

Abstract
The sodium/proton exchanger isoform 3 (NHE3) is expressed in the intestine and the kidney where it facilitates sodium (re)absorption and proton secretion. The importance of NHE3 in the kidney for sodium chloride homeostasis, relative to the intestine, is unknown. Constitutive tubule-specific NHE3 knockout mice (NHE3loxloxCre) did not show significant differences compared to control mice in body weight, blood pH or bicarbonate and plasma sodium, potassium or aldosterone levels. Fluid intake, urinary flow rate, urinary sodium/creatinine and pH were significantly elevated in NHE3loxloxCre mice while urine osmolality and GFR were significantly lower. Water deprivation revealed a small urinary concentrating defect in NHE3loxloxCre mice on a control diet; exaggerated on low sodium chloride. Ten days of low or high sodium chloride diet did not affect plasma sodium in control mice; however, NHE3loxloxCre mice were susceptible to low sodium chloride (about −4 mM) or high sodium chloride intake (about +2 mM) versus baseline, effects without differences in plasma aldosterone between groups. Blood pressure was significantly lower in NHE3loxloxCre mice and was sodium chloride-sensitive. In control mice, the expression of the sodium/phosphate co-transporter Npt2c was sodium chloride-sensitive. However, lack of tubular NHE3 blunted Npt2c expression. Alterations in the abundances of sodium/chloride cotransporter and its phosphorylation at threonine 58 as well as the abundances of the α-subunit of the epithelial sodium channel, and its cleaved form, were also apparent in NHE3loxloxCre mice. Thus, renal NHE3 is required to maintain blood pressure and steady state plasma sodium levels when dietary sodium chloride intake is modified.
Keywords: proximal tubule, aldosterone, distal tubule, hypernatremia, hyponatremia
Introduction
The kidneys play an important role in maintaining body sodium (Na+) and volume homeostasis and thus blood pressure regulation. If Na+ homeostasis cannot be achieved despite activation of various Na+ conserving mechanisms, Na+ loss and volume depletion results in hypotension, circulatory collapse and ultimately, death. Body Na+ balance is maintained, predominantly, by interplay between uptake in the gastrointestinal tract and reabsorption/excretion by the kidney. In the intestine, the luminal Na+/H+ exchanger isoform 3 (NHE3) is critical for Na+ absorption.1–3 NHE3 is also expressed in the luminal membrane of the renal proximal tubule, where the majority of renal Na+ reabsorption (60–75%) occurs.3 Perfusion studies in proximal tubules of whole body NHE3 knockout mice (NHE3−/−) determined that Na+ reabsorption was reduced by ~63% in vivo4 and ~46% in vitro.5 During microperfusion, inhibition of NHE3 in vivo using S3226 (inhibitor) determined that the maximum reduction of Na+ reabsorption was ~30%.6 This ~2-fold difference between Na+ reabsorption in genetically modified mice or subsequent to pharmacological inhibition was previously attributed to a contribution of NHE3 to both direct and indirect reabsorption of Na+ in the proximal tubule.
Due to the role of NHE3 in the intestine for Na+ absorption and volume regulation, NHE3−/− mice have diverse problems with regard to Na+ homeostasis. The mice suffer from severe diarrhea, dilation of the intestinal tract and a high mortality rate subsequent to weaning, with only ~30% of mice surviving into adulthood.3, 7 When fed a low NaCl diet, NHE3−/− mice experience severe dehydration resulting in hypovolemia, body weight loss, and death.2 When NHE3−/− mice are fed a high-NaCl diet (5% NaCl), the small intestine becomes severely swollen, consequently resulting in death within 2 days.8, 9 This was attributed to the osmotic effect exerted by the high NaCl levels in the intestine.
NHE3 has been linked directly to blood pressure regulation;10 however, the exact role of NHE3 is complex. In hereditary strains of hypertensive rats, NHE3 activity is increased before the onset of hypertension.11, 12 However, after hypertension is established, proximal tubular fluid reabsorption is decreased, consistent with the activation of pressure natriuresis.11 NHE3−/− mice with transgenic expression of NHE3 in the small intestine (tgNHE3−/−) show a blunted pressure natriuresis.9 Alterations in blood pressure may result in pressure natriuresis due to redistribution of NHE3 out of the microvilli to intermicrovillar clefts below the apical microvilli and then to endosomal pools.13 These effects, in part, may be due to angiotensin II (AngII). AngII has a biphasic effect on blood pressure; acutely applied pressure doses of AngII induce pressure natriuresis possibly via internalization of NHE3,14, 15 whereas long-term application of subpressor doses of AngII increase NHE3 expression and blood pressure.16–18 Vice versa, inhibiting angiotensin-converting enzyme induced a redistribution of NHE3 from enriched microvillar membranes to intermicrovillar membrane-enriched fractions.19 The role of NHE3 is supported by recent studies using NHE3−/− 20 and tgNHE3−/− 21 mice that have an absence of AngII-induced hypertension. Of note, in humans, NHE3 polymorphisms have not been linked to essential hypertension.22
Since the severe defects from global NHE3 deletion can arise due to lack of NHE3 in the intestine or the kidney,2, 3, 7 we examined whether renal tubular NHE3 plays a significant role in NaCl homeostasis and blood pressure regulation. We examined the effects of altered dietary NaCl on whole body NaCl homeostasis and blood pressure in control and constitutive tubule-specific NHE3 knockout mice.23 The results demonstrate that in the absence of tubular NHE3, dietary NaCl alterations directly impact total body NaCl levels by disturbing glomerulo-tubular balance. In addition, NHE3 contributes to the NaCl sensitivity of blood pressure and, possibly as a result of its expression in the medullary thick ascending limb (TAL), urinary concentrating ability. However, considering the overall mild phenotype observed during dietary NaCl challenges in the absence of tubular NHE3, an essential role for renal NHE3 for NaCl and acid-base homeostasis needs to be reconsidered.
RESULTS
Basal analysis of renal tubule-specific NHE3loxloxCre mice with free access to fluid and food
We utilized non-inducible, constitutive, tubule-specific NHE3loxloxCre mice.23 NHE3loxloxCre mice have absent cortical NHE3, and severely reduced (>85%) NHE3 labeling in the medullary TAL (Supplementary Figure 1). Labeling of NHE3 in the intestine identified a comparable signal between genotypes (Supplementary Figure 2). Physiological analysis of control (Con) and NHE3loxloxCre mice with free access to fluid and food in regular cages and metabolic cages indicated that fluid intake was significantly higher in NHE3loxloxCre mice associated with a higher urinary flow rate and lower urine osmolality (Figure 1 and Supplemental Figure 3). Body weight, plasma concentrations of Na+, K+ and aldosterone, plasma osmolality as well as bicarbonate and base excess were not significantly different between genotypes (Figure 1 and Supplementary Figures 3 and 4). NHE3loxloxCre mice had a higher urinary pH compared to Con mice and a ~20% lower GFR (Figure 1). In NHE3loxloxCre mice, food and Na+ intake tended to be higher compared to Con mice and were associated with a tendency for higher urinary Na+, K+ and Cl− excretion (Figure 1). Independent of genotype, ~80% of the ingested Na+ was excreted via the kidneys. In contrast, urinary Na+, K+ and Cl− concentrations were significantly lower in NHE3loxloxCre compared to Con mice (Supplemental Figure 3), a likely consequence of higher fluid intake/urine output. To test for a role of NHE3 in urinary concentrating ability an 18 hour water deprivation (WD) was performed on animals consuming a standard diet or after 7 days of low or high NaCl intake. On standard diet NHE3loxloxCre mice were not able to reach the same urine osmolality compared to Con mice in response to WD and remained ~400 mmol/kg lower (Figure 2). After 7 days of low dietary NaCl, NHE3loxloxCre mice have impaired urinary concentrating ability after 18 hour WD as evidenced by a lower urine osmolality (~750 mmol/kg) and greater increase in plasma osmolality (~15 mmol/kg) compared to Con mice (Figure 2). After 7 days of high NaCl intake and 18 hour WD no significant differences were observed between genotypes in urine and plasma osmolality (Supplementary Figure 5).
