

Abstract
Remission induction therapy for acute lymphoblastic leukemia (ALL) includes medications that may cause hepatotoxicity, including asparaginase. We used a genome-wide association study (GWAS) to identify loci associated with elevated alanine transaminase (ALT) levels after induction therapy in children with ALL enrolled on St. Jude Children’s Research Hospital (SJCRH) protocols. Germline DNA was genotyped using arrays and exome sequencing. Adjusting for age, body mass index, ancestry, asparaginase preparation and dosage, the PNPLA3 rs738409 (C>G) I148M variant, previously associated with fatty liver disease risk, had the strongest genetic association with ALT (P = 2.5×10−8). The PNPLA3 rs738409 variant explained 3.8% of the variability in ALT, and partly explained race-related differences in ALT. The PNPLA3 rs738409 association was replicated in an independent cohort of 2,285 patients treated on Children’s Oncology Group protocol AALL0232 (P = 0.024). This is an example of a pharmacogenetic variant overlapping with a disease risk variant.
Keywords: Acute Lymphoblastic Leukemia, Asparaginase, Alanine Aminotransferase, Hepatotoxicity, Pharmacogenomics
Introduction
Although cure rates for childhood acute lymphoblastic leukemia (ALL) are approaching 90%,(1) therapy is complicated by drug toxicity. Patients who tolerate less treatment due to adverse drug effects may be at a higher risk of relapse.(2, 3) Hepatotoxicity is a frequent complication leading to treatment interruptions,(4–6) and several drugs used in ALL therapy are associated with the development of hepatotoxicity, including asparaginase, mercaptopurine, thioguanine, methotrexate, etoposide, and glucocorticoid.(7)
Some treatment regimens for adults with ALL avoid using and/or dose modify asparaginase and high-dose methotrexate in order to minimize treatment-related hepatotoxicity,(8, 9) which may contribute to the lower cure rates for adults with ALL. Therefore, studies that can elucidate the mechanism of drug-induced hepatotoxicity during the treatment of ALL may lead to therapeutic strategies that can reduce the frequency of hepatotoxicity, identify patients who are most likely to benefit from treatment, and possibly improve cure rates by reducing treatment interruptions due to hepatotoxicity.
We investigated coding and non-coding genetic variants associated with the elevation of alanine aminotransferase (ALT) after remission induction therapy. A total of 715 St. Jude Children’s Research Hospital patients were evaluable with genotyping and post-induction chemotherapy ALT levels. We tested for replication in 2,285 patients treated on the Children’s Oncology (COG) AALL0232 trial.
Results
Patients enrolled on St. Jude Children’s Research Hospital (SJCRH) Total XV or Total XVI received similar induction therapy with a few notable differences (Table 1). The Total XV protocol included native E. coli asparaginase (Elspar), whereas the Total XVI protocol used PEGylated E. coli asparaginase (Oncaspar). Patients enrolled on Total XV received mercaptopurine, whereas most on Total XVI received thioguanine; those on Total XV received pre-induction window therapy with methotrexate, whereas those on Total XVI did not (Table 1). In both protocols, patients with higher minimal residual disease (MRD) at day 15–19 of induction received additional asparaginase compared to those without.
Table 1.
Chemotherapeutic agents used during Total XV and Total XVI remission induction therapy.
