

Abstract
Background
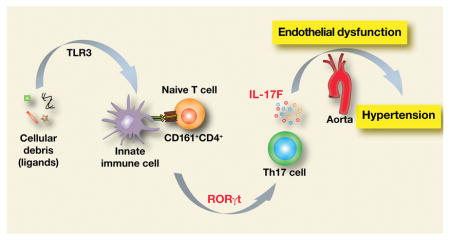
Hypertension is considered an immunological disorder. However, the role of IL-17 family in genetic hypertension of the spontaneously hypertensive rat (SHR) has not been investigated. Objective: We tested the hypothesis that enhanced Th17 programming and IL-17 expression in abundant CD161+ immune cells in SHR represent an abnormal proinflammatory adaptive immune response. Furthermore, we propose that this response is driven by the master regulator RORγt and a nicotinic proinflammatory innate immune response.
Methods
We measured the expression of CD161 surface marker on splenocytes in SHR and normotensive control Wistar-Kyoto (WKY) rats from birth to adulthood. We compared expression of IL-17A and IL-17F in splenic cells under different conditions. We then determined the functional effect of these cytokines on vascular reactivity. Finally, we tested whether pharmacological inhibition of RORγt can attenuate hypertension in SHR.
Results
The SHR exhibited an abnormally large population of CD161+ cells at birth that increased with age reaching more than 30% of the splenocyte population at 38 weeks. The SHR splenocytes constitutively expressed more RORγt than WKY and produced more IL-17F upon induction. Exposure of WKY aorta to IL-17F impaired endothelium-dependent vascular relaxation whereas IL-17A did not. Moreover, in vivo inhibition of RORγt by digoxin lowered systolic blood pressure in SHR. Conclusions: SHR has a markedly enhanced potential for RORγt –driven expression of proinflammatory, pro-hypertensive IL-17F in response to innate immune activation. Increased RORγt and IL-17F contribute to SHR hypertension and may be potential therapeutic targets.
Keywords: Hypertension, innate immune system, toll-like receptor, RORγt, digoxin, T cells, Th17, CD161, IL-17, SHR, gene expression
Graphical Abstract
Introduction
Multiple organ systems involving kidneys, vasculature, central nervous system and the immune system contribute to the genesis and maintenance of hypertension. Spontaneously hypertensive rat (SHR) is a widely studied rodent model of genetic hypertension that faithfully displays pathological changes seen in human disease. SHR shows progressive increase in blood pressure, elevated sympathetic tone, renal dysfunction and dysregulated immune system1, 2. The role of inflammation and the immune system in hypertension has long been known but the underlying mechanisms are just beginning to be understood.
SHR immune cells display an inherently enhanced inflammatory response. Cultured splenocytes from pre-hypertensive SHR produce greater amounts of proinflammatory cytokines than normotensive Wistar-Kyoto rats (WKY) when stimulated by TLR7/8 and TLR9 agonists in the presence of angiotensin II (Ang II) or nicotine 1. In contrast, nicotine suppresses the inflammatory cytokine release in WKY. In addition, nicotine treatment of cultured splenocytes from SHR increases the relative abundance of a CD161+ cell population. However, the distribution of CD161 cell surface markers in the immune system of the SHR and its potential role in hypertension related inflammation have not been examined.
Originally identified as a rodent homolog of a cell surface marker for human NK cells 3, CD161 marker correlates with activation of orphan nuclear receptor retinoic acid receptor-related orphan receptor gamma t (RORγt) that functions as a master regulator of polarization of interleukin-17 (IL-17) producing T helper 17 (Th17) cells.4–6 IL-17A and IL-17F are potent pro-inflammatory cytokines that drive inflammatory processes in autoimmune diseases and cardiovascular diseases including hypertension, 7, 8 and end organ damage 9, 10. Ang II-induced hypertension is not sustained in IL-17−/− mice 11. Dermal overexpression of IL-17A in psoriasis like skin disease induces endothelial dysfunction and arterial hypertension 12. In addition, infusion of IL-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension 13. Anti-hypertensive treatment using telmisartan and statin in patients with carotid atherosclerosis reduces blood pressure and IL-17 producing Th17 cells.14 DOCA-salt diet-induced organ damage is attenuated by blocking IL-17.15
IL-17 family of cytokines consists of six members (IL-17A, -B, -C, -D, -E, and – F). IL-17A (commonly known as IL-17) plays a key role in several autoimmune diseases and has been studied extensively. Relatively little is known about IL-17F that is the closest homolog of IL-17A and forms heterodimer with it. IL-17A and –F share a common receptor but are recognized to have non-overlapping functions.16 Despite a presumed role of immune system, inflammation and Th17 cells in hypertension, the effect of IL-17F in a hypertension-related physiological process has not been demonstrated.
The autonomic nervous system has immunomodulatory effects on expression of proinflammatory cytokines and proliferation of many immune cells. 17 This immunomodulation exerts a tonic inhibition of proinflammatory immune cells by acetylcholine binding to nicotinic acetylcholinergic receptors (nAChR).18, 19 However, the effects of nicotine, an agonist of nAChR, on different cell types may vary depending on the expressed subunit and pharmacology of the receptors.20–22 Th17 cells are differentiated from a CD4+ immune cell lineage that express nAChR and are subject to regulatory effects of nicotine.23–25 The inhibitory effect of nicotine on proinflammatory responses to TLR activation seen in splenocytes of normotensive WKY is abrogated in SHR splenocytes and replaced by a more pronounced proinflammatory response.1
Here we tested the hypothesis that enhanced Th17 programming by RORγt transcription factor, and IL-17F expression in CD161+ immune cells represent an abnormal proinflammatory adaptive immune response in SHR. Furthermore, we proposed that this response is driven by the master regulator RORγt and a nicotinic proinflammatory innate immune response.
Our results demonstrate that CD161 cell surface marker is widely and profusely expressed on SHR immune cells including CD4+ and CD8+ T lymphocytes. There are more infiltrating CD161+ cells in SHR kidney and aorta than in WKY. The CD4+CD161+ immune cells constitutively overexpress RORγt and induce IL-17F in response to inflammatory stimulation. Inhibition of RORγt expression with digoxin reduces systolic blood pressure and renal infiltration of CD3 cells in kidneys of SHR. We also report that nicotine, an anti-inflammatory agonist in WKY, actually increases CD4+CD161+ cells in SHR and potentiates their TLR3-mediated activation and IL17f expression. Finally, we identified a significant impairment of cholinergic endothelium-mediated vascular relaxation in response to IL-17F that was not seen with IL-17A.
