

Abstract
Podocytes are critical components of the nephron filtration barrier and are depleted in many kidney injuries and disease states. Terminally differentiated adult podocytes are highly specialized, post-mitotic cells, raising the question of whether the body has any ability to regenerate lost podocytes. Here, we review recent progress on this topic of significant interest and debate. The innovation of genetic labeling techniques enables fate tracing of individual podocytes, providing the strongest evidence yet that podocytes can be replaced by nearby progenitor cells. In particular, two progenitor pools have recently been identified in multiple studies: parietal epithelial cells (PECs), and cells of renin lineage (CoRL). These studies furthermore suggest that podocyte regeneration can be enhanced using ex vivo or pharmacological interventions. Based on these findings, we propose a set of criteria for evaluating podocyte regeneration, and suggest that restoration of podocyte number to a sub-sclerotic threshold be targeted as a potentially achievable clinical goal.
Keywords: glomerulus, glomerulosclerosis, progenitor, stem cell, renin cells, parietal epithelial cell
Introduction – Podocyte regeneration in the adult kidney
This review is intended to provide a balanced view on the broad topic of adult podocyte regeneration. Podocyte differentiation and regeneration during kidney development is beyond the scope of this review, and is well discussed by others [1, 2]. However for context, it is important to point out that nephron progenitor cells (NPCs), which give rise to all segments of the proximal nephron during kidney embryogenesis, cease to exist in mammalian kidneys shortly after birth [3, 4]. NPCs are also known as metanephric mesenchyme cells, and differentiate into distal and proximal tubular epithelia as well as podocytes. They express SIX2 and CITED1 and self-renew only transiently during kidney development, after which the pool of NPCs is completely exhausted [3, 4]. Thus, the embryonic progenitor cell that gives rise to the podocyte during development does not persist into adulthood in mammalian kidneys. Podocyte regeneration after organogenesis, if it occurs, must therefore originate from a different pool of cells.
We will use the dictionary definition of regeneration (biology), which is “the ability to recreate lost or damaged tissues, organs and limbs”. Applying this definition, we immediately note that a setting/scenario needs to first be established in which it is necessary and even required for adult podocytes to be regenerated. We would argue therefore in states where the kidney and/or adult podocytes are damaged, that a simple requirement for podocyte regeneration is an actual decrease in the number of podocytes. This typically occurs in several glomerular diseases, where the normal complement of podocytes has been reduced following injury. Stated differently, we do not expect podocyte regeneration in diseases where podocyte number has not been reduced. That said, the threshold at which regeneration might occur has not been proven. What is meant by ‘podocyte number’ will be discussed in the next section.
Absolute and relative podocyte number
This principle segues to the notion of absolute podocyte number versus relative podocyte number [5–7]. The latter, also known as podocyte density, is simply a measure of absolute podocyte number divided by the glomerular volume it occupies. However, the denominator glomerular volume, is influenced by age, nephron number, obesity and other factors, and in doing so, directly impacts podocyte density [8]. Podocyte density reflects the relative area of the underlying glomerular capillary that is covered by individual podocytes. Thus, either a decrease in absolute podocyte number (with no change in glomerular volume) and/or an increase in glomerular volume (with no change in absolute podocyte number) decreases podocyte density. Regardless, a decrease in podocyte number or density below a certain threshold has clearly been shown to underlie the onset of proteinuria, and the development of glomerulosclerosis.[9] Moreover, the lower the podocyte number/density, the more severe the glomerular scarring [9, 10]. For these reasons, the overall regulation of podocyte number in the adult kidney is of critical concern, and will be discussed in detail below.
Absolute and relative podocyte number has been studied in both man and rodents. Although numbers vary somewhat due to methodology, the mean number of podocytes per human glomerulus is 558 (range 263–983), and in mice, 72 (range 70–80) [11]. Recent human studies have shown that absolute podocyte number increases during the adolescent period by up to 20% [12]. In contrast, normal kidney aging is accompanied by reduced podocyte number in man[11] and rodents [13]. In contrast to absolute podocyte number, however, podocyte density is markedly decreased (>4 fold) in large glomeruli in adult humans, compared to children [12]. This illustrates the importance of considering both absolute podocyte number and podocyte density in the development of glomerular scarring. However, in the context of podocyte replacement and regeneration, we will argue that absolute podocyte number is the critical determinant.
Absolute podocyte number depletion
In glomerular diseases where podocytes are the initial cell injured, such as occurs in FSGS, or secondarily injured, such as occurs following mesangial cell injury in IgA nephropathy, studies have shown that several mechanisms can lead to podocyte depletion including apoptosis, detachment from the glomerular basement membrane (GBM) to which they are normally attached (for instance, due to weaknesses of the GBM in Alport’s syndrome), necrosis, DNA damage, and destabilization of the podocyte cytoskeleton (for instance, in α-actinin-4 mutants) [14–16]. Indeed, viable podocytes can be detected in the urine and can be cultured ex vivo [17, 18]. Because glomerular volume is unchanged (at least initially), absolute podocyte number decreases following the losses described above.
It is estimated that patients with kidney disease slough podocytes into their urine at > 10 cells/mg creatinine, including viable cells [17]. For an individual producing 1500 mg creatinine/day, this would add up to 15,000 cells/day in a disease state or approximately 300 glomeruli, which would mean that after ~ 20 years all podocytes would be gone from the body. Clinical progression is in many cases much slower and it has been observed that “these numbers are too high to be compatible with renal survival for 80 years, suggesting the existence of a regenerative mechanism” [19].
One might ask, why not simply replace lost podocytes through proliferation of the remaining cells? Studies have shown that in contrast to most other renal and non-renal epithelial cells, the adult terminally differentiated podocyte is unable to proliferate, thereby preventing adult podocytes from undergoing self-renewal in the face of ongoing losses [20]. Several mechanisms underlying the inability of adult podocytes to proliferate have been reported, including an increase in expression of cell cycle inhibitors (such as the cyclin kinase inhibitors p27 [21, 22] and p57 [22, 23]), DNA damage resulting in cell cycle arrest [24], mitotic catastrophe, and additional problems in mitosis with M-type cyclins [20]. Taken together, in states of acute or chronic podocyte loss such as occurs in several glomerular diseases, remaining adult podocytes cannot be relied on to replace those lost. Thus, loss without gain leads to adult podocyte depletion. This raises a challenge - to replace adult podocytes, other cell types such as stem/progenitor cells need to fulfill this biological role.
Relative podocyte number depletion
If the glomerular volume increases without any change in absolute podocyte number, podocyte density will decrease [25]. This occurs following a marked reduction in kidney mass such as nephrectomy, in the transplanted kidney, in aging, obesity and certain glomerular diseases following the scarring of injured glomeruli [8]. The increased glomerular volume leads to a mismatch in coverage by podocytes of underlying glomerular capillaries [8]. This is initially compensated for by hypertrophy of podocytes (i.e. an increase in their size without an increase in their number), that over time, becomes paradoxically maladaptive [8]. Under these circumstances, podocyte density is reduced, but podocyte number does not change. Whether this is sufficient to trigger podocyte regeneration is not yet clear and is an important question for further research. In the meantime, we propose that a decrease in absolute podocyte number be considered as a criterion for podocyte regeneration, as the loss of cells would more likely necessitate replacement than changes in cell density.
Evidence that adult podocytes can be partially or fully replaced in disease states
Several lines of evidence support the notion that adult podocytes can be replaced following their depletion. These include the following:
An increase in podocyte number in disease using cell specific markers: In studies using non-genetic cell fate mapping, the identification of podocytes relied on several markers such as WT-1 [26], p57 [23] and others [27]. Using these techniques, several studies have shown in experimental models of diabetic and non-diabetic kidney disease accompanied by reduced podocyte number, that podocyte number could be raised/increased when animals were given ACE-I, ARBs, steroids, retinoids or normalization of their diabetic state [28–34]. In all these studies, the increase in podocyte number occurred in the absence of podocyte proliferation. The authors of this review recognize that changes in podocyte-specific proteins can result secondary to injury, and thus the specificity of these changes need to considered when interpreting these results.