| Agents | Dosage and Routes and days of induction therapy | Number of doses | Differences between Total XV and Total XVI |
|---|---|---|---|
| Prednisonea | 40 mg/m2/day, PO, T.I.D.; Total XV: days 5–32 Total XVI: days 1–28 |
84 | - |
| Vincristine | 1.5 mg/m2/weekly, IV; Total XV: days 5, 12, 19, 26 Total XVI: days 1, 8, 15, 22 |
4 | - |
| MTXb | 1 g/m2, IV, over a 4 or 24 hour infusion | 1 | Window MTX was not included on the Total XVI protocol |
| Daunorubicin | 25 mg/m2/weekly, IV; Total XV: days 5 & 12 Total XVI: days 1 & 8 |
2 | - |
| ASNasec | Total XV: L-ASNase, 10,000 units/m2, IM, thrice weekly; days 6, 8, 10, 12, 14, 16 | L-ASNase: 6 | Preparation, dosage, and route |
| Total XVI: PEG-ASNase, 3,000 units/m2, IV; day 3 | PEG-ASNase: 1 | ||
| Cyclophosphamide | 1,000 mg/m2, IV; Total XV: day 26 Total XVI: day 22 |
1 | Cyclophosphamide was intensified for standard/high risk patients with ≥ 5% of blast on day 15 of induction for Total XVI patientsd |
| Cytarabine | 75 mg/m2/dose, IV; Total XV: days 27–30 & 34–37 Total XVI: days 23–26 & 30–33 |
8 | - |
| Thiopurine | Total XV: mercaptopurine, 60 mg/m2/day, PO; days 26–39 | 14 | Total XVI received thioguanine; Total XV received mercaptopurine |
| Total XVI: thioguaninee, 60 mg/m2/day, PO; days 22–35 |
Oral prednisone was substituted with methylprednisolone at 20 mg/m2/day, IV for patients that could not tolerate oral administration.
Total XV patients were randomized to receive short (4 hour) or prolonged (24 hour) methotrexate infusion.
Additional 3 doses of L-ASNase were given if day 19 MRD ≥ 5% on days 19, 21, 23 to Total XV patients. Additional PEG-ASNase dose was given if day 15 MRD ≥ 1% on day 15 to Total XVI patients.
Total XVI patients with day 15 MRD ≥ 5% received cyclophosphamide at 300 mg/m2, IV, q12 hours for 4 doses on days 22–23.
TPMT heterozygote or TPMT deficient Total XVI patients received mercaptopurine at 60 mg/m2/dose, PO instead of thioguanine.
Abbreviations: PO, by mouth; IV, intravenous; IM, intramuscular; T.I.D., three times a day; MRD, minimal residual disease; MTX, methotrexate; ASNase, asparaginase
Overall, 715 patients were evaluable for post-induction ALT data in the discovery cohort (Table 2, Figure S1). By univariate analysis (Table S1), treatment protocol (Figure 1A), additional asparaginase during induction (Figure 1B), race group (Figure 1C), age (Figure 1D), body mass index (BMI) percentile (Figure 1E), and gender were associated with post-induction ALT levels. In multivariate analysis (Table 3), higher ALT levels were associated with Total XVI treatment protocol (P = 0.041), receiving additional induction asparaginase (P = 2.7×10−4), non-black race (P = 8.7×10−13 for whites and P = 2.6×10−7 for Hispanics), older age (P = 1.4×10−6), and higher BMI (P = 2.9×10−3). Race group accounted for 7.2% of the variability in ALT levels (Table 3). Focusing on the National Cancer Institute’s common toxicity criteria (CTCAE) categories of ALT toxicity, Total XVI patients had more grade 2–3 ALT elevations compared to Total XV patients (P = 5.8×10−3, Figure S2A).
Table 2.
Demographics of ALL patients with serum ALT measurement data.
| Characteristics | Treatment protocol | ||
|---|---|---|---|
|
| |||
| SJCRH (Total XV) | SJRCH (Total XVI) | COG AALL0232d | |
| Sample Size (n) | 373 | 342 | 2285 |
|
| |||
| Age (n ≥ 10, mean in years) | 102 (27.4%),a 7.1 |
92 (26.9%),a 7.2 |
1,468 (64.3%),a 10.6 |
|
| |||
| Gender (n male) | 206 (55.2%)a | 198 (57.9%)a | 1,244 (54.4%)a |
|
| |||
| Race groupb | |||
| Whites | 261 (70.0%)a | 227 (66.4%)a | 1,275 (55.8%)a |
| Blacks | 64 (17.2%)a | 47 (13.7%)a | 139 (6.1%)a |
| Hispanics | 24 (6.4%)a | 36 (10.5%)a | 601 (26.3%)a |
| Others | 24 (6.4%)a | 32 (9.4%)a | 270 (11.8%)a |
|
| |||
| Diagnostic ALT(IU/L)c | |||
| Median (range) | 21 (3 – 3866) | 20 (4 – 2442) | NA |
| 95% confidence interval | 22.9 – 27.9 | 22.4 – 27.9 | NA |
|
| |||
| Post-induction ALT(IU/L) | |||
| Median (range) | 38 (4 – 416) | 44 (7 – 321) | NA |
| 95% confidence interval | 35.5 – 40.9 | 41.3 – 48.4 | NA |
The percentage of patients with the characteristic described in the first column.