We conclude that both the innate and the adaptive immune systems are inherently dysregulated in SHR. The dysregulation involves CD161+CD4+ immune cells RORγt transcription factor, a master regulator of IL-17A and IL-17F expression, that contributes to hypertension. The dysregulation also involves nicotine-mediated increase in CD4+CD161+ splenocytes and expression of IL-17F that disrupts cholinergic endothelial vasorelaxation.
Methods
Animals
WKY and SHR rats were obtained from Charles River Laboratory (USA). Male rats ranging in age from 1 day to 38 weeks were used. All experiments were performed under the guidelines set forth by the University of Iowa Institutional Animal Care and Use Committee (IACUC) and the National Institutes of Health. Rats were infused with saline (Sal) or nicotine (15 mg/kg body weight) using mini-osmotic pumps (Alzet 2001D) for 24 h.
Flow cytometry
Spleens from WKY and SHR were obtained after exsanguination under anesthesia (isoflurane) followed by decapitation. Spleen cells were disaggregated in pre-warmed Hank’s Balanced Salt Solution (HBSS, 37 °C). Following the erythrocyte lysis in a hypotonic buffer (155 mM ammonium chloride, 12 mM sodium bicarbonate and 0.1 mM EDTA), cells were washed with PBS, non-specific binding was blocked by incubation in Fc blocking solution (Phosphate buffered saline, PBS, with 5% fetal bovine serum and 1% mouse serum), and was subjected to antibody labeling using anti-rat FITC-CD161, PE-CD3, PerCP-CD8α, APC-CD4, and PE-Cy5-CD45RA. Cells were washed twice in the Fc blocking solution and resuspended in the same solution. Flow cytometry was performed using LSR II flow cytometer (Becton and Dickinson) and data were analyzed using either FlowJo or BD Aria software. For live-dead cell distinction, a vital fluorescent dye Hoechst 33258 (ThermoFisher Scientific, USA) was added to the cell samples immediately prior to flow cytometry.
Cell cultures
Splenocytes from WKY and SHR were plated in culture dishes containing ‘Complete’ RPMI1640 medium (RPMI 1640, 10% FBS, 10 mM Hepes, 1 mM sodium pyruvate supplemented with non-essential amino acids and penicillin/streptomycin). Cells and culture supernatants were collected and stored frozen at −80 °C until further use.
Cytokine induction
Poly-inosinic-poly-cytidylic acid (poly-IC, Sigma), the ligand of TLR3 was used for induction of splenocytes at a final concentration of 10 μg per ml added to the cultures, and cells were collected for RNA isolation. Th17 polarization program was induced by addition of anti-rat CD3 antibody (3 μg/ml, Affymetrix), rat transforming growth factor-β (TGF-β; 5 ng/ml, RD Biosciences) and rat interleukin-6 (IL-6; 20 ng/ml, RD Biosciences) for 4–7 days. Control cultures were left untreated.
RNA isolation and reverse transcription
RNA was isolated using Qiagen RNeasy RNA isolation kit according to the manufacturer’s protocol as described earlier.26 Equal amounts of RNA (0.5 to 2 μg each sample) were used for reverse transcription (RT) in 50 μl reactions. Oligo-dT primers were used for the first-strand synthesis with SuperScript-III reverse transcriptase enzyme. In some experiments, 2.5 μM of an 18s ribosomal RNA specific primer (5′-GAGCTGGAATTACCGCGGCT-3′) was added.
Quantitative real time PCR
Quantitative PCR was performed with either dye intercalation (SYBR green) or Taqman method using RT reactions as templates. Relative change in gene expression was quantified by ΔΔCT method using Gapdh or 18S ribosomal RNA as loading controls.27, 28
Enzyme linked immunosorbent assay (ELISA)
ELISA for detection of IL-17A and IL-17F was performed on splenocyte culture supernatants using Rat IL-17A Platinum ELISA kit (Affymetrix, USA) and IL-17F DuoSet ELISA development system (R&D Systems) according to manufacturers’ suggested protocols.
Digoxin treatment of SHR and blood pressure measurements
Prehypertensive male SHR (5 weeks old) were injected daily subcutaneously with digoxin (10 μg/g body weight in 20 μl volume, Acros Organics, USA) for 9 weeks. The dose for the drug was weekly adjusted to keep up with the gain in the body weight. Age and sex matched control SHR were injected with the vehicle (dimethyl sulfoxide, DMSO). Systolic blood pressures (SBP) were recorded twice every week by tailcuff plethysmography using Visitech 2000 system. Weekly SBP reported were averages of two recording sessions each week over a period of 3 months. The statistical analyses of SBP was performed using a Two-way ANOVA (Prism Software, version 7.0a) on the SBP data starting from the 7 week of age when the increase in pressure became evident.
Vascular reactivity
Thoracic aortas of adult WKY (14–16 weeks old) were used in the study. Vascular tension recordings were performed as described previously.29 Briefly, rats were euthanized by CO2 inhalation and dissected aorta were placed in ice-cold oxygenated Krebs buffer (118.3 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 25 mM NaHCO3, 1.2 mM MgSO4, 11 mM glucose, 0.0026 mM CaNa2EDTA). The vessels were cleared of connective tissue and cut into 2–3 mm rings and cultured overnight (16–20 h) in EGM2 medium (Lonza, USA) in the presence or absence of IL-17A or IL-17F. These aortic rings were then suspended between two wire stirrups (150 μm) in a four-chamber myograph system (DMT Instruments) in Krebs buffer (95% O2, 5% CO2, pH 7.4, 37 ºC). The mechanical force signal was amplified, digitalized, and recorded (PowerLab 8/30). The concentration-effect curves were recorded at the beginning of the optimum resting tone. Endothelium-dependent and –independent relaxation were determined by generating dose-response curves to acetylcholine (ACh 10−9–10−5 M) and sodium nitroprusside (SNP 10−9–10−5 M), respectively of the phenylephrine (PE, 10−6 M) induced pre-contracted vessel. Vasorelaxation evoked by ACh and SNP was expressed as percent relaxation, determined by calculating the percent inhibition of the pre-constricted tension.
Tissue and immunohistochemistry
At necroscopy, harvested tissues were fixed in 10% neutral buffered saline (3–5 days) followed by standard processing for paraffin embedding. Tissue sections (4 μm) were either stained with hematoxylin and eosin (HE) for histopathological examination or immunostained for CD3 or ED130 after heat induced antigen retrieval (Decloaking Chamber, BioCare Medical) using a rabbit polyclonal antibody (Cat. A0452, Dako Company) and counterstaining with Harris hematoxylin. A post-examination masking procedure described previously was used to avoid bias in tissue examination.31 Random areas of cortex in the sections of kidneys were photographed (100× magnification, CellSens software, Olympus BX51 microscope) and counted for cellular immnostaining (5 replicates per kidney) and total counts were normalized to unit area (counts of cells per mm2).