An increase in podocyte number in disease using genetic labeling techniques: In an era of increasing molecular sophistication, researchers are taking advantage of inducible conditional reporter labeling of podocytes for lineage tracing. For example, the mT/mG mouse crossed with a podocin-rtTA mouse permanently labels all cells in the body with a red color due to the tomato red reporter, including podocytes. However, importantly, when Cre is turned on by activating the podocin promoter with doxycycline, all podocytes become green in color, while other cells remain red [35, 36]. Using this type of experimental approach, two important studies strongly support the notion that podocytes can be replaced, albeit not back to the normal quota. Wanner and Huber used mT/mG mice crossed with inducible diptheria toxin receptor (iDTR) mice to partially deplete podocytes with the diphtheria toxin (DT) [37]. Podocytes were labeled green and expressed DTR following doxycycline administration; when DT was subsequently administered, there was a decrease in green cells i.e. podocytes. When followed over 4 weeks, red cells appeared on the glomerular tuft. An innovative FACS system was used to sort these cells, and showed that a new subpopulation of red cells on the tuft expressed podocyte proteins. The authors of the manuscript concluded that overall their results suggest a podocyte renewal of 38% of ablated cells, suggesting a distinct regenerative capacity of podocytes after acute loss.
Using the same mT/mG mice, Lasagni and Romagnani depleted podocytes with the podocyte toxin Adriamycin [38]. When followed over time, the percentage of newly generated podocytes was 6% when all podocytes were taken in to account. However, in mice that had remission of their proteinuria, regeneration of 30.4% of podocytes was observed. Using a conditional paired box gene 2 (PAX2) – confetti mouse, they labeled PAX2 cells in adult kidneys and induced Adriamycin nephropathy. Animals that underwent remission from proteinuria showed 0.4 PAX2-derived cells per section of glomerular tuft expressing podocyte proteins with ultrastructural features of podocytes (assessed by confocal microscopy), accounting for ~ 25% replacement of depleted podocytes. This translated to a ten-fold higher number of labeled parietal epithelial cells (PECs) that took on a podocyte phenotype in mice with regression of proteinuria compared to those that did not and 15% of glomeruli contained two or more PECs on the tuft. Overall, podocyte regeneration was further increased to 32.6% when the GSK-inhibitor BIO was administered [38].
Our group used an inducible podocin rtTA reporter, which permanently labels podocytes with ZsGreen only when mice are given doxycycline [39]. Following a doxycycline washout period, the total complement of podocytes, measured by the podocyte markers p57, podocin, and synaptopodin along with the podocyte reporter ZsGreen, was abruptly depleted by 30–40% with a cytopathic anti-podocyte antibody. This coincided with similar decreases in podocyte labeling. To prove that the decrease in p57, podocin, synaptopodin and ZsGreen were due to actual depletion of cells rather than reporter washout or decreased podocyte protein staining, DAPI staining was used to account for cell nuclei. Indeed, as expected, DAPI staining also decreased following the administration of the anti-podocyte antibody, consistent with cell depletion. Importantly, following this nadir in podocytes, there was a subsequent increase in the number of cells staining for p57, podocin and synaptopodin as well as DAPI staining. However, this was in the absences of the podocyte reporter ZsGreen. These results are consistent with the partial replacement of podocytes from a non-podocyte source. Repeated BrdU, administered to measure cumulative proliferation, was not detected in podocytes.
Finally, using live animal imaging, Hackl and Peti-Peterdi also showed that podocytes can be replaced in experimental models of glomerular disease [40]. Serial multiphoton microscopy on the same injured glomerulus revealed the appearance of a new podocyte (labeled with a fluorescent reporter) within 24 hours of the previous image, providing direct evidence for podocyte regeneration. Taken together, these results are consistent with the partial replacement of adult podocytes following depletion, and that the source had to be of non-podocyte origin given the absence of proliferation.
In summary, the studies above strongly support the notion that following a decrease in absolute podocyte number in experimental mouse models, podocytes can be at least partially replaced. Moreover, podocyte replacement can be further augmented by several therapeutics. The absence of podocyte proliferation is consistent with a non-podocyte source(s) for their replacement i.e. a podocyte stem/progenitor pools.
Adult Podocyte Progenitors
Based on the preceding literature, we recommend that the following criteria be considered as evidence that a stem/progenitor cell can transdifferentiate to an adult podocyte fate:
migration from their original location to a new location along the GBM in the glomerular tuft
de novo expression of several podocyte proteins, coincident with loss of their original cell specific proteins
expression of epithelial cell markers
acquisition of classic podocyte ultrastructural features (such as foot process, slit diaphragms)
displaying functionality (such as the production of VEGF and specific extracellular matrix proteins, angiotensin II responsiveness, limiting albumin permeability)
We also recommend a context for the need for podocytes to be replaced and for their progenitors to therefore be called upon i.e. a decrease in absolute podocyte number. Finally, it is our opinion that, like other cell types, adult podocyte stem/progenitors do not necessarily need to undergo intermediary steps, nor are they required to express certain stemness markers during their transdifferentiation to a podocyte fate.
Having argued that adult podocytes can be replaced either partially or fully by stem/progenitor cells, and that this number can be significantly increased under certain conditions such as RAAS blockade [33] and GSK3 inhibition [38], important questions arise including: what are the sources for podocyte replacement, what is the evidence in support of this, and is replacement the cumulative response from more than one source in a podocyte depleted glomerulus? Although it is tempting to speculate on opportunities for podocyte replacement from organoids [41, 42] and sources such as the bone marrow and amniotic fluid [43], we will address what is reported regarding resident endogenous stem/progenitor cells due to space limitations. To date, there are two potential endogenous sources normally resident in the kidney that need to be considered (and likely more will be discovered with time): glomerular parietal epithelial cells (PECs) and cells of renin lineage (CoRL).
Glomerular parietal epithelial cells (PECs) as adult podocyte progenitors
Several lines of evidence support the rationale that PECs possess biological programs necessary to differentiate into adult podocytes under certain circumstances. First, PECs and podocytes arise from a common restricted pool of mesenchymal cells during glomerulogenesis [44]. Second, a PEC subpopulation defined as cells at the tubular pole co-expressing the stemness markers CD133 and CD24 has been identified, and are referred to as multi-potent progenitors [45]. Third, in normal adult human, and rodent kidneys, a subpopulation of PECs lining Bowman’s capsule typically at the glomerular vascular pole co-express both PEC and podocyte proteins, and even show ultra-structural features of podocytes [46]. These cells have been named ‘glomerular epithelial transitional cells’. When such cells along Bowman’s capsule only express podocyte proteins under normal conditions, they have been referred to as ‘ectopic podocytes’ [47], and when they express WT-1, typical of adult podocytes [26], they have been called ‘parietal podocytes’ [47]. Moreover, the number and density of glomerular epithelial transitional cells increases under certain experimental and human glomerular diseases, oftentimes accompanying podocyte depletion, such as diabetic kidney disease [29], aging nephropathy [48], FSGS [49], and membranous nephropathy [31]. In addition, the number of transition cells can be increased by administration of retinoids [31], corticosteroids [32] and ACE-inhibition [30], as well as an improvement in the diabetic milieu [29].
However, the studies described above are all observational, and as such do not provide functional evidence that transition cells differentiate into podocytes. We therefore need to be cautious in the these interpretations, because we [39] and others [40, 50] have reported that podocytes can migrate from the glomerular tuft to Bowman’s capsule, where they might begin to co-express PEC proteins. Hence a cell on Bowman’s capsule that co-expresses both a podocyte protein(s) and a PEC protein(s) can be of either PEC or podocyte origin. Thus, we will discuss data supporting and not supporting PECs as adult podocyte progenitors, highlighting different experimental systems, species, disease models and other factors that might explain differences between different studies (see Table 1).
Table 1.