Figure 1. NHE3loxloxCre mice show higher fluid intake, lower GFR and lower urine osmolality in conjunction with higher urinary pH and lower Na+ excretion.

NHE3loxloxCre compared to Con fed a control diet (0.8% NaCl) show comparable body weight, plasma concentrations of Na+, K+, aldosterone, plasma osmolality and blood pH. Fluid intake and urine osmolality were significantly higher and lower, respectively, in NHE3loxloxCre mice. Urinary pH was significantly higher in NHE3loxloxCre mice. GFR was significantly lower, while Na+ intake and urinary Na+ excretion tended to be higher in NHE3loxloxCre compared to Con mice. Baseline fluid and food intake n = 31–32; for other parameters n = 9–32. *P < 0.05 versus Con.
Figure 2. Under baseline conditions and after 7 days on low NaCl intake NHE3loxloxCre mice have an impaired urinary concentrating ability in response to 18 hour water deprivation (WD).

(A) WD significantly increased urine osmolality in Con and NHE3loxloxCre mice and remained slightly but significantly lower after 18 hours in NHE3loxloxCre mice; however, (B) the total increase in urine osmolality was comparable between genotypes. (C) Plasma osmolality was comparable under baseline conditions and increased to a similar extent in both genotypes. (D) Body weight changes were comparable between Con and NHE3loxloxCre mice. After 7 days of low NaCl intake, WD significantly increased urine osmolality in both genotypes; however, the increase was significantly lower after 18 hours in NHE3loxloxCre mice. (E) The total increase in urine osmolality was significantly lower in NHE3loxloxCre mice. (F) Possibly as a consequence of the impaired urinary concentrating ability in NHE3loxloxCre mice, plasma osmolality increased to a significantly greater extent and (G) body weight loss tended to be higher in NHE3loxloxCre compared to Con mice. n=7–12/genotype; *P<0.05 versus Con same condition; #P<0.05 versus respective diet same mouse.
Response to varying NaCl intake
NHE3−/− mice do not tolerate a 0.01% NaCl diet; facing severe weight loss, indicative of volume depletion, and mortality.2 If the kidney is of major importance for these observations, we reasoned that NHE3loxloxCre mice would show a comparable phenotype when challenged by a low NaCl diet. In contrast to NHE3−/− mice,2 NHE3loxloxCre mice show a subtle but significant body weight loss (−1.5 g or 5% body weight versus baseline, Figure 3A). Fluid intake remained significantly higher in NHE3loxloxCre mice during low NaCl intake (Figure 3B). In both genotypes NaCl intake decreased significantly in response to low dietary NaCl (Figure 3C) in combination with significantly decreased urinary Na+/creatinine (Figure 3D). In metabolic cage experiments (Supplementary Figure 6), dietary Na+ intake (Figure 3E) was not significantly different between genotypes and associated with a comparable urinary Na+ excretion (Figure 3F). Urinary pH decreased in both genotypes; however, the pH remained significantly higher in NHE3loxloxCre mice during the 10 day experimental period (Figure 3G).
Figure 3. Response of NHE3loxloxCre and Con mice to low NaCl intake.

In response to low NaCl intake Con mice did not show a significant weight change; however, NHE3loxloxCre mice show a subtle but significant decrease in body weight over the 10 day experimental period (A). Fluid intake was significantly lower in NHE3loxloxCre mice compared to baseline during the experimental period, but remained significantly higher than Con mice throughout the treatment. In Con mice, fluid intake was only significantly different from baseline on day 1 of low dietary NaCl (B). Both genotypes show comparable reductions in NaCl intake which was not significantly different between genotypes during the experimental period (C). In response to low NaCl intake, urinary Na+/creatinine decreased and was not significantly different between genotypes (D). In metabolic cage experiments, Na+ intake (E) was comparable and urinary Na+ excretion (F) was not different between genotypes. Urinary pH decreased in both genotypes; however, remained significantly higher in NHE3loxloxCre mice (G). n = 6–7/genotype. *P < 0.05 versus Con, #P < 0.05 versus baseline (day 0) same genotype.
In response to high NaCl diet body weight was unaffected and not significantly different between genotypes (Figure 4A). Fluid intake increased in both genotypes, but was significantly higher in NHE3loxloxCre mice versus Con mice (Figure 4B). NaCl intake increased ~3-fold for the first 2 days in both genotypes and remained at this level in Con mice. NHE3loxloxCre mice slowly decreased NaCl intake throughout the experimental period, with Na+ intake being 30% lower at the end of the experiment (Figure 4C), an effect occurring without differences in urinary Na+/creatinine between genotypes (Figure 4D). Metabolic cage experiments (Supplementary Figure 7) confirmed a comparable Na+ intake (Figure 4E) and urinary Na+ excretion (Figure 4F) between genotypes. Urinary pH tended to decrease in Con mice but was not significantly different on day 10 versus baseline. In NHE3loxloxCre mice urinary pH decreased significantly in response to high NaCl; however, it remained significantly higher compared to Con mice (Figure 4G).
Figure 4. Higher fluid intake and urinary pH in NHE3loxloxCre mice following high NaCl dietary intake.

Body weight was not measurably sensitive to high NaCl intake in either genotype (A). High NaCl intake increased fluid intake in both genotypes but to a significantly greater extent in NHE3loxloxCre compared to Con mice (B). NaCl intake increased ~3-fold initially in both genotypes. Whereas Con mice stayed at this level for the remainder of the experimental period, NHE3loxloxCre mice slightly decreased their NaCl intake over time. (C). Urinary Na+/creatinine increased 3-fold in response to high NaCl diet but was not significantly different during the remained of the experimental period between genotypes (D). In metabolic cage experiments, Na+ intake (E) was comparable and urinary Na+ excretion (F) was not different between genotypes. Comparable to low NaCl, urinary pH decreased in response to high NaCl in both genotypes; however, remained at a significantly higher level in NHE3loxloxCre compared to Con mice (G). n = 5–7/genotype. *P < 0.05 versus Con, #P < 0.05 versus baseline (day 0) same genotype.
In Con mice, irrespective of dietary NaCl, plasma Na+ was unchanged (Figure 5A); however, plasma Na+ in NHE3loxloxCre mice was susceptible to the effects of low (−3.9±1.0 mM, P<0.05) and high NaCl diet (+2.2±0.6 mM, P<0.05; Figure 5B). Aldosterone plasma concentrations were not different between Con and NHE3loxloxCre mice on low or high NaCl intake (Figure 5E, 5F). Possibly as a consequence of increased aldosterone concentration in response to low NaCl intake, plasma K+ significantly decreased in Con mice (−0.5±0.2 mM, P<0.05; Figure 5C) but to a significantly greater extent in NHE3loxloxCre mice (−1.2±0.1 mM, P<0.05; Figure 5D). High NaCl intake did not significantly alter plasma K+ in Con mice, yet resulted in a significant decrease in NHE3loxloxCre mice (Figure 5C, 5D). Both genotypes responded with more alkaline and acidic blood pH (Figure 5G, 5H) and bicarbonate (Supplementary Figure 4) in response to low and high NaCl intake, respectively; which may be secondary to changes in plasma K+.24 GFR determined in conscious mice decreased in Con (−110±13 μl/min, P<0.05) and NHE3loxloxCre (−99±8 μl/min, P<0.05) mice to a similar extent following low NaCl intake. High NaCl did not show a clear effect on GFR irrespective of genotype (Figure 6A). To exclude structural defects of the glomeruli causing, or resulting from, the lower GFR in NHE3loxloxCre mice, we performed collagen IV staining in Con and NHE3loxloxCre mice on control diet. No observable differences in staining intensity or histology were observed between genotypes (Figure 6B).
Figure 5. Plasma Na+ in NHE3loxloxCre mice is susceptible to dietary NaCl.