Statistical analyses
Unpaired t-test or ANOVA were used for statistical analyses as shown in respective figures. P values < 0.05 were considered to be statistically significant differences. Results are presented as mean ± SEM.
Results
Age-related increase in CD161+ cells in SHR
Splenocytes from age-matched WKY and SHR ranging from 1 day to 38 weeks were analyzed by flow cytometry (Figure 1A and B). In 1-day old SHR splenocytes, the CD161+ cell population was significantly higher than in WKY (1.4 ± 0.1% in WKY vs. 14.0 ± 0.1% in SHR). In SHR, the CD161+ cell population progressively increased with age reaching 33.6 ± 0.8% of the splenic immune cell population at 38 weeks of age. In contrast, in WKY, CD161+ splenocytes were only 8.0 ± 0.7% by 38 weeks of age. The marked increase in the SHR cell population was not associated with an increase in spleen to body mass ratio of the mature SHR compared to WKY (1.5 ± 0.1 mg/g vs. 1.6 ± 0.1 mg/g, respectively; Figure 1C). The high abundance of CD161+ cells was not attributable to non-specific binding of surface marker antibody to dead cells (Suppl. Figure-1). Moreover, the lymphocyte populations of CD4+ and CD8+ cells in both WKY and SHR increased similarly with age (Figure 1D and 1E). Thus, SHR exhibit an extraordinarily high abundance of CD161+ immune cells in the spleen at birth; these cells developmentally increase with age to reach surprisingly high levels as hypertension develops. The increase appears to be caused by acquisition of the CD161 cell surface marker in SHR rather than a proliferation of the CD161+ cells.
Figure 1.

(A) Flow cytometry of CD161+ cells in the spleen of newborn (1 day old) and adult (21 weeks old) SHR and WKY. (B) Age-related increase in CD161+ cells in SHR spleen (n= 3 rats for each age), (C) Spleen weight to body weight ratio (g/kg body wt) of adult WKY and SHR, (D) Frequency of CD4+ lymphocytes and (E) CD8+ lymphocytes in the spleen at different ages of WKY and SHR.
In addition to the spleen, we also tested the abundance of CD161+ cells in kidney and aorta. There was greater infiltration of CD161+ cells in kidney and aorta of SHR than in WKY (Suppl. Figure 2).
CD161+ cell surface marker is overexpressed on SHR CD4+ and CD8+ T lymphocytes
In spleens of 5 day old rats, CD4+CD161+ cells were significantly more abundant in the SHR than in WKY (0.2 ± 0.02% in WKY vs. 2.1 ± 0.04% in SHR, P<0.0001) as were CD8+CD161+ (0.14 ±0.00% in WKY vs. 2.5 ± 0.15% in SHR, P<0.0001, Figures 2A and 2B).
Figure 2.

(A) Dot plot of flow cytometry results showing greater abundance of CD4+CD161+ cells as well as CD8+CD161+ cells in young pre-hypertensive (5 days old) and adult (12 weeks old) SHR. (B) SHR immune cells displayed greater age-related increase in both CD4+CD161+ and CD8+CD161+ cell populations. (n= 3 rats for each age).
With age, populations of these lymphocytes grew much more abundantly in SHR reaching twice the levels seen in WKY at 38 weeks. For CD4+CD161+ cells the values were 8.53 ± 0.62% in WKY vs. 14.70 ± 0.10% in SHR, and corresponding values for CD8+CD161+ cells were 8.76 ± 0.64% in WKY vs. 16.25 ± 0.15% in SHR (P= 0.0076). In contrast, the CD161+ marker was not expressed on CD45RA+ B lymphocytes (data not shown). Thus, SHR has a selective and specific abnormality of increased CD4+CD161+ and CD8+CD161+ lymphocytes at birth even before the onset of hypertension that increases progressively with age as does hypertension.
CD161+ cells express RORγt transcription factor
The CD161 marker is associated with IL-17 producing T-lymphocytes. 4, 6 Expression of IL-17 and CD161 surface markers is regulated by an orphan nuclear receptor RORγt (Rorc gene), the master regulator transcription factor that determines CD4+ T helper 17 (Th17) and CD8+ T cytotoxic 17 (Tc17) cell polarization and IL-17A and IL-17F expression. 5, 32 At baseline, the expression of Rorc in untreated SHR splenocytes was significantly greater than untreated WKY (approximately 5 fold greater in SHR, P= 0.03; Figure 3A). Upon Th17 polarization (anti-CD3 antibody, IL-6 and TGF-β), Rorc expression in cultured WKY splenocytes increased 4.5-folds. However, the large expression of Rorc in SHR splenocytes did not change (Figure 3A).
Figure 3.

(A) Expression of Rorc (RORγt) RNA in untreated (Untr) and Th17 polarized (Th17) WKY and SHR splenocytes. (B) Gating scheme for Fluorescence Activated Cell Sorting of SHR splenocytes. CD161−CD45R+ subpopulation was the negative control. (C) Summary diagram of FACS analyses showing the composition of CD161+ cell population in SHR. (D) Rorc RNA expression in FACS sorted subpopulation of the SHR splenocytes. Asterisks show P<0.05, N= 3 each.
We further investigated which subset of CD161+ cells in SHR expressed RORγt. Using fluorescence activated cell sorting (FACS) of splenocytes from adult SHR we separated SHR CD161+ cells into CD4+CD161+, CD8+CD161+ and CD4−CD8−CD161+ cells (Figure 3B). In addition, as a negative control, we also collected CD161−CD45R+ cells (Figure 3B). The different populations of CD161+ cells represent specific immune cell populations (Figure 3C). Reverse transcriptase-QPCR (RT-QPCR) based analysis showed that Rorc RNA was detected in CD161+ cells but not in cells lacking CD161 surface marker (CD161−CD45R+ cells; Figure 3D). Moreover, the expression of Rorc was highest in CD4+CD161+ cells followed by CD4−CD8−CD161+ cells and the lowest in CD8+CD161+ cells (~18 fold difference between CD4+CD161+ and CD8+CD161+ cells; Figure 3D). Thus, the expression of RORγt transcription factor in SHR is most abundantly expressed in CD4+CD161+ cells of the SHR splenocytes.
Enhanced IL-17F expression in SHR upon Th17 cell induction
Induction of T lymphocytes depends on interaction of the innate (antigen processing cells) and adaptive immune cells (T cells). The master transcription factor RORγt regulates Th17 differentiation of CD4+ T lymphocytes leading to expression of IL-17A and IL-17F in these cells (Figure 4A).5 We stimulated in vitro Th17 polarization in WKY and SHR splenic T cells using anti-CD3 agonist antibody (T cell receptor activation) in the presence Th17-promoting cytokines IL-6 and TGF-β. A significant and similar increase in Il17a RNA expression was seen in both WKY and SHR splenocytes (3.5 fold in WKY vs. 3.1 fold in SHR; Figure 4B). However, the induction of Il17f RNA expression was significantly greater in SHR splenocytes (4.7 fold increase) than in WKY splenocytes (1.4 fold; Figure 4C). These results suggest that SHR splenocytes have greater adaptive immune response through potentially greater Th17 polarization and expression of proinflammatory IL-17 cytokines.