Summary of published studies on parietal epithelial cells (PEC) as adult podocyte progenitors
| Model System | Data supporting PEC as adult podocyte progenitors | Reference | |
|---|---|---|---|
| Studies strongly supporting PECs as adolescent podocyte progenitors | PEC reporter mice |
|
[62] [37] |
| Human kidney |
|
[53] [45] [55] |
|
| Studies suggesting, but not definitively proving, that a subset of PECs are adult podocyte progenitors | Rat model |
|
[28] |
| Diabetic mouse |
|
[56] |
Studies supporting a role for PECs as adult podocyte progenitors
During normal adolescence, both Appel and Schulte from Moeller’s group reported that in PEC reporter mice, a subset of podocytes derive from PECs [51, 52]. They suggest that these are parietal podocytes, distinct from adult PECs, that are committed to a podocyte fate and constitute a ‘reserve’ of podocytes for development, but have not yet matured. This is not podocyte regeneration per se as there is no podocyte depletion, but these findings are certainly consistent with PECs serving as a reservoir for podocytes in the period following the completion of glomerulogenesis. Noteworthy is that the PEC derived podocyte reservoir during adolescence was validated recently by the laboratories of Huber [37] and Romagnani [38]. Both groups predict that this podocyte reservoir is responsible for 10% of podocytes. This fits very well with recent studies in humans by Puelles and Bertram, showing that the number of podocytes increases by up to 20% early in life [12].
Using normal human kidneys, Sagrinati and Ronconi from Romagnani’s laboratory isolated cells from Bowman’s capsule co-expressing PEC proteins and the surface markers CD133/CD24, and then propagated them ex vivo in a cell culture system [45], which required a decrease in Notch signaling [53]. These ‘renal progenitor cells’ as they were called by this group could be expanded under certain cell culture conditions, and began to co-express markers of podocytes and tubular cells when cultured in different media [54, 55]. Similarly, Darisipudi from Anders’ laboratory showed that blockade of the chemokine stromal-derived factor (SFD/CXCL12) enhanced the differentiation of renal progenitors towards a podocyte phenotype in culture [56]. Furthermore, when renal progenitors were administered intravenously to SCID mice with Adriamycin-induced nephropathy used to deplete podocytes, renal progenitors were found in the glomerulus, expressing podocyte proteins, acquiring several characteristic ultra-structural features of podocytes, and accompanied by significantly improved disease outcomes [45]. Collectively these studies support a biological role for this PEC sub-population as adult podocyte progenitors in a model of experimental glomerular disease.
More recently, several studies have highlighted factors that inhibit PEC progenitors. Peirid showed that the subpopulation of PECs expressing CD133/CD24 require retinoids for normal survival and function [57]. However, in albuminuric states, the filtered albumin in the urinary space binds to retinoic acid in this space, thereby limiting the exogenous pool of retinoids available to PECs. Moreover, when albumin was taken up by PECs, a phenomenon that we have shown previously [58], endogenous retinoid synthesis was impaired [57]. Importantly, the decrease in the exogenous and endogenous retinoids limited the capacity of adult human parietal epithelial multipotent progenitors to perform their normal progenitor function, which might explain in part why podocyte regeneration is limited in albuminuric states.
Benigni and Rizzo recently showed that a subpopulation of PECs co-expressing the differentiation marker NCAM [59] also express the angiotensin 1 receptor (AT1R), and that proliferation of these cells could be reduced by giving rats the angiotensin-converting-enzyme inhibitor (ACE inhibitor) Lisinopril [59, 60].
Migliorini from the Anders’ laboratory showed that α- and β-interferons reduced the capacity of PEC progenitors to induce nephrin mRNA expression suggesting these agents may limit the capacity of PEC progenitors to become podocytes [61].
We have reported that abrupt podocyte depletion induced by the administration of a cytotoxic anti-podocyte antibody is followed by partial podocyte replacement, in the absence of podocyte proliferation, and that RAAS inhibitors can further augment podocyte replacement [30, 33, 39]. This suggests partial podocyte replacement from a non-podocyte source. To determine if neighboring PECs might serve this role, podocytes were depleted in PEC-reverse tetracycline-transactivator/LC1/Rosa26 reporter (pPEC-rtTA/LC1/R26R) mice [62, 63], in which PECs are permanently labeled in a temporal manner following the administration of doxycycline. Our results showed that a subset of labeled PECs migrated from Bowman’s capsule onto the glomerular tuft, where they de novo began to express several proteins considered podocyte-specific (FIGURE 1) [64]. These results suggest a role in this model for a subset of PECs as adult podocyte progenitors.
Figure 1. Reporter labeled PECs migrate onto the glomerular tuft in FSGS, and express synaptopodin.
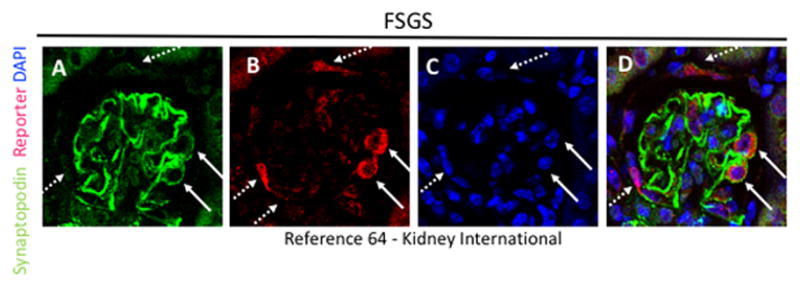
(A) Synaptopodin staining (green) is restricted to podocytes. (B) β-gal staining (red) is detected in cells lining Bowman’s capsule (dashed arrows) and inside the tuft (solid arrows). (C) DAPI staining is restricted to nuclei (blue). (D) Merge of all three stains, showing that a subset of reporter labeled PECs on the tuft co-stain with synaptopodin (solid arrows).
Finally, in their recent paper, Lasagni and Romagnani used two different conditional PEC reporter mice in which to lower podocyte number with Adriamycin [38]. In both instances, permanently labeled PECs moved onto the glomerular tuft, where they began to co-express three different podocyte proteins. The labeled PECs acquired many of the ultrastructural features that typify podocytes, including wrapping around the glomerular capillary loop and taking on a podocyte appearance by confocal microscopy. Noteworthy is that these events can be further increased by administering a GSK3 inhibitor [38]. Many of the studies described above fulfill the criteria we proposed above supporting that PECs serve as adult podocyte stem/progenitor cells.
Studies lacking a supporting a role for PECs as adult podocyte progenitors
An important recent paper by Puelles and Bertram showed that in humans, podocyte number increased during adolescence by up to 20% [12]. This suggested a pool of non-podocyte cells that served a progenitor role, as podocytes cannot adequately proliferate beyond glomerulogenesis. Using a transgenic mouse model expressing the tetracycline trans-activator protein rtTA, under regulation of a hybrid 3Kb fragment of the human and rabbit podocalyxin promoter to specifically label PECs permanently in vivo, Appel showed that a subset of PECs migrate to the glomerular tuft during mouse adolescence, where they begin to express podocyte proteins, and also take on their ultrastructural shape [62]. However, these authors have concluded in subsequent studies [51], using similar methods to those described above, that PECs do not serve as podocyte progenitors in adult mice in disease, a notion recently supported by Huber’s laboratory which found that podocyte replacement occurred after acute injury but not in age-related glomerulopathy [37]. While these studies were robustly performed, the authors of this review suggest that the three models studied, namely uninephrectomy, 5/6 nephrectomy and early middle aged mice 12 months of age, are typically not accompanied by a decrease in absolute podocyte number. Rather, these models can be associated with a decrease in podocyte density, due to an increase in glomerular volume. We therefore would not expect the need for any podocyte replacement/regeneration under these circumstances. Additional studies in models of absolute podocyte number depletion are therefore required to validate the claim that PECs do not serve as adult podocyte progenitors.