Dietary NaCl had no effect on plasma [Na+] in Con mice (A). In contrast, the amount of dietary NaCl directly impacted plasma [Na+] in NHE3loxloxCre mice (B). In response to low NaCl plasma [K+] was decreased to a significantly greater extent in NHE3loxloxCre compared to Con mice. High NaCl intake slightly reduced plasma [K+] in NHE3loxloxCre mice only (C, D). Aldosterone was increased and reduced in response to low and high NaCl intake, respectively; however, levels between genotypes were not significantly different (E, F). Low NaCl intake caused an increase of blood pH in both genotypes whereas high NaCl intake reduced blood pH (G, H). *P < 0.05 versus Con same diet, #P < 0.05 versus baseline same mouse, ¶P < 0.05 versus low NaCl diet same genotype.
Figure 6. Glomerular filtration rate (GFR) is significantly lower in NHE3loxloxCre compared to Con mice.

As shown in Figure 1, baseline GFR was significantly lower in NHE3loxloxCre compared to Con mice. GFR was measured following dietary NaCl challenges for a period of 10 days. (A) Low NaCl intake reduced GFR in both genotypes; in contrast, high NaCl intake was without effect on GFR in either genotype. (B) Representative imaging of Collagen IV staining surrounding the glomerulus did not highlight any clear differences in glomerular structure or size of Bowman’s space between genotypes. *P < 0.05 versus Con same diet, #P < 0.05 versus baseline same mouse, ¶P < 0.05 versus low NaCl diet same genotype.
Effect of dietary NaCl on renal protein expression and NCC/γENaC staining
In this study renal NHE3 was undetectable by Western blotting in NHE3loxloxCre mice, confirming NHE3 knockout, independent of dietary changes (Figure 7). Phosphorylation of NHE3 at serine 552 (pS552 NHE3), an inhibitory phosphorylation site, was also absent in NHE3loxloxCre mice. In Con mice, alterations in dietary NaCl were without significant effects on NHE3 or pS552 NHE3 abundance. The levels of the Na+/phosphate cotransporter, Npt2a, were not altered by dietary NaCl in either genotype. However, Npt2a expression was significantly lower (~30%) in NHE3loxloxCre mice compared to Con mice irrespective of dietary NaCl (Figure 7). In contrast, Npt2c expression (Figure 7) in Con mice showed significant NaCl dependence; low NaCl diet reduced, and high NaCl diet increased Npt2c expression. Of note, Npt2c expression in the NHE3loxloxCre mice was reduced to almost undetectable levels and was not affected by dietary NaCl. Abundance of the Na+/bicarbonate cotransporter, NBCe1, was not affected by dietary NaCl or genotype (Figure 7). The levels of the Na+/K+/2Cl− co-transporter NKCC2 were not different between genotypes and not affected by dietary NaCl (Figure 8). The expression of the Na+/Cl− co-transporter NCC was significantly greater (~40%) in NHE3loxloxCre mice compared to Con mice on high NaCl diet (Figure 8). In both genotypes, low NaCl significantly increased NCC abundance compared to high NaCl, which was confirmed using qualitative immunofluorescence (Figure 9). However, there were no clear differences between genotypes in NCC localization or staining intensity. Phosphorylation of NCC at threonine 58 (pT58 NCC), which stimulates NCC transport activity, was significantly greater (~25%) in NHE3loxloxCre mice compared to Con mice irrespective of diet. In both genotypes, low NaCl significantly increased pT58 NCC abundance compared to high NaCl; however, this increase was significantly greater (~25%) in NHE3loxloxCre compared to Con mice (Figure 8). The expression of the α-subunit of the epithelial Na+ channel (αENaC) was significantly greater (~25%) in NHE3loxloxCre compared to Con mice irrespective of diet. Whereas dietary NaCl did not affect full length αENaC levels in either genotype (Figure 8), low NaCl increased the abundance of the proteolytically cleaved form of αENaC irrespective of genotype. Cleaved αENaC was significantly higher in NHE3loxloxCre compared to Con mice on low dietary NaCl (~30%, Figure 8). On high NaCl diet, αENaC is undetectable by immunoflourescence.25 After 10 days on low NaCl intake, although αENaC labeling was clearly observed, the predominant apical distribution was similar between genotypes (Supplementary Figure 8). On low NaCl intake, γENaC localized to the apical membrane domain of connecting tubule (CNT) cells in Con and NHE3loxloxCre mice (Figure 10). Further downstream in the cortical collecting duct (CCD)/outer medullary collecting duct (OMCD), γENaC was localized predominantly intracellular in principal cells (PC). On high NaCl intake, Con and NHE3loxloxCre mice showed intracellular γENaC staining in CNT cells and PC of the CCD and OMCD which was more pronounced compared to low NaCl, but no clear differences were observed between genotypes. Similar ENaC staining in response to altered dietary NaCl intake has been observed previously.25
Figure 7. Independent of dietary NaCl content NHE3loxloxCre mice have lower Na+/phosphate co-transporter (Npt2a) and severely reduced Npt2c abundance.

NHE3 protein is absent in the kidney of NHE3loxloxCre mice. In Con mice, NHE3 expression was unaffected in response to dietary NaCl. Phosphorylation of NHE3 at serine residue 552 was not affected by dietary NaCl in Con mice and, consistent with the absence of NHE3, barely detectable in NHE3loxloxCre mice. Npt2a was unaffected by dietary NaCl in either genotype; however, significantly lower in NHE3loxloxCre compared to Con mice. In Con mice, dietary NaCl has significant impact on Npt2c abundance. In contrast to Con mice, Npt2c is almost completely absent in NHE3loxloxCre mice and not regulated by dietary NaCl. The Na+/bicarbonate cotransporter (NBCe1) was not affected by dietary NaCl or genotype. ND = not detectable. #P < 0.05 versus Con same diet.
Figure 8. Independent of dietary NaCl NHE3loxloxCre mice show greater phosphorylation of Na+/Cl− cotransporter (NCC) at threonine 58 (pT58 NCC).

The thick ascending limb Na+/K+/2Cl− co-transporter (NKCC2) was not significantly affected by dietary NaCl or genotype. Both genotypes responded with an increased expression of total NCC in response to low NaCl intake. However, under high dietary NaCl, total NCC expression was significantly higher in NHE3loxloxCre mice compared to Con mice. Phosphorylation of pT58 NCC was increased in response to low NaCl compared to high NaCl diet in both genotypes; however, NHE3loxloxCre mice show stronger phosphorylation under both low and high NaCl diet. Full-length α-subunit of the epithelial Na+ channel (αENaC) was significantly higher in NHE3loxloxCre mice compared to Con mice under either dietary condition. Cleaved αENaC expression was not different between genotypes on a high NaCl diet; however, it was significantly higher in NHE3loxloxCre compared to Con mice on low NaCl diet. *P < 0.05 versus high NaCl same genotype, #P < 0.05 versus Con same diet.
Figure 9. Qualitative immunofluorescent labeling identifies comparable NCC localization in Con and NHE3loxloxCre mice in response to low and high NaCl intake.

Panels show representative images of kidney sections from mice after 10 days on low or high NaCl intake. Differential interference contrast overlay is used to show kidney structure and localization of imaging. Under low and high NaCl intake, NCC localized to the apical plasma membrane domain of distal convoluted tubule (DCT) cells in Con and NHE3loxloxCre mice (arrows). The staining intensity tended to be higher under low compared with high NaCl intake in Con (A, B, E, and F) and NHE3loxloxCre mice (C, D, G, and H). No clear differences were observed between genotypes. n = 8/genotype.
Figure 10. Qualitative immunofluorescent labeling identifies γENaC localization in Con and NHE3loxloxCre mice in response to low and high NaCl intake.

Panels show representative images of kidney sections from mice after 10 days on low or high NaCl intake. Differential interference contrast overlay is used to show kidney structure and localization of imaging. On low NaCl intake, γENaC localized to the apical membrane domain in some connecting tubule (CNT) cells (arrows, cells with strong calbindin and γENaC staining) in Con and NHE3loxloxCre mice. (E–H) In principal cells (PC, cells with no/weak calbindin staining and γENaC staining) of the cortical collecting duct (CCD)/outer medullary collecting duct (OMCD), γENaC was localized intracellularly (arrow heads). (I–P) On high NaCl intake, Con and NHE3loxloxCre mice showed intracellular γENaC staining in CNT cells and PC of the CCD and OMCD (arrow heads). Furthermore, Con and NHE3loxloxCre mice showed more intense intracellular γENaC staining on high NaCl intake compared to low NaCl intake in both CNT cells and PC of the CCD/OMCD. No clear difference was observed between genotypes. n = 8/genotype.