Figure 4.

(A) Schematic showing the differentiation program of CD161+ T cells into IL-17A and IL-17F producing T helper 17 (Th17) cells by activation of the TLRs and TCR. (B) Il17a RNA expression in untreated (Untr) and Th17 polarized (Th17) splenocytes from WKY and SHR. (C) Il17f RNA expression in SHR splenocytes upon Th17 polarization. Asterisks show P<0.05, N= 3 each.
High expression of cytokine IL-17 in SHR splenocytes in response to TLR3 agonist is RORγt dependent
Innate immune signaling through TLR3 plays a key role in antigen presenting cells (APC) mediated activation of T cells and their polarization into Th17 lineage.4–6 Therefore, we treated splenocytes from adult WKY and SHR with a TLR3 agonist poly-IC and measured the expression of Il17a and Il17f RNA. Il17a RNA expression was significantly but modestly induced in both WKY (1.7 fold) and SHR (2.2 fold) splenocytes (Figure 5A). However, expression of Il17f RNA was much greater in SHR splenocytes (27 fold) compared to WKY (2.9 fold; Figure 5B). TLR3 mediated increase in Il17a and Il17f expression in SHR splenocytes was dependent on RORγt. since pretreatment of SHR splenocytes with digoxin, an inhibitor of RORγt 33, abolished the poly-IC-induced increase in Il17a and significantly decreased the increase in Il17f expression (Figure 5C).
Figure 5.

(A) Expression of Il17a RNA in untreated (Untr) and Poly-IC (PIC) treated splenocytes from WKY and SHR. (B) Il17f RNA expression in untreated (Untr) vs. PIC treated splenocytes from WKY and SHR. (C) Effect of RORγt inhibitor digoxin (2.5 μM) on PIC induced expression of Il17a and Il1fa. (D) ELISA-based quantification of IL-17A and IL-17F in culture supernatants of untreated (Untr) and poly-IC (PIC) treated splenocytes from WKY and SHR. Asterisks show P<0.05, N= 3 each.
We also measured the IL-17A and IL-17F cytokines in the poly-IC treated culture supernatants of WKY and SHR splenocytes (Figure 5D). The results from RNA expression analyses were paralleled by a similarly increased IL-17A in both WKY and SHR culture supernatants (226 pg/ml in WKY vs. 189 pg/ml in SHR, P=0.01). Moreover, there was a 4 fold greater abundance of IL-17F cytokine in SHR culture supernatant than in WKY (27 pg/ml in WKY vs. 104 pg/ml in SHR, P=0.0008). These results show that SHR have an abnormally large population of CD161+ cells that strongly induce the pro-inflammatory IL-17 cytokines in a RORγt dependent manner upon activation of the innate immune system.
Nicotine increases CD4+CD161+ immune cell population and its IL-17F response to TLR3 agonist (Poly-IC) in SHR
Nicotine infusion for 24 h caused a small but significant decrease in the CD4+CD161+ lymphocyte population of WKY splenocytes (5.6% in saline vs. 3.1% in nicotine) but significantly increased the population of CD4+CD161+ lymphocytes in SHR (10.3% in saline vs. 18.6% in nicotine, Figure 6A). In contrast, CD8+CD161+ cell population did not change after nicotine infusion in either WKY or SHR (Figure 6B). Nicotine also has a proinflammatory effect in SHR.1 The poly-IC induced expression of Il17a RNA by splenocytes was reduced with nicotine infusion in WKY but unchanged in SHR (Figures 6C and 6D). In contrast, the increased expression of Il17f RNA with poly-IC was unchanged by nicotine in WKY yet significantly enhanced by nicotine in SHR. Thus, in contrast to WKY, nicotine infusion in SHR resulted in increased CD4+CD161+ cell population. Nicotine also increased the expression of Il17f RNA by poly-IC that is dependent on RORγt in this population of SHR.
Figure 6.

(A) Change in CD4+CD161+ cell population in the spleens of nicotineinfused WKY and SHR. (B) No change in splenocyte population of CD8+CD161+ in either WKY or SHR. (C) Effect of PIC treatment on expression of Il17a and Il17f RNA in saline or nicotine infused WKY (splenocytes). (D) Effect of PIC treatment on expression of Il17a and Il17f RNA in saline or nicotine infused WKY (splenocytes). Asterisks show P<0.05, N= 3 each, ANOVA with Bonferroni post-hoc test.
Differential effects of IL-17A and IL-17F on vascular relaxation
We tested endothelium-dependent vascular relaxation in WKY rat aortic rings following treatment with IL-17A or IL-17F. IL-17F treatment impaired acetylcholine-induced, endothelium-dependent vasorelaxation in a dose-dependent manner (Figure 7A and 7C). However, sodium nitroprusside (SNP)-induced vasorelaxation was not affected in IL-17F treated aortic rings (Figure 7B and 7D). In contrast, IL-17A treatment did not affect endothelium-dependent vasorelaxation even at a higher concentration of 1000 ng/ml (Figure 7E and 7G). However, treatment with a higher concentration of IL-17A (1000 ng/ml) shifted the SNP dose-response curve for vasorelaxation to the left (Figure 7F and 7H). Thus, IL-17F, but not IL-17A, impairs endothelium-dependent vascular reactivity in aorta.
Figure 7.

Effect of IL-17F on WKY endothelium-dependent (A,C) or endothelium-independent (B, D) vasodilation of aortic rings preconstricted with phenylephrine. Effect of IL-17A on endothelium-dependent vasodilation (E, G) and endothelium-independent (F, H) vasodilation. Ach= acetylcholine, PE= phenylephrine, SNP= sodium nitroprusside). Asterisks show P<0.05, N= 4 aortic rings per group for IL-17F and N= 3 for IL-17A.
Digoxin lowers spontaneous increase in blood pressure in SHR
We treated 5 weeks old prehypertensive SHR with daily injections of digoxin (10 μg/g body weight). In the control SHR that were injected daily with the vehicle (DMSO), the systolic blood pressure rose from 7 weeks of age and continued until the end of experiment at 14 weels of age. However, in the prehypertensive SHR that were daily injected with digoxin, the systolic blood pressure was significantly lower (Figure 8A). We also observed a significant decrease in CD3+ lymphocytes in the kidneys of digoxin-injected SHR when compared to the controls (Figure 8B and 8C). However, there was no overt histological change observed (Suppl. Figure-3A) and no difference in ED1+ macrophage population in the kidneys (Suppl. Figure-3B and 3C). Thus, RORγt inhibition by digoxin reduced T lymphocyte infiltration and lowered the increase in systolic blood pressure.