Studies have shown that in several states of podocyte depletion, the number of glomerular epithelial transition cells (defined as cells co-expressing PEC and podocyte proteins) are increased both along Bowman’s capsule, and in the glomerular tuft [48, 65]. Guhr showed that the de novo expression of podocyte proteins in PECs was due to reduced ubiquitin-mediated degradation [66]. More recently, studies using reporter mice have suggested a very different paradigm. These studies reported that following injury, a subset of labeled podocytes could be detected having moved from the glomerular tuft and now lining Bowman’s capsule [40, 50, 52, 67][39]. In this location in some instances, the labeled podocytes co-expressed PEC proteins in addition to podocyte proteins. This podocyte-to-PEC migration may occur in response to injury or loss of PECs in experimental models such as ureteral obstruction [68]. These data suggest that one explanation for cells co-expressing PEC and podocyte proteins along Bowman’s capsule is that they derive from migrating cells of podocyte origin, and not from PEC origin.
Taken together, there are compelling data that support a biological role for PECs as adolescent and adult podocyte progenitors. Yet, there is not consistency across all the models and marker systems (TABLE 1). We need to consider several variables that might explain these differences such as species and the types of experimental and human glomerular diseases studied. We also need to be cautious. While a PEC might well differentiate into an adult podocyte, the magnitude of “regeneration” that results from this alone may be inadequate to fully replace the number of podocytes depleted, and the precise cues and mechanisms underlying such events are not yet well delineated. Finally, we need to keep an open mind that there may well be bi-directional repair/regenerative process ongoing, where PECs differentiate into podocytes, and podocytes differentiate into PECs. Also, we must be cautious that the animal models of PEC fate mapping have not been fully reproduced in independent studies, and that use of temporally expressed markers in tissue sections is not a surrogate for fate mapping and result at best in suggestive observations.
Cells of Renin Lineage (CoRL) as Adult Podocyte Progenitors
Cells of Renin Lineage (CoRL) are vascular smooth muscle cells which produce, or once produced renin [69]. Under normal conditions, they are restricted to the juxta-glomerular compartment in adults, and comprise approximately 0.01% of the total kidney cell number. They derive from FoxD1-positive stromal cells [70]. Total CoRL number decreases in aged mice [71]. CoRL exhibit marked plasticity for a variety of cell-types as follows: studies led by Gomez shows that CoRL can transdifferentiate into smooth muscle cells, mesangial cells, and possibly pericytes [72, 73]. Kurtz and colleagues showed that when VHL is selectively deleted in CoRL, they can differentiate into erythropoietin-producing cells [74]. Starke and Hugo showed that CoRL can transdifferentiate in to adult mesangial cells in glomerular disease [75], and we have shown that CoRL contribute to the interstitial pericyte pool following ureteral obstruction [76]. Adult CoRL themselves can be regenerated by adult renal mesenchymal-like cells that express CD44, c-Kit and/or CD105 markers, events that recapitulate developmental processes [77]. Taken together, these studies show that under different conditions, adult CoRL are multipotent with marked stemness, serving as progenitors for several kidney cell types, and that they themselves can be replaced by progenitors as well.
Studies supporting a role for CoRL as adult podocyte progenitors
In order to determine whether CoRL also can serve as progenitors for adult glomerular epithelial cells in disease, we utilized several constitutive and inducible reporter mice that permanently tag Renin-producing cells using Cre/loxP somatic DNA recombination [78]. As expected, administering tamoxifen labeled CoRL only in Ren1c-CreER; tdTomatoR animals, and labeled cells were confined to the juxta-glomerular compartment. Over 95% of labeled cells co-expressed renin, and none were detected in glomeruli of normal mice, as expected [33, 78]. We depleted podocyte number by 30–40% in reporter mice using a model of classic Focal Segmental Glomerulosclerosis (FSGS) induced by cytotoxic anti-podocyte antibodies [33, 78]. Based on these studies, several lines of evidence support a paradigm of CoRL serving as adult podocyte stem/progenitors.
First, labeled CoRL were detected in a subpopulation of injured glomeruli (FIGURE 2). In the majority of glomeruli containing labeled CoRL, cells in the glomerulus were distributed either within the glomerular tuft adherent to the glomerular basement membrane, or along Bowman’s capsule. Neither renin protein, nor renin mRNA, were detected in glomeruli of FSGS mice. Such de novo appearance of permanently tagged CoRL within the glomerulus can only be explained by the migration from the juxta-glomerular compartment, since in mice that were not exposed to tamoxifen and thus did not undergo genetic recombination in the Renin producing cells prior to disease, no cells were detected either in the juxta- or intra glomerular compartments [78]. The degree of scarring within individual glomeruli was also significantly lower in glomeruli that contained labeled CoRL. These data support the notion that following an abrupt decline in podocyte number, labeled CoRL migrate from the juxta- to the intra-glomerular location.
Figure 2. Reporter labeled CoRL migrate onto the glomerular tuft in FSGS and remnant kidney, and express synaptopodin and podocin.
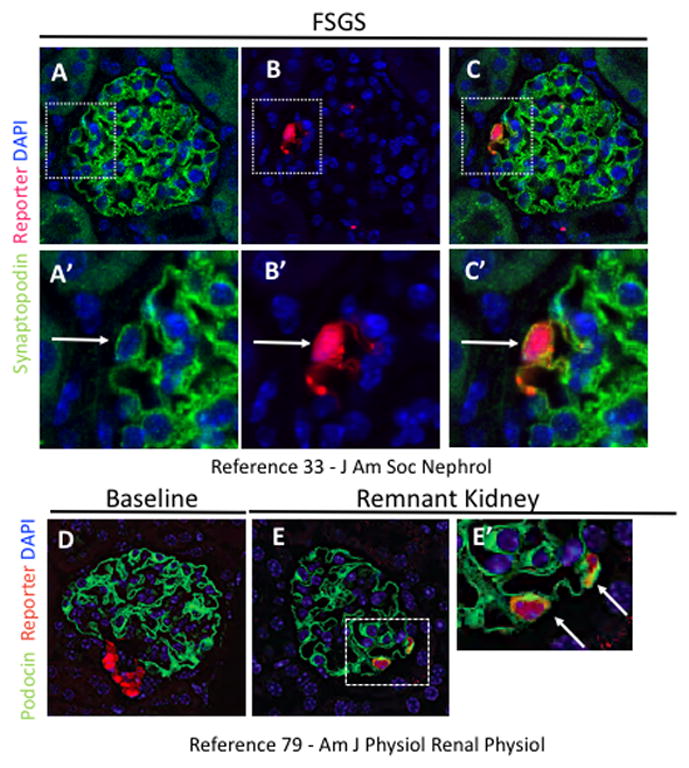
(A) Synaptopodin staining (green) is restricted to podocytes. (B) RFP staining (red) is detected within glomerular tuft (white arrow). (C) Synaptopodin and RFP staining colocalize (white arrow) in the glomerular tuft. (A′–C′) Higher magnification of insets shown above.
(D) At baseline, podocin staining (green) is restricted to podocytes and RFP staining (red) is restricted to CoRL in the juxtaglomerulus. (E) In the remnant kidney model, reporter labeled CoRL on the glomerular tuft co-stain with podocin. (E′) Higher magnification of inset shown in E (white arrows indicate co-staining).
Second, a subpopulation of intra-glomerular CoRL co-expressed one of the following podocyte proteins: WT-1, nephrin, podocin or synaptopodin [33, 78]. CoRL did not express nephrin, podocin, or synaptopodin when in a juxta-glomerular location in normal or diseased states. Some mapped CoRL cells that had moved into diseased glomeruli did not co-express markers of podocytes and therefore presumably had not differentiated into podocytes at the time of study.
Third, when CoRL reporter mice were given either the ACE-inhibitor Enalapril, or the angiotensin receptor blocker Losartan: (i) in the absence of disease, the pool of CoRL increased in the juxta-glomerular compartment, due to both recruitment and proliferation of existing CoRL; (ii) a subset of labeled CoRL migrated to the glomerulus prior to podocyte depletion; (iii) the number of CoRL that migrated to the glomerulus from the juxta-glomerular compartment following podocyte depletion was significantly augmented [33]. The latter was accompanied by overall kidney function improvement and a higher podocyte number, although direct proof that this was due to CoRL is lacking.