Whites were defined as having > 90% Northern European ancestry (CEU), blacks as having > 70% West African ancestry (YRI), Hispanics as having Native American ancestry > 10% and greater than their African ancestry percentage, and others as those whose ancestry was outside the above boundaries.
Diagnostic ALT (pre-induction therapy) was evaluable in 337 of 373 Total XV patients and 321 of 342 Total XVI patients, and ALT levels were lower at diagnosis than at end-of-induction (P < 2.2×10−16, paired t-test for 658 patients evaluable at both times, see also Figures S8&S9).
No discrete ALT levels were available for COG AALL0232 patients.
NA, not available
Figure 1. ALT level variation in pediatric ALL patients.

(A) Total XVI patients receiving PEGylated E. coli asparaginase had higher ALT levels compared to Total XV patients receiving native E. coli asparaginase (P = 3.0×10−3). (B) Among Total XV and Total XVI patients, those receiving additional doses of asparaginase during induction due to a higher ALL burden at day 15–19 had higher ALT levels compared to patients receiving fewer doses of asparaginase (P = 3.5×10−6). (C) Among Total XV and Total XVI patients, whites (P = 3.3×10−10) and Hispanics (P = 3.3×10−7) had higher ALT levels compared to blacks. (D) ALT levels were correlated with age (yrs) in the SJCRH discovery cohort (N = 715, r = 0.20, P = 9.4×10−8). (E) ALT levels were positively correlated with BMI percentile in the SJCRH discovery cohort (N = 715, r = 0.11, P = 5.2×10−3). The P values indicated are from univariate analyses (Table S1).
Table 3.
Multivariate analyses of covariates associated with alanine transaminase levels during Total XV and Total XVI remission induction therapy.
| Without inclusion of PNPLA3 rs738409 | Including PNPLA3 rs738409 | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Covariate | Effect sizea | P value | % of ALT level variability explained | Effect sizea | P value | % of ALT level variability explained |
|
|
|
|
||||
| Induction ASNase: Additional vs. standard schedule | 1.26 (1.11–1.42) | 2.7×10−4 | 1.7% | 1.27 (1.12–1.43) | 1.1×10−4 | 1.8% |
|
|
|
|
||||
| Gender: Male vs. female | 1.08 (0.97–1.20) | 0.16 | NA | NA | NA | NA |
|
|
|
|
||||
| Age: Continuous variable | 1.03 (1.02–1.04) | 1.4×10−6 | 2.9% | 1.03 (1.02–1.04) | 8.6×10−8 | 3.4% |
|
|
|
|
||||
| Race groupb: | ||||||
| Whites vs. blacks | 1.70 (1.48–1.96) | 8.7×10−13 | 7.2% | 1.62 (1.41–1.87) | 3.7×10−11 | 5.3% |
| Hispanic vs. blacks | 1.77 (1.43–2.20) | 2.6×10−7 | 1.57 (1.26–1.95) | 6.6×10−5 | ||
| Others vs. blacks | 1.52 (1.21–1.90) | 3.2×10−4 | 1.41 (1.14–1.76) | 2.0×10−3 | ||
|
|
|
|
||||
| Protocol: Total XVI vs. Total XV | 1.11 (1.00–1.24) | 0.041 | 0.49% | 1.12 (1.01–1.24) | 0.025 | 0.58% |
|
|
|
|
||||
| BMI percentile: Continuous variable | 1.29 (1.09–1.52) | 2.9×10−3 | 1.3% | 1.28 (1.09–1.50) | 2.7×10−3 | 1.0% |
|
|
|
|
||||
| PNPLA3 rs738409 genotype: Numeric variablec | NA | NA | NA | 1.29 (1.18–1.40) | 1.2×10−8 | 3.8% |
The effect size refers to the exponent of the beta coefficient estimated from the linear regression. Numbers in parentheses indicate 95% confidence interval.