Figure 8.

(A) Effect of digoxin treatment (10 μg/g body weight) on spontaneously developed systolic blood pressure (SBP) in SHR. Treatment was from age 5 weeks to 13 weeks. N= 4 vehicle injected (DMSO), 6 digoxin injected. (B) CD3 surface marker immunostaining of T lymphocytes in kidney sections of control and digoxin injected SHR. (C) Summary data of CD3 infiltration in kidneys of control and digoxin injected SHR (n= 4 control, 6 digoxin). Asterisk shows P<0.05.
Discussion
T cells play a significant role in hypertension that has been recognized by several research groups.7, 8, 34–36 In SHR, the hypertension is viewed as a genetically determined and environmentally induced pathology. However, despite numerous studies of SHR, the immunological and molecular basis of SHR hypertension remains elusive. In this study, we have identified a large population of CD161+ innate and adaptive immune cells in SHR that have enhanced potential for the production of proinflammatory cytokines IL-17A and IL-17F. We show that IL-17F specifically impairs the vascular function, and inhibition of RORγt, the master regulator transcription factor for IL-17 producing immune cells, lowers the developing blood pressure in SHR.
Abundance of CD161+ immune cells
Our results show that normotensive WKY have a small population of CD161+ immune cells, whereas SHR have a much greater abundance of potentially proinflammatory CD161+ immune cells at birth and prior to the onset of hypertension. These cells dramatically increase with aging and are associated with the progressive increase in arterial pressure. This surface molecule CD161 was originally identified as the human homolog of the NKRP1 glycoproteins expressed on rodent natural killer cells (NK cells).3 Later, it was found to be expressed on several different cell types including CD4+ and CD8+ T cells as well as on CD4−CD8− double negative T cells.6 Furthermore, this increase in CD161+ marker is related to their greater potential for production of proinflammatory cytokines IL-17A and IL-17F. To our knowledge, such expansion of the CD161+ cell surface marker in vivo is unprecedented. Interestingly, the increase in CD161+ cells is not from hyperplasia as the spleen size and the relative abundance of CD4+ or CD8+ lymphocytes over time in SHR were similar to that of WKY. Instead, a larger population of immune cells in SHR seemed to have arisen from a dysregulated expression of the CD161+ cell surface marker.
RORγt overexpression in CD4+CD161+ T cells
CD161 is considered as one of the defining markers of Th17 cells. This marker is significantly upregulated in CD4+ Th17 cells that express RORγt and secrete IL-17A and IL-17F.4, 5 In fact, all IL-17 producing cells also express CD161, and a compelling correlation exists between IL-17 production and CD161 expression under the control of the transcription factor RORγt, which may be activated under Th17-differentiation conditions.6 Our results show a possible genetic defect in regulation of RORγt in SHR. Unlike the WKY splenocytes that have low basal expression of RORγt but induce its expression under Th17 polarization conditions, the SHR splenocytes express high basal RORγt transcription factor. Furthermore, fluorescence activated cell sorting followed by RT-QPCR also showed that RORγt expression was highest in the CD4+ cell population, a precursor for Th17 lineage.4 These results are consistent with other studies showing greater constitutive expression of RORγt RNA in CD4+CD161+ cells4, and suggested that the higher RORγt transcription factor expression might contribute to greater expression of IL-17 in SHR splenocytes.
RORγt, IL-17 and hypertension
Inhibition of RORγt by digoxin reduced the developmental increase of systolic blood pressure in SHR. In this study, we demonstrated that the SHR has a genetic predisposition of generating abundant RORγt expressing CD161+ cells that produced IL-17A and IL-17F in RORγt-dependent manner. IL-17A has been shown to be required for angiotensin II-induced hypertension.34 Moreover, increased IL-17A is a key inflammatory cytokine in the patients with systemic lupus erythematosus (SLE)37 who also have a high prevalence of hypertension.38 However, a role of IL-17F, seen in our study, in vascular relaxation has not been demonstrated. Thus, our results for the first time implicate RORγt-dependent IL-17F in SHR hypertension and also support the concept of hypertension as an autoimmune disease. Autoimmunity is thought to be at the root of both primary and secondary hypertension.8, 39, 40 Both IL-17 and RORγtexpressing CD161+Th17 cells have been implicated in autoimmune diseases including multiple sclerosis, psoriasis, and Crohn’s disease as well as in the mouse experimental model of MS- experimental autoimmune encephalomyelitis (EAE).41 We also observed a decrease in CD3+ T lymphocytes in the kidneys of digoxin-treated SHR. Kidneys are a critical organ in hypertension that endure inflammation during hypertension. RORγt has been shown to cause renal inflammation by increased leukocytes and cytokine production in a model of glomerulonephritis.42 Thus, inhibition of RORγt is likely to have contributed to lowering SBP by digoxin. Interestingly, despite greater potential for IL-17 production, there are no reports of skin lesions or EAE in the SHR.
It is also pertinent to note that hypertension is related to the deficiency of regulatory T cells (Treg cells), which are anti-inflammatory.43–45 Lineage determination of Th17 and Treg cells is held in a precarious balance. Th17 cells can convert to Tregs and vice versa or Th1-type cells. This can occur by a process of plasticity46 resulting in part by T-bet transcription factor-mediated repression of RORγt 47, the master regulator transcription factor of Th17 cells.
IL-17A vs. IL-17F
We observed a differential effect of IL-17A and IL-17F on rat aortic vasorelaxation. Despite a similar increase in Il17a expression in both WKY and SHR, Il17f expression was significantly greater in the SHR. IL-17A and IL-17F belong to the six-member proinflammatory IL-17 cytokine family and share significant sequence homology. Both IL-17A and IL-17F are secreted as homodimers but can also form IL-17A/IL-17F heterodimers. Importantly, these cytokines have distinct as well as overlapping roles in highly pathogenic inflammatory diseases.16, 48, 49 Their similar or distinct effects arise from their binding to shared or distinct receptors on the target cells.50, 51 However, little is known about either the expression of IL-17F or its effects in rat. We observed a dramatically specific role of IL-17F on the vascular function of rat. In our study, IL-17F had a profound and specific effect on impairment of endothelium-dependent vascular function whereas IL-17A had essentially no effect. This IL-17F effect was likely through interference in nitric oxide production by endothelium or by enhancing turnover of nitric oxide. IL-17F did not affect vascular smooth muscle cells as the nitric oxide donor sodium nitroprusside had similar effect on both control and treated aortic rings. Interestingly, at higher concentration, IL-17A had an opposite to IL-17F effect on vascular relaxation; it sensitized the aortic vasorelaxation by SNP. We currently do not know the mechanism of this phenomenon but our results are consistent with the concept that increased inflammatory status is accompanied by reduced NO production and endothelial function.52 However, considering that IL-17A and IL-17F share receptor subunit, our finding is both novel and important as it suggests receptor specificity in aorta for these cytokines.
