Fn14 system and PGC-1alpha in skeletal muscle atrophy program. FASEB J. 2014;28:1398–1411. doi: 10.1096/fj.13-242123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sala D, Sacco A. Signal transducer and activator of transcription 3 signaling as a potential target to treat muscle wasting diseases. Curr Opin Clin Nutr Metab Care. 2016;19:171–176. doi: 10.1097/MCO.0000000000000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonetto A, Aydogdu T, Jin X, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab. 2012;303:E410–E421. doi: 10.1152/ajpendo.00039.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sartori R, Gregorevic P, Sandri M. TGFbeta and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends Endocrinol Metab. 2014;25:464–471. doi: 10.1016/j.tem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Sartori R, Schirwis E, Blaauw B, et al. BMP signaling controls muscle mass. Nat Genet. 2013;45:1309–1318. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- 23.Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Kim Y, Genoud N, et al. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol Cell. 2006;24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knockaert M, Sapkota G, Alarcon C, et al. Unique players in the BMP pathway: small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc Natl Acad Sci U S A. 2006;103:11940–11945. doi: 10.1073/pnas.0605133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sapkota G, Knockaert M, Alarcon C, et al. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281:40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Xiao M, Sun B, et al. C-terminal domain (CTD) small phosphatase-like 2 modulates the canonical bone morphogenetic protein (BMP) signaling and mesenchymal differentiation via Smad dephosphorylation. J Biol Chem. 2014;289:26441–26450. doi: 10.1074/jbc.M114.568964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Pan J, Dong Y, et al. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013;18:368–379. doi: 10.1016/j.cmet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas SS, Dong Y, Zhang L, Mitch WE. Signal regulatory protein-alpha interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int. 2013;84:308–316. doi: 10.1038/ki.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas SS, Mitch WE. Mechanisms stimulating muscle wasting in chronic kidney disease: the roles of the ubiquitin-proteasome system and myostatin. Clin Exp Nephrol. 2013;17:174–182. doi: 10.1007/s10157-012-0729-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–2129. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodine SC, Furlow JD. Glucocorticoids and Skeletal Muscle. Adv Exp Med Biol. 2015;872:145–176. doi: 10.1007/978-1-4939-2895-8_7. [DOI] [PubMed] [Google Scholar]
- 33.Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab. 2014;307:E469–E484. doi: 10.1152/ajpendo.00204.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez AM, Candau RB, Bernardi H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci. 2014;71:1657–1671. doi: 10.1007/s00018-013-1513-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Yan L, Lavin VA, Moser LR, et al. PP2A regulates the pro-apoptotic activity of FOXO1. J Biol Chem. 2008;283:7411–7420. doi: 10.1074/jbc.M708083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabony AM, Yoshida T, Sukhanov S, Delafontaine P. Protein phosphatase 2C-alpha knockdown reduces angiotensin II-mediated skeletal muscle wasting via restoration of mitochondrial recycling and function. Skelet Muscle. 2014;4:20. doi: 10.1186/2044-5040-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du J, Mitch WE, Wang X, Price SR. Glucocorticoids induce proteasome C3 subunit expression in L6 muscle cells by opposing the suppression of its transcription by NF-kappa B. J Biol Chem. 2000;275:19661–19666. doi: 10.1074/jbc.M907258199. [DOI] [PubMed] [Google Scholar]
- 39.Yu R, Chen JA, Xu J, et al. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J Cachexia Sarcopenia Muscle. 2016 doi: 10.1002/jcsm.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou X, Li R, Liu X, et al. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci Rep. 2016;6:29669. doi: 10.1038/srep29669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwee DT, Gomes AV, Bodine SC. Cardiac proteasome activity in muscle ring finger-1 null mice at rest and following synthetic glucocorticoid treatment. Am J Physiol Endocrinol Metab. 2011;301:E967–E977. doi: 10.1152/ajpendo.00165.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hwee DT, Baehr LM, Philp A, et al. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell. 2014;13:92–101. doi: 10.1111/acel.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.