Levels of the Na+/K+-ATPase α-subunit was not affected by dietary NaCl or genotype (Figure 11). Levels of the Cl−/bicarbonate exchanger pendrin expressed in type B and non-A, non-B intercalated cells was significantly lower (~25%) in NHE3loxloxCre mice compared to Con mice, but was not affected by dietary NaCl irrespective of genotype (Figure 11). The levels of the H+-ATPase β1-subunit, located in type A, type B, and non-A, non-B intercalated cells, were significantly higher (~25%) in NHE3loxloxCre compared to Con mice, but not affected by dietary NaCl (Figure 11). The abundance of aquaporin-2 (AQP2) was slightly but significantly greater (~15%) on high compared to low NaCl in NHE3loxloxCre mice, whereas levels of AQP2 phosphorylated at serine 256 (pS256 AQP2) were not affected by genotype or dietary NaCl.
Figure 11. Lower abundance of the Cl−/bicarbonate exchanger (pendrin) and higher abundance of H+-ATPase β1-subunit expression in NHE3loxloxCre mice.

Expression of the Na+/K+-ATPase α-subunit was unaffected by dietary NaCl or genotype. In NHE3loxloxCre mice, pendrin levels were significantly lower compared to Con mice but unaffected by dietary NaCl intake. Vice versa, the H+-ATPase β1-subunit levels were significantly higher in NHE3loxloxCre compared to Con mice, but also unaffected by dietary NaCl. Aquaporin-2 (AQP2) expression was significantly greater in NHE3loxloxCre on high compared to low NaCl intake, an effect absent in Con mice. Expression of phosphorylated AQP2 at serine 256 (pS256 AQP2) was not affected by genotype or dietary NaCl. *P < 0.05 versus high NaCl same genotype, #P < 0.05 versus Con same diet.
Effect of dietary NaCl on systolic blood pressure and heart rate
The role of dietary NaCl on systolic blood pressure and heart rate in respect to genotype was measured via direct and continuous measurements by a fluid filled catheter implanted into the femoral artery under anesthesia. No significant differences in blood pressure were detected between Con mice after 10 days on low or high NaCl diet (Figure 12). In contrast, NHE3loxloxCre mice showed lower systolic blood pressure independent of NaCl. Of note, blood pressure in NHE3loxloxCre mice was NaCl-sensitive, with a 10 mmHg increase upon high NaCl diet, while heart rate was not affected by dietary NaCl or genotype.
Figure 12. Lower systolic blood pressure independent of dietary NaCl intake in NHE3loxloxCre mice.
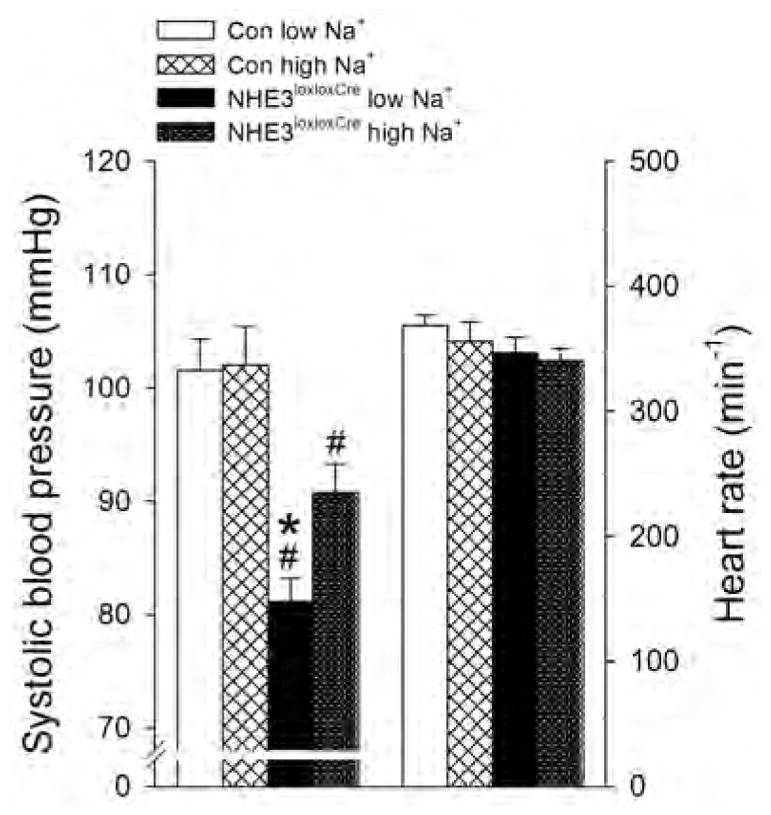
After 10 days on either low or high NaCl intake systolic blood pressure was not different in Con mice. In NHE3loxloxCre mice blood pressure was significantly lower regardless of NaCl intake compared to Con mice; however, systolic blood pressure was significantly lower on low compared to high NaCl. Heart rate was not affected by genotype or NaCl intake. n = 4–5/group, *P < 0.05 versus high NaCl same genotype, #P < 0.05 versus Con same diet.
DISCUSSION
NHE3 plays an important role in regulating Na+ absorption and blood pressure. Previous studies of NHE3 function after total body deletion in mice were hampered due to the severe phenotype observed under baseline conditions or when challenged by alterations in dietary NaCl. These defects were attributed to a critical role of NHE3 in the intestine and kidney but could so far not be clearly delineated. Lack of renal NHE3 in NHE3loxloxCre mice has no observable impact on intestinal NHE3 abundance or localization, providing us with an excellent model to assess if there is a critical role of NHE3 in the kidney tubules for NaCl homeostasis and maintaining blood pressure either under baseline conditions or during alterations in dietary NaCl. The results suggest that NHE3 is required to maintain steady state plasma Na+ levels when dietary NaCl is modified. Despite a concerted interplay of decreased GFR and increased activation of distal tubule Na+ conserving mechanisms via NCC and ENaC, plasma Na+ in NHE3loxloxCre mice is directly impacted by dietary NaCl. Furthermore, under baseline conditions NHE3loxloxCre mice have a higher urinary Na+/creatinine ratio, and a tendency for higher urinary Na+ excretion. However, our detailed balance studies indicate that the amount of Na+ wasting is relatively minor in the absence of renal NHE3. Our data from NHE3loxloxCre mice unraveled a contribution of NHE3 to urinary concentrating ability that was inversely correlated with Na+ intake. Of note, the compensatory mechanisms in NHE3loxloxCre mice are unable to restore blood pressure to levels found in Con mice.