The IL-17 receptor complex is heteromeric that consists of IL-17RA and other IL-17 receptors. Both IL-17A and IL-17F bind to the IL-17RA and IL-17RC heterodimeric complex to transduce downstream signaling. The binding affinity of IL-17A and IL-17F are different for IL-17 receptors. For example, mouse IL-17RC preferably binds to IL-17F.53, 54 Whereas IL-17RA is expressed ubiquitously, IL-17RC expression is low in hematopoietic tissues and high in non-immune cells, such as liver and kidney.55, 56 An additional level of complexity is that some members of the IL-17R family are highly spliced at sites in the extracellular domain, potentially giving rise to both agonistic and antagonistic (soluble) forms of the receptors. There are more than 90 splice isoforms of IL-17RC identified in human prostate cancer lines.57 Moreover, the main responsive cells to IL-17 are epithelial cells, endothelial cells and fibroblasts 53, 56, which is consistent with our results of IL-17F affecting endothelial cells in rat aorta.
Selective increase in Il17f expression in SHR with TLR3 activation and Th17 differentiation
IL-17 producing Th17 cells exclusively originate from a CD161+CD4+ T cell precursor.4 We observed a similar induction of Il17a RNA expression in both WKY and SHR, but a remarkably greater Il17f RNA expression in SHR. Inhibition of both Il17a and Il17f RNA expression by RORγt inhibitor digoxin33 confirmed that expression of these genes was driven by RORγt and hence related to Th17 differentiation. Yet, they are known to be regulated differentially.58–60 Despite their gene location in the same region of the chromosome, their regulatory sequences vary and their chromatin modifications are different. Thus, our results suggest that SHR may have altered genetic and epigenetic marks or signals that enhance Il17f expression.
Antipodal effect of nicotine on SHR CD4+CD161+ cells
In most hypertensive humans and animal models of hypertension, sympathetic nerve activity is exaggerated, 61–63 an effect that may result in increased inflammatory responses. In contrast, the parasympathetic autonomic nervous system, through activation of the α7-nAChR exerts an anti-inflammatory effect on the T cells.64–66 Activation of α7-nAChR expression on the surface of CD4+ T cells67 by nicotine reduces the Th17 response.68 Interestingly, CD4+ lymphocytes also express significantly greater α3- and β4-nAChR than CD8+ cells.69 Nicotine promotes an increase in CD3+CD4+ cells via activation of the α4 nAChR and regulation of a G protein subunit.70 nicotine infusions of 24 hours and 2 weeks were found to induce an increase in renal macrophage/monocyte migration and premature hypertension in SHR.30 Thus, nicotine may have opposite effects on CD4+ Th17 cell mediated inflammation in different contexts.71 In this study, SHR splenocytes responded by increasing their production of pro-inflammatory cytokines when induced by TLR agonists in the presence of nicotine. In addition, nicotine also increased the CD161+ cell population in vitro. Our in vivo results are consistent with the observed proliferation of cultured CD161+ cells in SHR.1 The underlying molecular mechanism of this opposite effect of nicotine in SHR is not fully understood. It could result from differential expression of nicotinic cholinergic receptors or through impaired intracellular signaling, and deserves further study.
In summary, we have shown that in SHR, onset of hypertension is preceded by an abnormally large population of CD161+ cells that increase with age. Furthermore, a Th17-like CD4+CD161+ cell population expresses high levels of proinflammatory IL-17F that may be further enhanced by nicotine. IL-17F impaired the endothelium-mediated vasorelaxation in rat aorta. Moreover, inhibition of RORγt, a transcription factor that regulates IL-17F expression, reduced systolic blood pressure in the SHR. These findings implicate a role of dysregulated immune system with increased potential for IL-17F production by RORγt transcription factor in the genetic hypertension of the SHR.
Supplementary Material
Key Messages.
Inflammation and immune system play a major role in the onset and maintenance of hypertension.
Our finding demonstrates an inherent abnormality in the immune system of a genetic model of hypertension that potentially produces cytokines involved in several autoimmune diseases.
These findings may have therapeutic potential to ameliorate hypertension and associated end-organ damage by eliminating the responsible cells or neutralizing the proinflammatory cytokines.
Acknowledgments
We thank the Flow Cytometry Core Facility of the University of Iowa for technical assistance and cell sorting. We also acknowledge discussions with and suggestions from Dr. John Colgan.
Source(s) of Funding
This research study was funded by the National Institutes of Health Program Project Grant to FMA (HL 14388), VA Merit Review Award to MWC (I01 BX001414) and American Heart Association Innovative Research Grant to MVS (16IRG27260323).