Fourth, we next asked if these events also occur following progressive podocyte depletion (rather than abrupt loss) in two additional models (5/6 nephrectomy and aged kidney). First, a 5/6 nephrectomy remnant kidney model was induced surgically in CoRL reporter mice [79]. As expected, mice developed mild FSGS [79]. However, in a subpopulation of glomeruli that did not have obvious segmental scarring, labeled CoRL could be detected, and a subset of these cells co-expressed several podocyte proteins (FIGURE 2) [79]. Glomerulosclerosis was substantially reduced in glomeruli with labeled CoRL. The results suggested that CoRL might also be serving as adult podocyte progenitors during chronic depletion. However, the ability of CoRL to fully replace or normalize podocyte number was not sufficient to prevent glomerulosclerosis.
Fifth, we studied the aged mouse kidney, as it is characterized by a progressive segmental and global glomerulosclerosis, accompanied by, and largely likely to a slow decline in podocyte number with age [80]. In CoRL reporter mice aged 1 year, a decrease in podocyte number was oftentimes accompanied by the presence of CoRL in the intra-glomerular compartment, and some of these cells co-expressed podocyte proteins [71]. Noteworthy was that electron microscopy studies of CoRL in the intra-glomerular compartment, showed characteristic ultra-morphological features of podocytes, namely foot processes and slit diaphragms [71]. With increasing age, the density of labeled CoRL within the juxtaglomerular compartment decreased significantly compared to younger ages.
Taken together, in three settings of podocyte depletion (one acute and two chronic), labeled CoRL were detected in glomeruli, where a subset co-expressed podocyte proteins, and some acquired ultrastructural morphological features of adult podocytes. These findings support the notion that CoRL might serve as adult podocyte progenitors and for partial regeneration in states of podocytes depletion.
Evidence Not Supporting a Role for CoRL as adult Glomerular Epithelial Cell Progenitors
As stated above, there is increasing evidence that CoRL have multi-cellular differentiation potential, that is likely context dependent. One view might be that there are simply too few cells of renin lineage to be able to provide any meaningful contribution to podocyte replacement. While this might be correct if adult podocyte was solely reliant on CoRL, we believe that multiple progenitors likely function to replace podocytes, albeit not quantitatively to the same degree. Moreover, the number of CoRL does increase under certain circumstances such as when mice are given ACE-inhibitors or angiotensin receptor blockers, thus providing an increased reservoir size.
What might be the goals for adult podocyte regeneration?
Eloquent experimental studies by Kim and Wharram from Wiggins’ laboratory have shown the close correlation between the magnitude of podocyte depletion, and the ensuing development of glomerular scarring [8, 9, 81]. This leads to the question “how much podocyte replacement is required to recover/repair normal structure and function?” Because such studies have not been reported, and are not easily accomplished, we offer a speculative opinion, using simple mathematical modeling as discussed in Figure 3 legend. We argue that answering this question through clinical lenses is that a reasonable podocyte replacement goal should be to simply increase podocyte number to that above the critical scarring threshold (20% podocyte loss), which limits/prevents segmental sclerosis progressing to global, as the former has a better prognosis than the latter.
Figure 3. Podocyte loss and regeneration, and their relationship to structural glomerular changes.
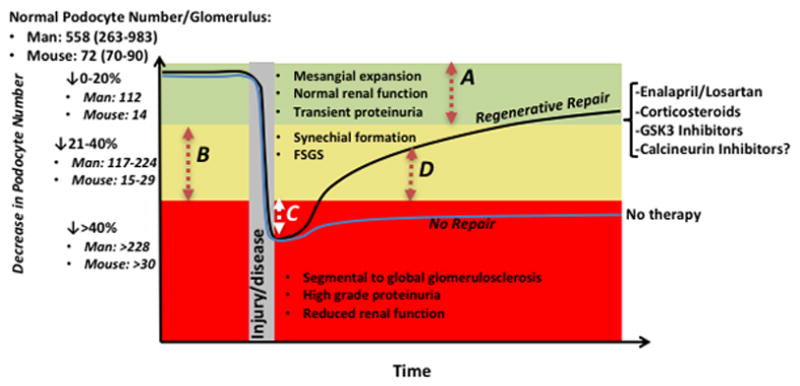
The Y axis shows the normal podocyte number and range per glomerulus in man and mouse. Three scenarios represented by different colors are shown for podocyte depletion, based on studies by Wharram.8 A decrease in podocyte number by 20% (represented by the green area) corresponds to a decrease in podocyte number per glomerulus by 112 in man, and 14 in mice. A decrease in podocyte number by 21–40% (represented by the yellow area) corresponds to a decrease in podocyte number per glomerulus by 117–224 in man, and 15–29 in mice. A decrease in podocyte number by greater than 40% (represented by the red area) corresponds to a decrease in podocyte number per glomerulus by >228 in man, and >30 in mice.
A decrease in podocyte number by 20% (represented by the red dashed arrow A), is not accompanied by any serious decline in kidney function, at least over the short term, unless there is subsequent podocyte depletion. In a steady state, we argue therefore that there is no major biological need to replace these podocytes. There is however a need to replace podocytes when their number is depleted between 21–40%, in order to limit further synchial formation and focal segmental glomerulosclerosis (FSGS), and to even reverse these processes. However, podocyte replacement does NOT need to return to the normal number in order for this to happen. Rather, we propose that the goal to replace podocyte number is above the threshold for scarring. As shown in the graph, this is typically less than 20% of normal (represented by the dashed arrow B). For example, to reach 20% of normal from a nadir of 40% of normal to prevent/reverse scarring in an individual glomerulus, requires only a 20% increase in podocyte number for that glomerulus (red arrow B). The mean number of glomeruli in a mouse kidney is 12,083±1009 per kidney, or 24,166 glomeruli total per mouse.80 In FSGS for example, if 20% of glomeruli are affected, the number of podocytes that need to be replaced in one kidney to return to the original baseline is 67,665 (20% of glomeruli * 24,166 glomeruli * 14 podocytes lost per glomerulus). If podocyte number drops by 40% (decrease of 29 podocytes in a single glomerulus), global glomerulosclerosis will ensue. Thus, still using the example of 20% of glomeruli affected implies that 20 % of glomeruli *24,166 glomeruli *29 podocytes lost per glomerulus= 140,162 total podocytes needed. Thus, for repair from a 40% drop in podocytes, to a 20% drop in podocytes (above which scarring does not occur), 72,497 podocytes are needed. While complete restoration would require 140,162 podocytes. If podocyte number drops below the 40% threshold to 50% for example, then an immediate goal for replacement would be to increase podocyte number by 10% (white arrow C) in order to limit the progression from focal to global sclerosis, and to reverse global to focal sclerosis. The second goal is to increase podocyte number by another 20% to limit focal sclerosis (red arrow B). Finally, several therapies (shown on the right side of the graph) increase podocyte number and in doing so, limit and/or reverse the progression of scarring by regenerative repair (black line)(example is red arrow D), whereas no therapy leads to non-regenerative repair (blue line)
In conclusion, following disease induced podocyte depletion, we propose that a goal should not be to replace podocyte number to normal (pre-disease) levels, but rather to restore podocyte number to a level/threshold which prevents the onset of glomerulosclerosis, and even reverses sclerosis. Most of the work that has been performed thus far in animal models has focused on the existence of cells capable of migrating into the glomerular tuft and assuming the podocyte phenotype. Now that we know such cells exist, it is important to investigate more thoroughly how they affect podocyte injury states and whether their enhancement can functionally improve outcomes such as albuminuria. Improved understanding of the mechanisms involved in the activation of these cells will guide the development of interventions, such as small molecules, that can be used to increase the number of podocyte replacement cells above the threshold required to prevent the onset of glomerulosclerosis.
Table 2.