Baseline analyses (current study and23) show no significant differences between Con and NHE3loxloxCre mice in body weight, plasma concentrations of Na+, K+, aldosterone as well as osmolality and blood pH. Consistent with a higher fluid intake and higher urinary flow rate, urine osmolality was significantly lower in NHE3loxloxCre mice indicating that NHE3loxloxCre mice can maintain fluid and electrolyte homeostasis. Eighteen hour WD unraveled the importance of NHE3 for urinary concentrating ability on low NaCl diet. Under these conditions, NHE3loxloxCre mice have a problem conserving fluid resulting in a lower urine osmolality and consequently a greater increase in plasma osmolality. This data likely indicates that NHE3 in the medullary TAL contributes to some extent towards establishing the medullary interstitial gradient. Differences in urea handling could also potentially contribute to the impaired urinary concentrating ability. Under control, but more importantly under low NaCl intake, K+ and urea are the dominating osmolytes. Alterations in their handling may partly account for differences in anti-diuresis and changes in plasma Na+ (Figure 5) of NHE3loxloxCre compared to Con mice. A mild urinary concentrating defect was also apparent on control NaCl diet, whereas on high NaCl no differences were observed. Of note, other compensatory mechanisms (see below) are unable to fully compensate for these defects. In contrast to global NHE3−/− mice, which have elevated plasma aldosterone levels,3 NHE3loxloxCre mice show plasma aldosterone levels comparable to Con mice. Dietary NaCl challenges did not result in different plasma aldosterone concentrations between genotypes, while phosphorylated NCC, αENaC and cleaved αENaC expression were significantly higher in NHE3loxloxCre versus Con mice. NCC phosphorylation can be mediated by AngII independent of aldosterone,26 vasopressin,27, 28 insulin29 and/or adrenergic factors.30 Vasopressin31 and insulin32 also contribute to ENaC regulation. Furthermore, proteolysis-mediated activation of αENaC is regulated by various proteases,33 the activity of which may be regulated. Regarding NCC, hypokalemia, which was more pronounced in NHE3loxloxCre mice, may also be important for increased NCC activity.34 The higher αENaC/cleaved αENaC expression in NHE3loxloxCre mice possibly provides the driving force for increased K+ secretion via the renal outer medullary K+ channel and/or big-conductance K+ channels,35 consequently resulting in a more prominent decrease in plasma K+ compared to Con mice. Ultimately, other signals that contribute to the increased NCC phosphorylation and activation of αENaC via proteolytic cleavage in NHE3loxloxCre mice, which may help to maintain Na+ homeostasis, remain to be determined.
In contrast to NHE3−/− mice, which show lethality when exposed to low/high NaCl diet,2 NHE3loxloxCre mice do not have a problem adapting to either NaCl diet and appear healthy. While NHE3 in the proximal tubule was postulated to be essential for Na+ reabsorption and Na+ balance, our data suggest otherwise. Dietary NaCl does not affect NHE3 or pS552 NHE3 expression in Con mice, which is comparable to other studies.36–39 Consequently, the adaptive mechanisms that occur in Con mice in response to changes in dietary NaCl should be exaggerated in NHE3loxloxCre mice in order to maintain plasma Na+ levels. Indeed, we found that NHE3loxloxCre mice have significantly increased pT58 NCC, αENaC and cleaved αENaC abundances following dietary NaCl restriction compared to Con mice; supporting that a larger magnitude of compensatory mechanisms are required in NHE3loxloxCre mice to maintain Na+ balance during NaCl restriction. Although our immunofluorescent labeling did not show clear differences in localization of αENaC, γENaC or NCC between the genotypes, we cannot rule out that subtle differences exist that we were not able to detect via immunofluorescence.
Consistent with other studies,36, 40 we do not show an effect of dietary NaCl on NKCC2 expression. Although NKCC2 levels tended to be lower in NHE3loxloxCre mice relative to Con mice following high dietary NaCl intake, these differences did not reach statistically significance. These results would be in line with NHE3−/− mice that have reduced NKCC2 expression.41, 42 Why NKCC2 expression in the NHE3loxloxCre and NHE3−/− mice are reduced rather than increased (a response considered anti-homeostatic) remains elusive. Of note, treatment of normal mice with acetazolamide for 3 days does not affect NKCC2 expression.42 These findings argue against an increased fluid and bicarbonate load to the TAL mediating this effect.
Intra-arterial blood pressure was significantly lower in NHE3loxloxCre mice and sensitive to dietary NaCl. While under low and high dietary NaCl intake, although no significant differences in urinary Na+ excretion were detectable between genotypes, we still observed differences in blood pressure. This possibly indicates that renal NHE3 could affect other proteins that do not primarily transport Na+ but could affect blood pressure regulation. Of note, the only electrolyte excreted in the urine at a greater amount under low dietary NaCl was Cl−. A contributing role of Cl− for determining NaCl-sensitivity of blood pressure has been postulated over 30 years ago.43 A lower blood pressure was described for NHE3−/− and tgNHE3−/− mice.3, 8 When replacing tap water with a 0.9% NaCl solution, blood pressure in tgNHE3−/− increased to the levels of WT mice,9 possibly due to increased consumption of 0.9% NaCl in the tgNHE3−/− mice compared to controls. However, similar to the observations here, blood pressure in tgNHE3−/− remained significantly lower compared to WT mice when feeding a 1% or 5% NaCl diet.8 Our experiments did not show an effect of dietary NaCl or genotype on heart rate, which may be linked to an abolished aortic depressor nerve stimulation that is common under barbiturate anesthesia.44 The current results warrant future studies to determine effects of dietary Na+ and/or Cl− on blood pressure and heart rate in conscious NHE3loxloxCre mice via radio-telemetry.
Surprisingly, a dietary-independent ~30% reduction in Npt2a abundance was observed in NHE3loxloxCre mice. This is in contrast to NHE3−/− mice that have ~350% greater Npt2a expression,41 which may be an adaptive response to defective phosphate absorption from the small intestine. Measured PTH levels in NHE3−/− mice were not significantly different from WT mice, suggesting that alterations in PTH are not the primary cause of increased Npt2a expression.45 NHE3 tethers to NHE regulatory factor 146 which also regulates Npt2a.47 It needs to be determined if, in the absence of NHE3, this physical tethering is disrupted and consequently affects Npt2a expression. Hypertension is associated with internalization of Npt2a;48, 49 however, NHE3loxloxCre mice have lower and not higher blood pressure, arguing against blood pressure causing decreased Npt2a expression. In line with the effects on Npt2a, levels of Npt2c are severely reduced in the NHE3loxloxCre mice. Furthermore, we observed that Npt2c expression was NaCl-sensitive in Con mice, with changes parallel to dietary NaCl, whereas NHE3loxloxCre mice were unresponsive. This suggests a novel link between Npt2c and NHE3, with NHE3 possibly required for the effects of dietary NaCl on Npt2c expression. While pressure natriuresis reduces the expression of Npt2, NHE3loxloxCre mice have lower and not higher blood pressure arguing against such a mechanism being responsible for the reduced Npt2a and absent Npt2c expression.
Our data also suggest that Npt2c, in addition to NHE3,5 H+-ATPase5 and AQP1,50 possibly contributes to functional glomerulotubular balance. Microperfusion experiments in proximal tubules employing bafilomycin, an H+-ATPase inhibitor, indicated that ~20% of bicarbonate reabsorption is mediated via H+-ATPase.4, 51 Our experiments indicate that in the absence of NHE3-dependent bicarbonate reabsorption (NHE3loxloxCre mice), an increased NHE3-independent component contributes to bicarbonate reabsorption, as evidenced by a higher, dietary-independent, H+-ATPase β1-subunit expression. In NHE3−/− mice, bicarbonate reabsorption in the collecting duct was found to be increased possibly via higher H+-ATPase expression.52 We previously reported a higher urinary pH in NHE3loxloxCre mice,23 confirmed in the current study under baseline conditions alongside a comparable blood pH. During dietary NaCl challenges, urinary pH remained significantly higher in NHE3loxloxCre mice paralleled by significantly higher H+-ATPase β1-subunit abundance. Type-B and non-A, non-B intercalated cells express pendrin,53 which is not altered by dietary NaCl in Con mice. However, independent of dietary NaCl, NHE3loxloxCre mice show a higher urinary pH alongside lower pendrin expression. The lower pendrin expression may be a mechanism to limit bicarbonate loss and maintain blood pH.
Conscious NHE3loxloxCre mice show a ~20% lower GFR, a finding consistent with NHE3−/− and tgNHE3−/− mice that have a 20–30% lower GFR compared to wild-type mice.8, 41 Low NaCl intake decreased GFR in both genotypes while high NaCl was without significant effects. Various labs have described no change in GFR in response to dietary NaCl restriction54, 55 or a decrease in GFR.56, 57 In response to high dietary NaCl, GFR was described to be unaffected58, 59 or increased.54, 60 Of note, a recent study found GFR to be NaCl-sensitive.61 All NHE3loxloxCre mice and Con mice in this study clearly respond to low NaCl intake with a reduction in GFR, possibly indicating intact resetting of the TGF response in order to restore efficiency.62 The reduced GFR in NHE3loxloxCre mice is possibly another mechanism that contributes to the maintenance of NaCl homeostasis. The lower GFR is unlikely due to structural changes in the kidney and rather a physiological response to changes in Na+/K+/Cl− delivery to the macula densa.