Whites were defined as having > 90% Northern European ancestry, blacks as having > 70% West African ancestry, Hispanics as having Native American ancestry greater than 10% and Native American ancestry was greater than their percent African ancestry, and others were those whose ancestry was outside the above boundaries.
Genotype of SNP was treated as numeric variable, which equals to the number of minor allele in the genotype of each patient.
NA, not applicable
To identify genetic variants associated with post-induction ALT levels in the SJCRH discovery cohort, we used a multivariate linear regression model including treatment protocol, use of additional asparaginase doses, percentage of Northern European ancestry, West African ancestry and Native American ancestry, age, and BMI percentile as covariates along with genotypes. The strongest association (P = 2.5×10−8) with ALT levels was with the PNPLA3 rs738409 (C>G) I148M variant (Figure 2A, Table 4), a missense polymorphism known as a quantitative trait locus (QTL) for ALT in the general population.(10) The median ALT level for patients of CC, GC, and GG diplotypes was 35, 45, and 58 IU/L, respectively. When ALT was translated into CTCAE categories of ALT toxicity, the frequency of higher ALT grade (grade > 2) was 2.6%, 3.9% and 5.3% respectively among patients of CC, GC, and GG diplotypes (Figure S2B). The association of PNPLA3 rs738409 with ALT treated as a continuous variable was observed in both Total XV (P = 5.5×10−6) and Total XVI (P = 2.7×10−4) (Figure S3A), and differed within race groups (Figure 3A, Figure S3B). Interestingly, the minor allele frequency (MAF) of PNPLA3 rs738409 was highest among Hispanics (44.2%), followed by whites (23.1%), and lowest in blacks (13.1%) in our cohort, as observed in previous studies,(11) and followed the same trend as their post-induction ALT levels (Figure 3B). Principal component analysis (PCA) within each race group did not uncover any stratification among the patients carrying the different PNPLA3 rs738409 genotypes (Figure S4A–C).
Figure 2. Manhattan plots of inverse P values for genome-wide SNP associations with ALT levels.

(A) Association between SNPs and natural logarithm ALT levels versus each chromosome for Total XV and XVI patients (N = 715). The analysis identified a variant in PNPLA3 (rs738409) associated with ALT levels. (B) Association between SNPs and ALT levels versus each chromosome for black patients in Total XV and XVI patients (n = 111). A variant near PIGV and a variant in 3′-UTR of GREB1 were associated with ALT levels with genome-wide significance.
Table 4.
Top 20 variants associated with post-induction ALT in the combined SJCRH Total XV and Total XVI cohort.
| rsID | Hg19 position | Major allelea | Minor allelea | Platform | P value | Effect sizeb | Confidence interval | Gene | Function annotation |
|---|---|---|---|---|---|---|---|---|---|
| rs738409 | 22:44324727 | C | G | exomechip | 2.52E-08 | 1.29 | 1.04–1.58 | PNPLA3 | missense |