Summary of published studies on cells of renin lineage as adult podocyte progenitors
| Model System | Data supporting cells of renin lineage (CoRL) as adult podocyte progenitors | Reference | |
|---|---|---|---|
| Studies supporting CoRL as adult podocyte progenitors | CoRL reporter mice following abrupt podocyte depletion |
|
[33] [79] [78] |
| CoRL reporter mice following chronic podocyte depletion |
|
[79] [71] |
|
| Human kidney |
|
Acknowledgments
Grant Support: 5 R01 DK 056799-10, 5 R01 DK 056799-12, 1 R01 DK097598-01A1, K01 DK102826, American Society of Nephrology Career Development Award
Abbreviations
- CoRL
cells of renin lineage
- PEC
glomerular parietal epithelial cell
Footnotes
COI: None of the authors have any financial or other conflicts of interest.
References
References published within the past 18 months only are bulleted and annotated.
-References have one bullet (*) for special interest and two bullets (**) for outstanding interest.
-Annotations provide a brief description of the paper’s importance.
- 1.Little MH. The Life Cycle of the Nephron Progenitor. Dev Cell. 2015;35(1):5–6. doi: 10.1016/j.devcel.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 2.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4(5) doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol. 2007;310(2):379–387. doi: 10.1016/j.ydbio.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3(2):169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, Paterson AD, Nitschke P, Bole-Feysot C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *6.Puelles VG, van der Wolde JW, Schulze KE, Short KM, Wong MN, Bensley JG, Cullen-McEwen LA, Caruana G, Hokke SN, Li J, Firth SD, Harper IS, Nikolic-Paterson DJ, et al. Validation of a Three-Dimensional Method for Counting and Sizing Podocytes in Whole Glomeruli. J Am Soc Nephrol. 2016;27(10):3093–3104. doi: 10.1681/ASN.2015121340. Methods for quantifying podocytes are limited. In this study, the authors introduce a new method that utilizes optical clearing and immunofluorescence to enable counting of podocytes in intact, whole glomeruli. The technique is validated in dose-dependent mouse models of podocyte depletion, establishing a new tool for studies of podocyte regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemley KV, Bertram JF, Nicholas SB, White K. Estimation of glomerular podocyte number: a selection of valid methods. J Am Soc Nephrol. 2013;24(8):1193–1202. doi: 10.1681/ASN.2012111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiggins The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney International. 2007;71(12):1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 9.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16(10):2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, Wickman LT, Wiggins JE, Muchayi T, Fingar D, Shedden KA, Inoki K, Wiggins RC. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23(8):1351–1363. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puelles VG, Cullen-McEwen LA, Taylor GE, Li J, Hughson MD, Kerr PG, Hoy WEAF, Bertram JF. Human podocyte depletion in association with older age and hypertension. Am J Physiol Renal Physiol. 2016 doi: 10.1152/ajprenal.00497.2015. ajprenal 00497 02015. [DOI] [PubMed]
- **12.Puelles VG, Douglas-Denton RN, Cullen-McEwen LA, Li J, Hughson MD, Hoy WE, Kerr PG, Bertram JF. Podocyte Number in Children and Adults: Associations with Glomerular Size and Numbers of Other Glomerular Resident Cells. J Am Soc Nephrol. 2015;26(9):2277–2288. doi: 10.1681/ASN.2014070641. The authors utilize a combination of microscopy techniques to measure the number and density of glomerular cell populations in human children and adults. The results reveal that children have significantly lower absolute podocyte numbers but much greater podocyte density, compared to adults. This suggests age-related podocyte replacement from an unknown source that serves to partially offset a net loss in podocyte density over time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roeder SS, Stefanska A, Eng DG, Kaverina N, Sunseri MW, McNicholas BA, Rabinovitch P, Engel FB, Daniel C, Amann K, Lichtnekert J, Pippin JW, Shankland SJ. Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol. 2015;309(2):F164–178. doi: 10.1152/ajprenal.00144.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tharaux PL, Huber TB. How many ways can a podocyte die? Semin Nephrol. 2012;32(4):394–404. doi: 10.1016/j.semnephrol.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Lim BJ, Yang JW, Do WS, Fogo AB. Pathogenesis of Focal Segmental Glomerulosclerosis. J Pathol Transl Med. 2016;50(6):405–410. doi: 10.4132/jptm.2016.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fogo AB. Decade in review--glomerular disease: The glomerulus reveals some secrets. Nat Rev Nephrol. 2015;11(11):633–634. doi: 10.1038/nrneph.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol. 2003;285(1):F40–48. doi: 10.1152/ajprenal.00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petermann AT, Krofft R, Blonski M, Hiromura K, Vaughn M, Pichler R, Griffin S, Wada T, Pippin J, Durvasula R, Shankland SJ. Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 2003;64(4):1222–1231. doi: 10.1046/j.1523-1755.2003.00217.x. [DOI] [PubMed] [Google Scholar]
- 19.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20(2):333–343. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall CB, Shankland SJ. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol. 2007;106(2):e51–59. doi: 10.1159/000101793. [DOI] [PubMed] [Google Scholar]
- 21.Combs HL, Shankland SJ, Setzer SV, Hudkins KL, Alpers CE. Expression of the cyclin kinase inhibitor, p27kip1, in developing and mature human kidney. Kidney Int. 1998;53(4):892–896. doi: 10.1111/j.1523-1755.1998.00842.x. [DOI] [PubMed] [Google Scholar]
- 22.Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D’Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int. 2000;58(2):674–683. doi: 10.1046/j.1523-1755.2000.00213.x. [DOI] [PubMed] [Google Scholar]
- 23.Hiromura K, Haseley LA, Zhang P, Monkawa T, Durvasula R, Petermann AT, Alpers CE, Mundel P, Shankland SJ. Podocyte expression of the CDK-inhibitor p57 during development and disease. Kidney Int. 2001;60(6):2235–2246. doi: 10.1046/j.1523-1755.2001.00057.x. [DOI] [PubMed] [Google Scholar]
- 24.Shankland SJ, Pippin JW, Couser WG. Complement (C5b-9) induces glomerular epithelial cell DNA synthesis but not proliferation in vitro. Kidney International. 1999;56(2):538–548. doi: 10.1046/j.1523-1755.1999.00560.x. [DOI] [PubMed] [Google Scholar]
- 25.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development. 1993;119(4):1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- 27.Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, Wiggins RC. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol. 2014;25(5):1118–1129. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benigni A, Morigi M, Rizzo P, Gagliardini E, Rota C, Abbate M, Ghezzi S, Remuzzi A, Remuzzi G. Inhibiting angiotensin-converting enzyme promotes renal repair by limiting progenitor cell proliferation and restoring the glomerular architecture. American Journal Of Pathology. 2011;179(2):628–638. doi: 10.1016/j.ajpath.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pichaiwong W, Hudkins KL, Wietecha T, Nguyen TQ, Tachaudomdach C, Li W, Askari B, Kobayashi T, O’Brien KD, Pippin JW, Shankland SJ, Alpers CE. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol. 2013;24(7):1088–1102. doi: 10.1681/ASN.2012050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Yanez D, Floege A, Lichtnekert J, Krofft RD, Liu ZH, Pippin JW, Shankland SJ. ACE-inhibition increases podocyte number in experimental glomerular disease independent of proliferation. Journal of the renin-angiotensin-aldosterone system: JRAAS. 2015;16(2):234–248. doi: 10.1177/1470320314543910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, Nelson PJ, Liu Z-H, Shankland SJ. Retinoids Augment the Expression of Podocyte Proteins by Glomerular Parietal Epithelial Cells in Experimental Glomerular Disease. Nephron Exp Nephrol. 2012;121(1–2):e23–e37. doi: 10.1159/000342808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Pippin JW, Krofft RD, Naito S, Liu Z, Shankland SJ. Podocyte Repopulation by Renal Progenitor Cells Following Glucocorticoids Treatment in Experimental FSGS. Am J Physiol Renal Physiol. 2013;304(11):F1375–1389. doi: 10.1152/ajprenal.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **33.Lichtnekert J, Kaverina NV, Eng DG, Gross KW, Kutz JN, Pippin JW, Shankland SJ. Renin-Angiotensin-Aldosterone System Inhibition Increases Podocyte Derivation from Cells of Renin Lineage. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2015080877. Several new insights in to cells of renin biology are noted. First, in addition to the well described notion of recruitment to increase the population of cells of renin lineage in the juxta-glomerular compartment in response to RAAS blockade, this study uses frequent BrdU injections to measure proliferation, and shows that cells of renin lineage also undergo proliferation when mice are given Enalapril or Losartan. Second, both these RAAS inhibitors increase the migration of cells of renin lineage to the intra-glomerular compartment following podocyte depletion. Third, this leads to an overall increase in the transdifferentiation of cells of renin lineage to podocytes. [DOI] [PMC free article] [PubMed]