In summary, our data indicate that renal NHE3 is required to maintain steady state plasma Na+ levels when dietary NaCl intake is modified. We also provide new evidence that Npt2c is part of an adaptation response to dietary NaCl and Npt2c expression is modulated by functional renal NHE3. Our data provide novel evidence that increased NCC and ENaC abundances are part of a compensatory response in the absence of NHE3 along the tubule. Despite these adaptive responses, blood pressure remains significantly lower and is NaCl-sensitive in tubule-specific NHE3 knockout mice. The overall “mild” phenotype in response to NaCl challenges in tubule-specific NHE3 knockout mice indicates that the severe phenotype of NHE3−/− mice is most likely a result of NHE3 deletion in the intestine.
METHODS
Animals
All animal experimentation was conducted in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD) and was approved by the local Institutional Animal Care and Use Committee. The generation of tubule-specific, non-inducible, constitutive NHE3 knockout mice using Pax8-Cre transgenic mice was described previously.23 Mice were housed under a 12:12 hour light:dark cycle in standard rodent cages with free access to standard rodent chow (0.8% NaCl, TD.7001, Harlan Teklad, Madison, WI) and tap water. Age-matched, 3–6-month old male control (Con, NHE3loxlox) or tubule-specific NHE3 knockout mice (NHE3loxloxCre) were used for experiments.
Food and fluid intake, blood and urine collection/analysis
Food and fluid intake was determined while the mice were maintained in their regular cages.63, 64 Urine was obtained via reflex urination. Blood was collected from the retrobulbar plexus under brief isoflurane anesthesia.23 Metabolic cage experiments were performed as described previously.35, 65 Blood chemistry was determined by an OPTI® CCA blood gas analyzer (OPTIMedical, Roswell, GA). Concentrations of plasma aldosterone were determined by radioimmunoassay (Beckman Coulter, Brea, CA).35 Urinary pH was determined using a pH electrode (9810BN, Thermo Fisher Scientific, Carlsbad, CA). Urinary Na+ and creatinine were analyzed as described previously.63
Measurement of glomerular filtration rate (GFR) in conscious mice
GFR measurements were performed by determining the plasma kinetics of the GFR marker fluorescein isothiocyanate-sinistrin (Fresenius-Kabi, Linz, Austria) following a bolus i.v. injection.23, 64, 66
Low and high NaCl diet manipulations
GFR was assessed (3 days before dietary changes) under when mice were on standard diet (TD.7001). Fluid and food intake was determined over a 3 day period and averaged (day 0), a blood collection performed and mice subsequently randomized to either low NaCl diet (Harlan Teklad, <0.01% NaCl, TD.08601) or high NaCl diet (Harlan Teklad, 4% NaCl, TD.140038).54 Apart from NaCl, diets were otherwise identical in composition to the standard diet. Food and fluid intake was determined daily (and averaged for days 3–4, 5–6, 7–8 and 9–10) while the mice were maintained in their regular cages. After 10 days on the new diet, GFR measurements were performed as described above. One day later mice were anesthetized for blood collection and kidneys removed and processed for Western blotting.
Immunoblot analysis and immunohistochemical analysis
Western blotting was performed as described previously.27, 63, 64 Blots were probed with the following antibodies: total NHE367, pS552 NHE3 (Novus Biologicals, Littleton, CO), total NCC68, pT58 NCC28, NKCC269, NBCe123, Npt2a63, Npt2c63, Na+/K+-ATPase α-subunit (Merck Millipore, Darmstadt, Germany), H+-ATPase β1-subunit70, Pendrin,71 αENaC72, AQP273 and pS256-AQP274. Chemiluminescent detection and densitrometric analyses were performed using Image Studio Lite® (Qiagen, Valencia, CA). For immunohistochemistry tissue preparation, kidney and intestinal sectioning, and labeling were performed as previously described27, 64 utilizing antibodies against collagen IV (ICN Biochemicals, Inc., Auroa, Ohio, USA) αENaC72, γENaC75 NCC (SPC402D, StressMarq Biosciences, Cadboro Bay, Canada) and NHE3.67
Intra-arterial blood pressure and heart rate measurements
Mice received low or high NaCl diet for 10 days. Subsequently mice were anesthetized for terminal blood pressure measurements as described previously.63, 76
Statistical analysis
Data are expressed as mean±S.E.M. Unpaired and paired Student’s t-test or Mann-Whitney U-test were performed as appropriate to analyze for statistical differences between groups. One-way ANOVA or two-way repeated-measures ANOVA followed by Student-Newman-Keuls and Tukey’s multiple comparison tests, respectively, were used to test for significant differences between genotype and experimental conditions. Significance was considered at P < 0.05.
Supplementary Material
Acknowledgments
FUNDING
This work was supported by an APS STRIDE Undergraduate Summer Research Fellowship 1R25HL115473-01 (to S.d.l.M.C), a Bastyr University Faculty Research Seed Grant (to J.D.R.), National Institute of Diabetes and Digestive and Kidney Diseases Grant 1R01DK110621-01 (T.R), the O’Brien Center for Acute Kidney Injury Research Grant P30DK079337 (to T.R.), Diabetes Endocrinology Research Center P30DK063491 (to T.R.), American Heart Association 15BGIA22410018 (to T.R.), Satellite Healthcare (a not-for-profit renal care provider) (to T.R.), Department of Veterans Affairs Merit Review Award (to M.S.), and Center on Genetics of Transport and Epithelial Biology at University of Cincinnati. Funding (to R.A.F) was provided by the Lundbeck Foundation, the Danish Medical Research Council, and the Novo Nordisk Foundation.
Footnotes
DISCLOSURE
The authors declare no competing interests.