| rs144104656 | 2:119418867 | A | G | imputed | 1.60E-07 | 8.04 | 1.36–47.52 | EN1 | intergenic |
| rs2294915 | 22:44340904 | C | T | imputed | 5.72E-07 | 1.26 | 1.03–1.53 | PNPLA3 | intronic |
| rs147481775 | 22:32225363 | C | A | imputed | 5.77E-07 | 4.66 | 1.22–17.88 | DEPDC5 | intronic |
| rs74709575 | 13:38615256 | A | C | imputed | 1.23E-06 | 4.52 | 1.19–17.19 | TRPC4 | intergenic |
| rs17857135 | 17:78262161 | T | C | exomeseq | 1.68E-06 | 0.78 | 0.63–0.97 | RNF213 | missense |
| rs112769843 | 12:62308665 | C | G | imputed | 1.76E-06 | 0.54 | 0.32–0.94 | FAM19A2 | intronic |
| rs11696756 | 20:22982087 | G | A | imputed | 2.59E-06 | 1.39 | 1.03–1.86 | SSTR4 | intergenic |
| rs12530134 | 6:170919470 | G | A | exomechip | 3.37E-06 | 1.36 | 1.03–1.80 | PDCD2 | intergenic |
| rs117483095 | 9:132784775 | C | T | imputed | 3.43E-06 | 1.62 | 1.05–2.50 | FNBP1 | intronic |
| rs113941845 | 2:239501832 | C | A | imputed | 4.01E-06 | 1.24 | 1.02–1.51 | ASB1 | intergenic |
| rs145145722 | 7:17243557 | T | C | imputed | 4.03E-06 | 0.42 | 0.19–0.92 | AHR | intergenic |
| rs11020478 | 11:93409448 | A | G | imputed | 4.30E-06 | 1.21 | 1.02–1.43 | KIAA1731 | intronic |
| rs1348850 | 2:178418575 | G | A | exomechip | 4.31E-06 | 1.22 | 1.02–1.46 | TTC30B | intergenic |
| rs7024024 | 9:92210595 | A | T | imputed | 4.37E-06 | 0.82 | 0.68–0.98 | GADD45G | intergenic |
| rs149940960 | 2:11781618 | T | C | exomeseq | 4.41E-06 | 0.16 | 0.03–0.85 | GREB1 | 3′-UTR |
| rs143969105 | 12:58201465 | A | G | exomeseq | 4.48E-06 | 8.68 | 1.21–62.08 | TSFM | missense |
| rs75870332 | 14:27753539 | T | A | imputed | 4.88E-06 | 1.51 | 1.04–2.19 | intergenic | |
| rs58800477 | 6:20759577 | A | G | imputed | 4.94E-06 | 4.10 | 1.13–14.91 | CDKAL1 | intronic |
| rs738409 | 22:44324727 | C | G | exomechip | 2.52E-08 | 1.29 | 1.04–1.58 | PNPLA3 | missense |
Alleles were assigned as major or minor ones based on their frequency in the cohort of our study.
The effect size refers to the exponent of the beta coefficient of the minor allele estimated from the linear regression in PLINK. An effect size greater than 1 indicates the minor allele is the risk allele.
Figure 3. Post-induction ALT levels varied by genotype in 715 SJCRH Total XV and XVI patients.
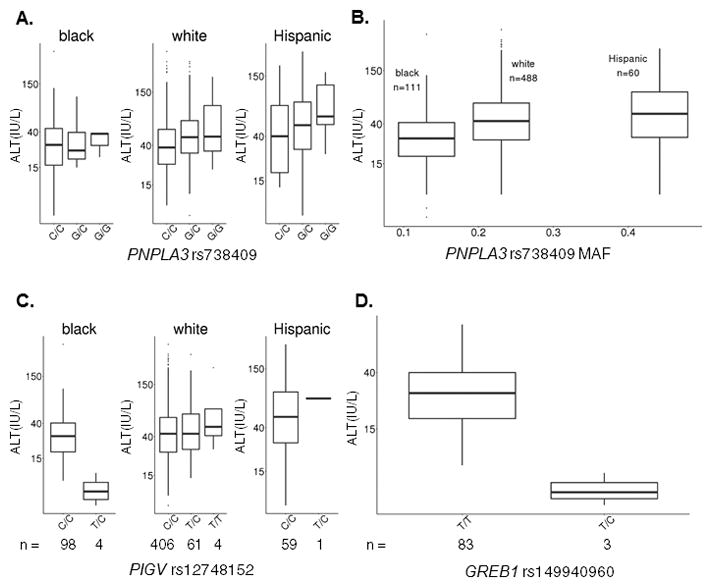
(A) Boxplots of ALT levels by PNPLA3 rs738409 genotype in blacks (n = 111, P = 0.20), whites (n = 488, P = 1.5×10−6), and Hispanics (n = 60, P = 0.058) from the SJCRH discovery cohort. (B) Boxplots of ALT levels by PNPLA3 rs738409 MAF in blacks, whites and Hispanics from the SJCRH discovery cohort. (P values of whites and Hispanics vs. blacks are 3.3×10−10 and 3.3×10−7 respectively in univariate analysis.) (C) Boxplots of ALT levels by PIGV rs12748152 genotype in blacks (n = 102, P = 1.3×10−8), whites (n = 471, P = 0.14), and Hispanics (n = 60, P = 0.59) from the SJCRH discovery cohort. (D) Boxplots of ALT levels by GREB1 rs149940960 genotype in blacks (n = 86, P = 2.0×10−8) from the SJCRH discovery cohort. The boxes depict median with upper and lower quartiles.