- 34.Wang L, Tang Y, Howell DN, Ruiz P, Spurney RF. A novel mouse model of podocyte depletion. Nephron Exp Nephrol. 2012;121(1–2):e10–22. doi: 10.1159/000342369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romagnani P, Rinkevich Y, Dekel B. The use of lineage tracing to study kidney injury and regeneration. Nat Rev Nephrol. 2015;11(7):420–431. doi: 10.1038/nrneph.2015.67. [DOI] [PubMed] [Google Scholar]
- 36.Lombardi D, Lasagni L. Transgenic Strategies to Study Podocyte Loss and Regeneration. Stem Cells Int. 2015;2015:678347. doi: 10.1155/2015/678347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **37.Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R, Walz G, Moeller MJ, Grahammer F, Huber TB. Unraveling the Role of Podocyte Turnover in Glomerular Aging and Injury. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013050452. The authors genetically labelled glomerular parietal epithelial cells (PECs) in inducible hPODXL1.rtTA;tetO.Cre;mT/mG mice exposed to doxycycline from embryonic day (E) 8.5 to postnatal day (P) 28. They confirmed studies by the group of Moeller that during adolescence, PECs serve as podocyte progenitors under physiological conditions. Using a model of acute podocyte depletion by genetically inserting the diphtheria toxin receptor into podocytes, the authors noted podocyte regeneration, but the results suggested a source other than PECs. PECs did not serve a role for podocyte regeneration in the unilateral nephrectomy model, which is typically not accompanied by a marked decrease in podocyte number. Finally, using young middle aged mice (12m old), the authors noted no role for PECs progenitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **38.Lasagni L, Angelotti ML, Ronconi E, Lombardi D, Nardi S, Peired A, Becherucci F, Mazzinghi B, Sisti A, Romoli S, Burger A, Schaefer B, Buccoliero A, et al. Podocyte Regeneration Driven by Renal Progenitors Determines Glomerular Disease Remission and Can Be Pharmacologically Enhanced. Stem Cell Reports. 2015;5(2):248–263. doi: 10.1016/j.stemcr.2015.07.003. The authors evaluated whether the regenerative response to podocyte injury influences chronic kidney disease outcome. In models of focal segmental glomerulosclerosis performed in inducible transgenic mice where podocytes are tagged, the authors used the amount of regenerated podocytes as a measure of remission or progression of disease. When the same model was established in inducible transgenic mice where renal progenitors are tagged, the disease remitted if renal progenitors successfully differentiated into podocytes, while it persisted if differentiation was ineffective, resulting in glomerulosclerosis. Treatment with BIO, a GSK3 inhibitor, significantly increased disease remission by enhancing renal progenitor sensitivity to the differentiation effect of endogenous retinoic acid. These results establish renal progenitors as critical determinants of glomerular disease outcome and a pharmacological enhancement of their differentiation as a possible therapeutic strategy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *39.Kaverina NV, Eng DG, Schneider RR, Pippin JW, Shankland SJ. Partial podocyte replenishment in experimental FSGS derives from nonpodocyte sources. Am J Physiol Renal Physiol. 2016;310(11):F1397–1413. doi: 10.1152/ajprenal.00369.2015. Using a cytotoxic antibody model of FSGS in combination with fluorescence-labeled podocytes, the authors show that podocytes fail to proliferate after loss but can be partially replaced from a non-podocyte origi. In addition, this process can be augmented with the use of the ACE inhibitor enalapril, and is accompanied by migration of podocytes into the parietal epithelium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ElHackl MJ, Burford JL, Villanueva K, Lam L, Susztak K, Schermer B, Benzing T, Peti-Peterdi J. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med. 2013;19(12):1661–1666. doi: 10.1038/nm.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takasato M, Little MH. A strategy for generating kidney organoids: Recapitulating the development in human pluripotent stem cells. Dev Biol. 2016;420(2):210–220. doi: 10.1016/j.ydbio.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, Saad AF, Li MK, Hughes MR, Werff RV, Peters DT, Lu J, Baccei A, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Da Sacco S, Lemley KV, Sedrakyan S, Zanusso I, Petrosyan A, Peti-Peterdi J, Burford J, De Filippo RE, Perin L. A novel source of cultured podocytes. PLoS One. 2013;8(12):e81812. doi: 10.1371/journal.pone.0081812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romagnani P, Lasagni L, Remuzzi G. Renal progenitors: an evolutionary conserved strategy for kidney regeneration. Nat Rev Nephrol. 2013;9(3):137–146. doi: 10.1038/nrneph.2012.290. [DOI] [PubMed] [Google Scholar]
- 45.Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20(2):322–332. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes in normal human glomeruli. J Am Soc Nephrol. 2006;17(10):2770–2780. doi: 10.1681/ASN.2006040325. [DOI] [PubMed] [Google Scholar]
- 47.Shankland SJ, Smeets B, Pippin JW, Moeller MJ. The emergence of the glomerular parietal epithelial cell. Nat Rev Nephrol. 2014 doi: 10.1038/nrneph.2014.1. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Hansen KM, Pippin JW, Chang AM, Taniguchi Y, Krofft RD, Pickering SG, Liu Z, Abrass CK, Shankland SJ. De novo expression of podocyte proteins in parietal epithelial cells in experimental aging nephropathy. Am J Physiol Renal Physiol. 2012;302(5):F571–F580. doi: 10.1152/ajprenal.00516.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Pippin JW, Krofft RD, Naito S, Liu ZH, Shankland SJ. Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am J Physiol Renal Physiol. 2013;304(11):F1375–1389. doi: 10.1152/ajprenal.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyazaki Y, Shimizu A, Ichikawa I, Hosoya T, Pastan I, Matsusaka T. Mice are unable to endogenously regenerate podocytes during the repair of immunotoxin-induced glomerular injury. Nephrol Dial Transplant. 2013 doi: 10.1093/ndt/gft413. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- *51.Berger K, Schulte K, Boor P, Kuppe C, van Kuppevelt TH, Floege J, Smeets B, Moeller MJ. The regenerative potential of parietal epithelial cells in adult mice. J Am Soc Nephrol. 2014;25(4):693–705. doi: 10.1681/ASN.2013050481. The authors showed that in PEC reporter mice, a subset of PECs serve as podocyte progenitors during adolescence, but not in adult mice, nor during glomerular hypertrophy following partial nephrectomy. In humans aged 2 weeks to 2 years of age, some cells on Bowman’s capsule express the podocyte marker synaptopodin, which was not detected at 7 years of age. The authors conclude that a small fraction of committed podocytes resides on Bowman’s capsule close to the vascular stalk and is recruited onto the glomerular tuft during infancy to adolescence in mice and humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulte K, Berger K, Boor P, Jirak P, Gelman IH, Arkill KP, Neal CR, Kriz W, Floege J, Smeets B, Moeller MJ. Origin of parietal podocytes in atubular glomeruli mapped by lineage tracing. J Am Soc Nephrol. 2014;25(1):129–141. doi: 10.1681/ASN.2013040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, Peired A, Ronconi E, Becherucci F, Bani D, Gacci M, Carini M, Lazzeri E, et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells. 2010;28(9):1674–1685. doi: 10.1002/stem.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Angelotti ML, Ronconi E, Ballerini L, Peired A, Mazzinghi B, Sagrinati C, Parente E, Gacci M, Carini M, Rotondi M, Fogo AB, Lazzeri E, Lasagni L, et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells. 2012;30(8):1714–1725. doi: 10.1002/stem.1130. [DOI] [PubMed] [Google Scholar]