AUTHORSHIP
R.A.F. and T.R. made the conception and design of the work, all animal experiments were performed in the lab of T.R.; R.A.F., S.C., M.S., S.B.P., J.D.R. and T.R. contributed to the acquisition, analysis, or interpretation of data for the work. R.A.F., S.C., M.S., S.B.P., J.D.R. and T.R. drafted the work or revised it critically for important intellectual content. All authors approved the final version of the manuscript, agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gawenis LR, Stien X, Shull GE, et al. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2002;282:G776–784. doi: 10.1152/ajpgi.00297.2001. [DOI] [PubMed] [Google Scholar]
- 2.Ledoussal C, Lorenz JN, Nieman ML, et al. Renal salt wasting in mice lacking NHE3 Na+/H+ exchanger but not in mice lacking NHE2. American journal of physiology Renal physiology. 2001;281:F718–727. doi: 10.1152/ajprenal.2001.281.4.F718. [DOI] [PubMed] [Google Scholar]
- 3.Schultheis PJ, Clarke LL, Meneton P, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nature genetics. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 4.Wang T, Yang CL, Abbiati T, et al. Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. The American journal of physiology. 1999;277:F298–302. doi: 10.1152/ajprenal.1999.277.2.F298. [DOI] [PubMed] [Google Scholar]
- 5.Du Z, Yan Q, Duan Y, et al. Axial flow modulates proximal tubule NHE3 and H-ATPase activities by changing microvillus bending moments. American journal of physiology Renal physiology. 2006;290:F289–296. doi: 10.1152/ajprenal.00255.2005. [DOI] [PubMed] [Google Scholar]
- 6.Vallon V, Schwark JR, Richter K, et al. Role of Na(+)/H(+) exchanger NHE3 in nephron function: micropuncture studies with S3226, an inhibitor of NHE3. American journal of physiology Renal physiology. 2000;278:F375–379. doi: 10.1152/ajprenal.2000.278.3.F375. [DOI] [PubMed] [Google Scholar]
- 7.Bradford EM, Sartor MA, Gawenis LR, et al. Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G886–898. doi: 10.1152/ajpgi.90520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo AL, Noonan WT, Schultheis PJ, et al. Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. American journal of physiology Renal physiology. 2003;284:F1190–1198. doi: 10.1152/ajprenal.00418.2002. [DOI] [PubMed] [Google Scholar]
- 9.Noonan WT, Woo AL, Nieman ML, et al. Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. American journal of physiology Regulatory, integrative and comparative physiology. 2005;288:R685–691. doi: 10.1152/ajpregu.00209.2004. [DOI] [PubMed] [Google Scholar]
- 10.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. American journal of physiology Regulatory, integrative and comparative physiology. 2010;298:R851–861. doi: 10.1152/ajpregu.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas D, Harris PJ, Morgan TO. Age-related changes in angiotensin II-stimulated proximal tubule fluid reabsorption in the spontaneously hypertensive rat. J Hypertens Suppl. 1988;6:S449–451. doi: 10.1097/00004872-198812040-00141. [DOI] [PubMed] [Google Scholar]
- 12.LaPointe MS, Sodhi C, Sahai A, et al. Na+/H+ exchange activity and NHE-3 expression in renal tubules from the spontaneously hypertensive rat. Kidney international. 2002;62:157–165. doi: 10.1046/j.1523-1755.2002.00406.x. [DOI] [PubMed] [Google Scholar]
- 13.McDonough AA, Leong PK, Yang LE. Mechanisms of pressure natriuresis: how blood pressure regulates renal sodium transport. Annals of the New York Academy of Sciences. 2003;986:669–677. doi: 10.1111/j.1749-6632.2003.tb07281.x. [DOI] [PubMed] [Google Scholar]
- 14.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, et al. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. American journal of physiology Renal physiology. 2010;298:F177–186. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leong PK, Yang LE, Holstein-Rathlou NH, et al. Angiotensin II clamp prevents the second step in renal apical NHE3 internalization during acute hypertension. American journal of physiology Renal physiology. 2002;283:F1142–1150. doi: 10.1152/ajprenal.00178.2002. [DOI] [PubMed] [Google Scholar]
- 16.Banday AA, Lokhandwala MF. Angiotensin II-mediated biphasic regulation of proximal tubular Na+/H+ exchanger 3 is impaired during oxidative stress. American journal of physiology Renal physiology. 2011;301:F364–370. doi: 10.1152/ajprenal.00121.2011. [DOI] [PubMed] [Google Scholar]
- 17.Reilly AM, Harris PJ, Williams DA. Biphasic effect of angiotensin II on intracellular sodium concentration in rat proximal tubules. The American journal of physiology. 1995;269:F374–380. doi: 10.1152/ajprenal.1995.269.3.F374. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen MT, Han J, Ralph DL, et al. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep. 2015:3. doi: 10.14814/phy2.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leong PK, Devillez A, Sandberg MB, et al. Effects of ACE inhibition on proximal tubule sodium transport. American journal of physiology Renal physiology. 2006;290:F854–863. doi: 10.1152/ajprenal.00353.2005. [DOI] [PubMed] [Google Scholar]
- 20.Li XC, Shull GE, Miguel-Qin E, et al. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiological genomics. 2015;47:479–487. doi: 10.1152/physiolgenomics.00056.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li XC, Shull GE, Miguel-Qin E, et al. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol Rep. 2015:3. doi: 10.14814/phy2.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Sagnella GA, Dong Y, et al. Molecular variants of the sodium/hydrogen exchanger type 3 gene and essential hypertension. J Hypertens. 2004;22:1269–1275. doi: 10.1097/01.hjh.0000125428.28861.11. [DOI] [PubMed] [Google Scholar]
- 23.Fenton RA, Poulsen SB, de la Mora Chavez S, et al. Caffeine-induced diuresis and natriuresis is independent of renal tubular NHE3. American journal of physiology Renal physiology. 2015;308:F1409–1420. doi: 10.1152/ajprenal.00129.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aronson PS, Giebisch G. Effects of pH on potassium: new explanations for old observations. Journal of the American Society of Nephrology : JASN. 2011;22:1981–1989. doi: 10.1681/ASN.2011040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loffing J, Pietri L, Aregger F, et al. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. American journal of physiology Renal physiology. 2000;279:F252–258. doi: 10.1152/ajprenal.2000.279.2.F252. [DOI] [PubMed] [Google Scholar]
- 26.van der Lubbe N, Lim CH, Fenton RA, et al. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney international. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 27.Rieg T, Tang T, Uchida S, et al. Adenylyl cyclase 6 enhances NKCC2 expression and mediates vasopressin-induced phosphorylation of NKCC2 and NCC. The American journal of pathology. 2013;182:96–106. doi: 10.1016/j.ajpath.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedersen NB, Hofmeister MV, Rosenbaek LL, et al. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney international. 2010;78:160–169. doi: 10.1038/ki.2010.130. [DOI] [PubMed] [Google Scholar]
- 29.Chavez-Canales M, Arroyo JP, Ko B, et al. Insulin increases the functional activity of the renal NaCl cotransporter. J Hypertens. 2013;31:303–311. doi: 10.1097/HJH.0b013e32835bbb83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terker AS, Yang CL, McCormick JA, et al. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension. 2014;64:178–184. doi: 10.1161/HYPERTENSIONAHA.114.03335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ecelbarger CA, Kim GH, Terris J, et al. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. American journal of physiology Renal physiology. 2000;279:F46–53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- 32.Pavlov TS, Ilatovskaya DV, Levchenko V, et al. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:2723–2732. doi: 10.1096/fj.12-223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. The Journal of biological chemistry. 2009;284:20447–20451. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, et al. Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. American journal of physiology Renal physiology. 2014;306:F1507–1519. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rieg T, Vallon V, Sausbier M, et al. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney international. 2007;72:566–573. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 36.Masilamani S, Wang X, Kim GH, et al. Time course of renal Na-K-ATPase, NHE3, NKCC2, NCC, and ENaC abundance changes with dietary NaCl restriction. American journal of physiology Renal physiology. 2002;283:F648–657. doi: 10.1152/ajprenal.00016.2002. [DOI] [PubMed] [Google Scholar]
- 37.Song J, Hu X, Shi M, et al. Effects of dietary fat, NaCl, and fructose on renal sodium and water transporter abundances and systemic blood pressure. American journal of physiology Renal physiology. 2004;287:F1204–1212. doi: 10.1152/ajprenal.00063.2004. [DOI] [PubMed] [Google Scholar]
- 38.Eladari D, Leviel F, Pezy F, et al. Rat proximal NHE3 adapts to chronic acid-base disorders but not to chronic changes in dietary NaCl intake. American journal of physiology Renal physiology. 2002;282:F835–843. doi: 10.1152/ajprenal.00188.2001. [DOI] [PubMed] [Google Scholar]