Abbreviations
- Ang II
Angiotensin II
- APC
Antigen presenting cells
- EAE
Experimental autoimmune encephalitis
- ELISA
Enzyme linked immunosorbent assay
- FACS
Fluorescence activated cell sorting
- IL-6
Interleukin 6
- IL-17
Interleukin 17
- nAChR
Nicotinic acetyl cholinergic receptor
- NK cell
Natural killer cell
- Poly-IC/PIC
Poly-inosinic-poly-cytidylic acid
- RORγt
Retinoic acid receptor related orphan receptor gamma t
- SHR
Spontaneously hypertensive rat
- SLE
Systemic lupus erythematosus
- SNA
Sympathetic nerve activity
- Tc17
T cytotoxic cells
- TGF-β
Transforming growth factor-β
- Th1
T helper 1 cells
- Th17
T helper 17 cells
- TLR
Toll-like receptor
- Treg
Regulatory T cells
- WKY
Wistar-Kyoto rat
Footnotes
Conflict(s) of Interest/Disclosure(s)
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–1197. doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ofosu-Appiah W, Ruggiero C. Abnormal activation and loss of suppressor t cells in the spontaneously hypertensive rat. Cell Immunol. 1992;145:130–145. doi: 10.1016/0008-8749(92)90318-j. [DOI] [PubMed] [Google Scholar]
- 3.Lanier LL, Chang C, Phillips JH. Human nkr-p1a. A disulfide-linked homodimer of the c-type lectin superfamily expressed by a subset of nk and t lymphocytes. J Immunol. 1994;153:2417–2428. [PubMed] [Google Scholar]
- 4.Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, Berrino L, Fambrini M, Caproni M, Tonelli F, Lazzeri E, Parronchi P, Liotta F, Maggi E, Romagnani S, Annunziato F. Human interleukin 17-producing cells originate from a cd161+cd4+ t cell precursor. J Exp Med. 2008;205:1903–1916. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ t helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 6.Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, Querci V, Fambrini M, Liotta F, Levings MK, Maggi E, Cosmi L, Romagnani S, Annunziato F. Cd161 is a marker of all human il-17-producing t-cell subsets and is induced by rorc. Eur J Immunol. 2010;40:2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 7.Harrison DG. The immune system in hypertension. Transactions of the American Clinical and Climatological Association. 2014;125:130–138. discussion 138–140. [PMC free article] [PubMed] [Google Scholar]
- 8.Schiffrin EL. The immune system: Role in hypertension. The Canadian journal of cardiology. 2013;29:543–548. doi: 10.1016/j.cjca.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Guzik TJ, Mikolajczyk T. In search of the t cell involved in hypertension and target organ damage. Hypertension. 2014;64:224–226. doi: 10.1161/HYPERTENSIONAHA.114.03340. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J, Ma X, Yin Z, Du J. Gammadeltat cell-derived interleukin-17a via an interleukin-1beta-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension. 2014;64:305–314. doi: 10.1161/HYPERTENSIONAHA.113.02604. [DOI] [PubMed] [Google Scholar]
- 11.Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:1565–1572. doi: 10.1161/ATVBAHA.111.227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, Koukes L, Yogev N, Nikolaev A, Reissig S, Ullmann A, Knorr M, Waldner M, Neurath MF, Li H, Wu Z, Brochhausen C, Scheller J, Rose-John S, Piotrowski C, Bechmann I, Radsak M, Wild P, Daiber A, von Stebut E, Wenzel P, Waisman A, Munzel T. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. 2014;34:2658–2668. doi: 10.1161/ATVBAHA.114.304108. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Zhao Y, Wei F, Ye L, Lu F, Zhang H, Diao Y, Song H, Qi Z. Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating th17/treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis. 2014;233:291–299. doi: 10.1016/j.atherosclerosis.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Amador CA, Barrientos V, Pena J, Herrada AA, Gonzalez M, Valdes S, Carrasco L, Alzamora R, Figueroa F, Kalergis AM, Michea L. Spironolactone decreases doca-salt-induced organ damage by blocking the activation of t helper 17 and the downregulation of regulatory t lymphocytes. Hypertension. 2014;63:797–803. doi: 10.1161/HYPERTENSIONAHA.113.02883. [DOI] [PubMed] [Google Scholar]
- 16.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17a and −17f in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 2003;74:675–696. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 18.De Rosa MJ, Dionisio L, Agriello E, Bouzat C, del Esandi MC. Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 2009;85:444–449. doi: 10.1016/j.lfs.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Pavlov VA, Tracey KJ. Neural circuitry and immunity. Immunologic research. 2015;63:38–57. doi: 10.1007/s12026-015-8718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karimi K, Bienenstock J, Wang L, Forsythe P. The vagus nerve modulates cd4+ t cell activity. Brain, behavior, and immunity. 2010;24:316–323. doi: 10.1016/j.bbi.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to legionella pneumophila infection by nicotine. J Immunol. 2001;167:6518–6524. doi: 10.4049/jimmunol.167.11.6518. [DOI] [PubMed] [Google Scholar]
- 22.Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, Nandi JS, Mishra NC, Singh SP, Pena-Philippides JC, Langley R, Sopori ML. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional tcr and leukocyte-specific protein tyrosine kinase for nicotine-induced ca2+ response. J Immunol. 2007;179:2889–2898. doi: 10.4049/jimmunol.179.5.2889. [DOI] [PubMed] [Google Scholar]
- 23.Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA. Plasticity of the murine spleen t-cell cholinergic receptors and their role in in vitro differentiation of naive cd4 t cells toward the th1, th2 and th17 lineages. Genes Immun. 2011;12:222–230. doi: 10.1038/gene.2010.72. [DOI] [PubMed] [Google Scholar]
- 24.Filippini P, Cesario A, Fini M, Locatelli F, Rutella S. The yin and yang of non-neuronal alpha7-nicotinic receptors in inflammation and autoimmunity. Curr Drug Targets. 2012;13:644–655. doi: 10.2174/138945012800399008. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Yang YH, Rajaiah R, Moudgil KD. Nicotine-induced differential modulation of autoimmune arthritis in the lewis rat involves changes in interleukin-17 and anti-cyclic citrullinated peptide antibodies. Arthritis and rheumatism. 2011;63:981–991. doi: 10.1002/art.30219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM. Dual activation of trif and myd88 adaptor proteins by angiotensin ii evokes opposing effects on pressure, cardiac hypertrophy, and inflammatory gene expression. Hypertension. 2015;66:647–656. doi: 10.1161/HYPERTENSIONAHA.115.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lardizabal MN, Nocito AL, Daniele SM, Ornella LA, Palatnik JF, Veggi LM. Reference genes for real-time pcr quantification of micrornas and messenger rnas in rat models of hepatotoxicity. PLoS One. 2012;7:e36323. doi: 10.1371/journal.pone.0036323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu LJ, Altmann SW. Mrna and 18s-rna coapplication-reverse transcription for quantitative gene expression analysis. Anal Biochem. 2005;345:102–109. doi: 10.1016/j.ab.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 29.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. Sirt1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harwani SC, Ratcliff J, Sutterwala FS, Ballas ZK, Meyerholz DK, Chapleau MW, Abboud F. Nicotine mediates cd161a+ renal macrophage infiltration and premature hypertension in the spontaneously hypertensive rat. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.309402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson-Corley KN, Olivier AK, Meyerholz DK. Principles for valid histopathologic scoring in research. Vet Pathol. 2013;50:1007–1015. doi: 10.1177/0300985813485099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of th17 cell differentiation. Seminars in immunology. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, Cuesta A, Santori FR, Lafaille JJ, Xu HE, Gin DY, Rastinejad F, Littman DR. Digoxin and its derivatives suppress th17 cell differentiation by antagonizing rorgammat activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin ii-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tipton AJ, Baban B, Sullivan JC. Female spontaneously hypertensive rats have a compensatory increase in renal regulatory t cells in response to elevations in blood pressure. Hypertension. 2014;64:557–564. doi: 10.1161/HYPERTENSIONAHA.114.03512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Crowley SD. Role of t lymphocytes in hypertension. Current opinion in pharmacology. 2015;21:14–19. doi: 10.1016/j.coph.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin JC, Baeten DL, Josien R. Emerging role of il-17 and th17 cells in systemic lupus erythematosus. Clinical immunology. 2014;154:1–12. doi: 10.1016/j.clim.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Ryan MJ. The pathophysiology of hypertension in systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1258–1267. doi: 10.1152/ajpregu.90864.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison DG, Guzik TJ, Goronzy J, Weyand C. Is hypertension an immunologic disease? Current cardiology reports. 2008;10:464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 40.Pober JS. Is hypertension an autoimmune disease? J Clin Invest. 2014;124:4234–4236. doi: 10.1172/JCI77766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toussirot E. The il23/th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflamm Allergy Drug Targets. 2012;11:159–168. doi: 10.2174/187152812800392805. [DOI] [PubMed] [Google Scholar]
- 42.Steinmetz OM, Summers SA, Gan PY, Semple T, Holdsworth SR, Kitching AR. The th17-defining transcription factor rorgammat promotes glomerulonephritis. Journal of the American Society of Nephrology : JASN. 2011;22:472–483. doi: 10.1681/ASN.2010040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin ii-induced hypertension and vascular injury. Hypertension. 2011;57:469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 44.Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL. Deficiency of t-regulatory cells exaggerates angiotensin ii-induced microvascular injury by enhancing immune responses. Journal of hypertension. 2016;34:97–108. doi: 10.1097/HJH.0000000000000761. [DOI] [PubMed] [Google Scholar]
- 45.Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 2014;126:267–274. doi: 10.1042/CS20130407. [DOI] [PubMed] [Google Scholar]
- 46.Zhu J, Paul WE. Heterogeneity and plasticity of t helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH. T-bet represses t(h)17 differentiation by preventing runx1-mediated activation of the gene encoding rorgammat. Nat Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF. Rorgamma-expressing th17 cells induce murine chronic intestinal inflammation via redundant effects of il-17a and il-17f. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 49.Tan W, Huang W, Gu X, Zhong Q, Liu B, Schwarzenberger P. Il-17f/il-17r interaction stimulates granulopoiesis in mice. Experimental hematology. 2008;36:1417–1427. doi: 10.1016/j.exphem.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, Tomkinson KN, Fitz LJ, Wolfman NM, Collins M, Dunussi-Joannopoulos K, Chatterjee-Kishore M, Carreno BM. The human il-17f/il-17a heterodimeric cytokine signals through the il-17ra/il-17rc receptor complex. J Immunol. 2008;181:2799–2805. doi: 10.4049/jimmunol.181.4.2799. [DOI] [PubMed] [Google Scholar]
- 51.Ely LK, Fischer S, Garcia KC. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol. 2009;10:1245–1251. doi: 10.1038/ni.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iantorno M, Campia U, Di Daniele N, Nistico S, Forleo GB, Cardillo C, Tesauro M. Obesity, inflammation and endothelial dysfunction. J Biol Regul Homeost Agents. 2014;28:169–176. [PubMed] [Google Scholar]
- 53.Gaffen SL. Structure and signalling in the il-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 55.Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, Aujla SJ, Reardon B, Moore M, Shea P, Schreckhise R, Bukowski TR, Presnell S, Guerra-Lewis P, Parrish-Novak J, Ellsworth JL, Jaspers S, Lewis KE, Appleby M, Kolls JK, Rixon M, West JW, Gao Z, Levin SD. Identification of the il-17 receptor related molecule il-17rc as the receptor for il-17f. J Immunol. 2007;179:5462–5473. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen F, Gaffen SL. Structure-function relationships in the il-17 receptor: Implications for signal transduction and therapy. Cytokine. 2008;41:92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haudenschild DR, Curtiss SB, Moseley TA, Reddi AH. Generation of interleukin-17 receptor-like protein (il-17rl) in prostate by alternative splicing of rna. Prostate. 2006;66:1268–1274. doi: 10.1002/pros.20422. [DOI] [PubMed] [Google Scholar]
- 58.Adamik J, Henkel M, Ray A, Auron PE, Duerr R, Barrie A. The il17a and il17f loci have divergent histone modifications and are differentially regulated by prostaglandin e2 in th17 cells. Cytokine. 2013;64:404–412. doi: 10.1016/j.cyto.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (il-17)-il-17f cytokine gene locus during inflammatory helper t cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 60.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, August A, Schwartzberg PL. Differential expression of interleukin-17a and - 17f is coupled to t cell receptor signaling via inducible t cell kinase. Immunity. 2009;31:587–597. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: Implications for hypertension and cardiovascular disease. Hypertension. 2012;59:755–762. doi: 10.1161/HYPERTENSIONAHA.111.186833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Esler M. The sympathetic nervous system through the ages: From thomas willis to resistant hypertension. Experimental physiology. 2011;96:611–622. doi: 10.1113/expphysiol.2010.052332. [DOI] [PubMed] [Google Scholar]
- 63.Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation. 2004;109:120–124. doi: 10.1161/01.CIR.0000105721.71640.DA. [DOI] [PubMed] [Google Scholar]
- 64.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 65.Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Molecular medicine (Cambridge, Mass) 2003;9:125–134. [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S, Petro TM. The effect of nicotine on murine cd4 t cell responses. International journal of immunopharmacology. 1996;18:467–478. doi: 10.1016/s0192-0561(96)00054-9. [DOI] [PubMed] [Google Scholar]
- 67.Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of th1 and th17 responses. J Immunol. 2009;183:6681–6688. doi: 10.4049/jimmunol.0902212. [DOI] [PubMed] [Google Scholar]
- 68.Liu Z, Han B, Li P, Wang Z, Fan Q. Activation of alpha7nachr by nicotine reduced the th17 response in cd4(+)t lymphocytes. Immunol Invest. 2014;43:667–674. doi: 10.3109/08820139.2014.914532. [DOI] [PubMed] [Google Scholar]
- 69.Mihovilovic M, Denning S, Mai Y, Whichard LP, Patel DD, Roses AD. Thymocytes and cultured thymic epithelial cells express transcripts encoding alpha-3, alpha-5 and beta-4 subunits of neuronal nicotinic acetylcholine receptors: Preferential transcription of the alpha-3 and beta-4 genes by immature cd4 + 8 + thymocytes. J Neuroimmunol. 1997;79:176–184. doi: 10.1016/s0165-5728(97)00120-3. [DOI] [PubMed] [Google Scholar]
- 70.Nordman JC, Muldoon P, Clark S, Damaj MI, Kabbani N. The alpha4 nicotinic receptor promotes cd4+ t-cell proliferation and a helper t-cell immune response. Molecular pharmacology. 2014;85:50–61. doi: 10.1124/mol.113.088484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galitovskiy V, Qian J, Chernyavsky AI, Marchenko S, Gindi V, Edwards RA, Grando SA. Cytokine-induced alterations of alpha7 nicotinic receptor in colonic cd4 t cells mediate dichotomous response to nicotine in murine models of th1/th17- versus th2-mediated colitis. J Immunol. 2011;187:2677–2687. doi: 10.4049/jimmunol.1002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.