- 55.Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. Journal of the American Society of Nephrology: JASN. 2006;17(9):2443–2456. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- 56.Darisipudi MN, Kulkarni OP, Sayyed SG, Ryu M, Migliorini A, Sagrinati C, Parente E, Vater A, Eulberg D, Klussmann S, Romagnani P, Anders HJ. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am J Pathol. 2011;179(1):116–124. doi: 10.1016/j.ajpath.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peired A, Angelotti ML, Ronconi E, la Marca G, Mazzinghi B, Sisti A, Lombardi D, Giocaliere E, Della Bona M, Villanelli F, Parente E, Ballerini L, Sagrinati C, et al. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J Am Soc Nephrol. 2013;24(11):1756–1768. doi: 10.1681/ASN.2012090950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohse T, Chang AM, Pippin JW, Jarad G, Hudkins KL, Alpers CE, Miner JH, Shankland SJ. A new function for parietal epithelial cells: a second glomerular barrier. American Journal of Physiology-Renal Physiology. 2009;297(6):F1566–1574. doi: 10.1152/ajprenal.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benigni A, Morigi M, Rizzo P, Gagliardini E, Rota C, Abbate M, Ghezzi S, Remuzzi A, Remuzzi G. Inhibiting angiotensin-converting enzyme promotes renal repair by limiting progenitor cell proliferation and restoring the glomerular architecture. Am J Pathol. 2011;179(2):628–638. doi: 10.1016/j.ajpath.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rizzo P, Perico N, Gagliardini E, Novelli R, Alison MR, Remuzzi G, Benigni A. Nature and Mediators of Parietal Epithelial Cell Activation in Glomerulonephritides of Human and Rat. Am J Pathol. 2013;183(6):1769–1778. doi: 10.1016/j.ajpath.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Migliorini A, Angelotti ML, Mulay SR, Kulkarni OO, Demleitner J, Dietrich A, Sagrinati C, Ballerini L, Peired A, Shankland SJ, Liapis H, Romagnani P, Anders H–J. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. American Journal Of Pathology. 2013;183(2):431–440. doi: 10.1016/j.ajpath.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 62.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ. Recruitment of podocytes from glomerular parietal epithelial cells. Journal of the American Society of Nephrology: JASN. 2009;20(2):333–343. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JFM, Floege J, Moeller MJ. Tracing the Origin of Glomerular Extracapillary Lesions from Parietal Epithelial Cells. Journal of the American Society of Nephrology. 2009;20(12):2604–2615. doi: 10.1681/ASN.2009010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *64.Eng DG, Sunseri MW, Kaverina NV, Roeder SS, Pippin JW, Shankland SJ. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015;88(5):999–1012. doi: 10.1038/ki.2015.152. This study used inducible glomerular parietal epithelial cell (PEC) mice, in which PECs are permanently labelled following the administration of doxycycline to these reporter mice. Podocyte depletion was then induced in a model of FSGS. Labeled PECs were then detected on the glomerular tuft, consistent with their migration from Bowman’s capsule. A subset of these labelled began to co-express several different podocyte markers, consistent with the notion that they were transdifferentiating towards a podocyte fate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohse T, Vaughan MR, Kopp JB, Krofft RD, Marshall CB, Chang AM, Hudkins KL, Alpers CE, Pippin JW, Shankland SJ. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. American Journal of Physiology-Renal Physiology. 2010;298(3):F702–711. doi: 10.1152/ajprenal.00428.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guhr SS, Sachs M, Wegner A, Becker JU, Meyer TN, Kietzmann L, Schlossarek S, Carrier L, Braig M, Jat PS, Stahl RA, Meyer-Schwesinger C. The expression of podocyte-specific proteins in parietal epithelial cells is regulated by protein degradation. Kidney Int. 2013;84(3):532–544. doi: 10.1038/ki.2013.115. [DOI] [PubMed] [Google Scholar]
- 67.Sakamoto K, Ueno T, Kobayashi N, Hara S, Takashima Y, Pastan I, Matsusaka T, Nagata M. The direction and role of phenotypic transition between podocytes and parietal epithelial cells in focal segmental glomerulosclerosis. Am J Physiol Renal Physiol. 2014;306(1):F98–F104. doi: 10.1152/ajprenal.00228.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol. 2011;301(1):F110–117. doi: 10.1152/ajprenal.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sauter A, Machura K, Neubauer B, Kurtz A, Wagner C. Development of renin expression in the mouse kidney. Kidney Int. 2008;73(1):43–51. doi: 10.1038/sj.ki.5002571. [DOI] [PubMed] [Google Scholar]
- 70.Sequeira Lopez ML, Gomez RA. Development of the renal arterioles. J Am Soc Nephrol. 2011;22(12):2156–2165. doi: 10.1681/ASN.2011080818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pippin JW, Glenn ST, Krofft RD, Rusiniak ME, Alpers CE, Hudkins K, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage take on a podocyte phenotype in aging nephropathy. Am J Physiol Renal Physiol. 2014;306(10):F1198–1209. doi: 10.1152/ajprenal.00699.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell. 2004;6(5):719–728. doi: 10.1016/s1534-5807(04)00134-0. [DOI] [PubMed] [Google Scholar]
- 73.Gomez RA, Belyea B, Medrano S, Pentz ES, Sequeira-Lopez ML. Fate and plasticity of renin precursors in development and disease. Pediatr Nephrol. 2013 doi: 10.1007/s00467-013-2688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurt B, Paliege A, Willam C, Schwarzensteiner I, Schucht K, Neymeyer H, Sequeira-Lopez ML, Bachmann S, Gomez RA, Eckardt KU, Kurtz A. Deletion of von Hippel-Lindau protein converts renin-producing cells into erythropoietin-producing cells. J Am Soc Nephrol. 2013;24(3):433–444. doi: 10.1681/ASN.2012080791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Starke C, Betz H, Hickmann L, Lachmann P, Neubauer B, Kopp JB, Sequeira-Lopez ML, Gomez RA, Hohenstein B, Todorov VT, Hugo CP. Renin Lineage Cells Repopulate the Glomerular Mesangium after Injury. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014030265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stefanska A, Eng D, Kaverina N, Pippin JW, Gross KW, Duffield JS, Shankland SJ. Cells of renin lineage express hypoxia inducible factor 2alpha following experimental ureteral obstruction. BMC Nephrol. 2016;17:5. doi: 10.1186/s12882-015-0216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang H, Gomez JA, Klein S, Zhang Z, Seidler B, Yang Y, Schmeckpeper J, Zhang L, Muramoto GG, Chute J, Pratt RE, Saur D, Mirotsou M, et al. Adult renal mesenchymal stem cell-like cells contribute to juxtaglomerular cell recruitment. J Am Soc Nephrol. 2013;24(8):1263–1273. doi: 10.1681/ASN.2012060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pippin JW, Sparks MA, Glenn ST, Buitrago S, Coffman TM, Duffield JS, Gross KW, Shankland SJ. Cells of Renin Lineage Are Progenitors of Podocytes and Parietal Epithelial Cells in Experimental Glomerular Disease. Am J Pathol. 2013;183(2):542–557. doi: 10.1016/j.ajpath.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pippin JW, Kaverina NV, Eng DG, Krofft RD, Glenn ST, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol. 2015;309(4):F341–358. doi: 10.1152/ajprenal.00438.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, Yang Y, Meadowbrooke C, Chowdhury M, Kikuchi M, Wiggins JE, Wiggins RC. Glomerular Aging and Focal Global Glomerulosclerosis: A Podometric Perspective. J Am Soc Nephrol. 2015;26(12):3162–3178. doi: 10.1681/ASN.2014080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001;60(3):957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]