- 39.Yang LE, Sandberg MB, Can AD, et al. Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. American journal of physiology Renal physiology. 2008;295:F1003–1016. doi: 10.1152/ajprenal.90235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schiessl IM, Rosenauer A, Kattler V, et al. Dietary salt intake modulates differential splicing of the Na-K-2Cl cotransporter NKCC2. American journal of physiology Renal physiology. 2013;305:F1139–1148. doi: 10.1152/ajprenal.00259.2013. [DOI] [PubMed] [Google Scholar]
- 41.Brooks HL, Sorensen AM, Terris J, et al. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. The Journal of physiology. 2001;530:359–366. doi: 10.1111/j.1469-7793.2001.0359k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amlal H, Ledoussal C, Sheriff S, et al. Downregulation of renal AQP2 water channel and NKCC2 in mice lacking the apical Na+-H+ exchanger NHE3. The Journal of physiology. 2003;553:511–522. doi: 10.1113/jphysiol.2003.053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitescarver SA, Ott CE, Jackson BA, et al. Salt-sensitive hypertension: contribution of chloride. Science. 1984;223:1430–1432. doi: 10.1126/science.6322303. [DOI] [PubMed] [Google Scholar]
- 44.Ma X, Abboud FM, Chapleau MW. Analysis of afferent, central, and efferent components of the baroreceptor reflex in mice. American journal of physiology Regulatory, integrative and comparative physiology. 2002;283:R1033–1040. doi: 10.1152/ajpregu.00768.2001. [DOI] [PubMed] [Google Scholar]
- 45.Pan W, Borovac J, Spicer Z, et al. The epithelial sodium/proton exchanger, NHE3, is necessary for renal and intestinal calcium (re)absorption. American journal of physiology Renal physiology. 2012;302:F943–956. doi: 10.1152/ajprenal.00504.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weinman EJ, Steplock D, Shenolikar S. NHERF-1 uniquely transduces the cAMP signals that inhibit sodium-hydrogen exchange in mouse renal apical membranes. FEBS letters. 2003;536:141–144. doi: 10.1016/s0014-5793(03)00043-7. [DOI] [PubMed] [Google Scholar]
- 47.Giral H, Lanzano L, Caldas Y, et al. Role of PDZK1 protein in apical membrane expression of renal sodium-coupled phosphate transporters. The Journal of biological chemistry. 2011;286:15032–15042. doi: 10.1074/jbc.M110.199752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang LE, Leong PK, McDonough AA. Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. American journal of physiology Renal physiology. 2007;293:F1197–1208. doi: 10.1152/ajprenal.00040.2007. [DOI] [PubMed] [Google Scholar]
- 49.Magyar CE, Zhang Y, Holstein-Rathlou NH, et al. Proximal tubule Na transporter responses are the same during acute and chronic hypertension. American journal of physiology Renal physiology. 2000;279:F358–369. doi: 10.1152/ajprenal.2000.279.2.F358. [DOI] [PubMed] [Google Scholar]
- 50.Pohl M, Shan Q, Petsch T, et al. Short-term functional adaptation of aquaporin-1 surface expression in the proximal tubule, a component of glomerulotubular balance. Journal of the American Society of Nephrology : JASN. 2015;26:1269–1278. doi: 10.1681/ASN.2014020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ulate G, Fernandez R, Malnic G. Effect of bafilomycin on proximal bicarbonate absorption in the rat. Braz J Med Biol Res. 1993;26:773–777. [PubMed] [Google Scholar]
- 52.Nakamura S, Amlal H, Schultheis PJ, et al. HCO-3 reabsorption in renal collecting duct of NHE-3-deficient mouse: a compensatory response. The American journal of physiology. 1999;276:F914–921. doi: 10.1152/ajprenal.1999.276.6.F914. [DOI] [PubMed] [Google Scholar]
- 53.Soleimani M. The multiple roles of pendrin in the kidney. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2015;30:1257–1266. doi: 10.1093/ndt/gfu307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallon V, Schroth J, Satriano J, et al. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiology. 2009;111:p30–38. doi: 10.1159/000208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tucker BJ, Blantz RC. Mechanism of altered glomerular hemodynamics during chronic sodium depletion. The American journal of physiology. 1983;244:F11–18. doi: 10.1152/ajprenal.1983.244.1.F11. [DOI] [PubMed] [Google Scholar]
- 56.Wiggins WS, Manry CH, Lyons RH, et al. The effect of salt loading and salt depletion on renal function and electrolyte excretion in man. Circulation. 1951;3:275–281. doi: 10.1161/01.cir.3.2.275. [DOI] [PubMed] [Google Scholar]
- 57.Shalmi M, Petersen JS, Thomsen K, et al. Distal lithium reabsorption in the sodium-restricted rat is not dependent on the urinary sodium to lithium concentration ratio. Clin Sci (Lond) 1990;79:109–112. doi: 10.1042/cs0790109. [DOI] [PubMed] [Google Scholar]
- 58.Molstrom S, Larsen NH, Simonsen JA, et al. Normotensive sodium loading in normal man: regulation of renin secretion during beta-receptor blockade. American journal of physiology Regulatory, integrative and comparative physiology. 2009;296:R436–445. doi: 10.1152/ajpregu.90754.2008. [DOI] [PubMed] [Google Scholar]
- 59.Schor N, Ichikawa I, Brenner BM. Glomerular adaptations to chronic dietary salt restriction or excess. The American journal of physiology. 1980;238:F428–436. doi: 10.1152/ajprenal.1980.238.5.F428. [DOI] [PubMed] [Google Scholar]
- 60.Haberle DA, Davis JM. Chronic salt loading: effects on plasma volume and regulation of glomerular filtration rate in Wistar rats. Klin Wochenschr. 1982;60:1245–1248. doi: 10.1007/BF01716731. [DOI] [PubMed] [Google Scholar]
- 61.Isaksson GL, Stubbe J, Lyngs Hansen P, et al. Salt sensitivity of renin secretion, glomerular filtration rate and blood pressure in conscious Sprague-Dawley rats. Acta Physiol (Oxf) 2014;210:446–454. doi: 10.1111/apha.12191. [DOI] [PubMed] [Google Scholar]
- 62.Thomson SC, Vallon V, Blantz RC. Resetting protects efficiency of tubuloglomerular feedback. Kidney Int Suppl. 1998;67:S65–70. doi: 10.1046/j.1523-1755.1998.06713.x. [DOI] [PubMed] [Google Scholar]
- 63.Fenton RA, Murray F, Dominguez Rieg JA, et al. Renal phosphate wasting in the absence of adenylyl cyclase 6. Journal of the American Society of Nephrology : JASN. 2014;25:2822–2834. doi: 10.1681/ASN.2013101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rieg T, Tang T, Murray F, et al. Adenylate cyclase 6 determines cAMP formation and aquaporin-2 phosphorylation and trafficking in inner medulla. Journal of the American Society of Nephrology : JASN. 2010;21:2059–2068. doi: 10.1681/ASN.2010040409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rieg T, Bundey RA, Chen Y, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 66.Rieg T. A High-throughput method for measurement of glomerular filtration rate in conscious mice. Journal of visualized experiments : JoVE. 2013:e50330. doi: 10.3791/50330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim GH, Ecelbarger C, Knepper MA, et al. Regulation of thick ascending limb ion transporter abundance in response to altered acid/base intake. Journal of the American Society of Nephrology : JASN. 1999;10:935–942. doi: 10.1681/ASN.V105935. [DOI] [PubMed] [Google Scholar]
- 68.Kortenoeven ML, Pedersen NB, Miller RL, et al. Genetic ablation of aquaporin-2 in the mouse connecting tubules results in defective renal water handling. The Journal of physiology. 2013;591:2205–2219. doi: 10.1113/jphysiol.2012.250852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim GH, Ecelbarger CA, Mitchell C, et al. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle’s loop. The American journal of physiology. 1999;276:F96–F103. doi: 10.1152/ajprenal.1999.276.1.F96. [DOI] [PubMed] [Google Scholar]
- 70.Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. American journal of physiology Renal physiology. 2006;291:F39–48. doi: 10.1152/ajprenal.00383.2005. [DOI] [PubMed] [Google Scholar]
- 71.Kim YH, Kwon TH, Frische S, et al. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. American journal of physiology Renal physiology. 2002;283:F744–754. doi: 10.1152/ajprenal.00037.2002. [DOI] [PubMed] [Google Scholar]
- 72.Sorensen MV, Grossmann S, Roesinger M, et al. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney international. 2013;83:811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 73.Moeller HB, Aroankins TS, Slengerik-Hansen J, et al. Phosphorylation and ubiquitylation are opposing processes that regulate endocytosis of the water channel aquaporin-2. J Cell Sci. 2014;127:3174–3183. doi: 10.1242/jcs.150680. [DOI] [PubMed] [Google Scholar]
- 74.Hoffert JD, Fenton RA, Moeller HB, et al. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. The Journal of biological chemistry. 2008;283:24617–24627. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Masilamani S, Kim G-H, Mitchell C, et al. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. The Journal of clinical investigation. 1999;104:R19–R23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dominguez Rieg JA, Burt JM, Ruth P, et al. P2Y(2) receptor activation decreases blood pressure via intermediate conductance potassium channels and connexin 37. Acta Physiol (Oxf) 2015;213:628–641. doi: 10.1111/apha.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.