Because the impact of PNPLA3 rs738409 on ALT elevation differed by race (Figure 3A, Figure S3B), and because ALT levels differed among races in the same direction that the PNPLA3 rs738409 MAF did (Figure 3B), we questioned whether the association between race and ALT elevation was accounted for mostly by this variant. After adding PNPLA3 rs738409 genotype to a multivariate analysis for determinants of ALT levels, the PNPLA3 SNP accounted for 3.8% of ALT variability, and race group still remained an important risk factor (P = 3.7×10−11, 6.6×10−5 and 2.0×10−3 for whites, Hispanics and others vs. blacks, respectively), and explained 5.3% of the variability in ALT elevation after incorporating the PNPLA3 SNP (Table 3). Therefore, we performed three separate ancestry-specific GWAS among whites, blacks and Hispanics (Tables S2–S4) to identify additional possible race-specific genetic determinants of ALT, including the percentage of West African and Native American ancestries as covariates in the GWASs within blacks and Hispanics, respectively. Two variants, PIGV rs12748152 and GREB1 rs149940960, had P values approaching genome-wide significance and only in blacks (P = 1.3×10−8 and 2.0×10−8 respectively, Figure 2B, 3C&D). Coincidentally, the three subjects harboring the minor allele of GREB1 rs149940960 also had the minor allele of PIGV rs12748152. Both variants were associated with lower ALT only in blacks (P = 0.14 in whites and P = 0.59 in Hispanics for PIGV rs12748152, Figure 3C; the GREB1 rs149940960 was detected in blacks only). The PIGV rs12748152 had a higher MAF in whites (7.1%) than blacks (1.8%) and Hispanics (0.8%), whereas the GREB1 rs149940960 had MAF = 1.7% in blacks, in accordance with the general population frequency (1000 Genomes). In addition, when race structure was calculated with PCA and the first five principal components were used as covariates in each race-specific GWAS, the top SNPs and P values for association with ALT did not change substantially (Tables S2–S4), and PCA within blacks did not uncover any stratification among the patients carrying the different PIGV rs12748152 genotypes (Figure S4D). PNPLA3 rs6006460 (G>T) was previously reported to protect blacks from the development of nonalcoholic fatty liver disease (NAFLD).(12) The PNPLA3 rs6006460 minor allele was among the protective variants in our discovery cohort as well (P = 3.2×10−3, Figure S5A), and the effect was significant when restricted only to blacks in our cohort (P = 0.041, Figure S5B). The frequency of this protective variant was highest in the blacks of our cohort (MAF = 11%) and thus may have counteracted the adverse effects of carrying the risk allele of PNPLA3 rs738409, therefore helping to explain why PNPLA3 rs738409 was not significantly associated with increased ALT among blacks (P = 0.20, Figures 3A&S3B). In blacks, the PNPLA3 rs738409 explained 1.3% of the variability in ALT level (P = 0.065) while PNPLA3 rs6006460 explained another 5.8% (P = 0.014). In contrast, among non-blacks in the discovery cohort, PNPLA3 rs738409 explained 4.2% of the variability whereas PNPLA3 rs6006460 added only 1.2% (P < 2.2×10−16 for both).
A total of 109 variants within the PNPLA3 gene were found in the SJCRH cohort, 65 of which had nominal P values of < 0.05 for their association with ALT, with 61/65 having the minor allele associated with higher ALT (Table S5). A gene-level GWAS including the same covariates as the single SNP GWAS (protocol, asparaginase dosage, percent ancestry, age, and BMI percentile) identified PNPLA3 as an overall detrimental gene for ALT levels (P = 0.0093), reflecting the collective impact of PNPLA3 variants.
In the replication cohort AALL0232, ALT elevation was classified into grade > 2 vs. grade ≤ 2. Accounting for the significant covariates of age, race, gender, and the induction steroid (Table S6), PNPLA3 rs738409 was associated with the frequency of grade > 2 ALT elevations in the entire AALL0232 cohort (P = 0.024) (Figure S6) and when the analysis was limited to Hispanics (P = 0.01), thus independently replicating the polymorphism’s association.
Because the PNPLA3 variant rs738409 has been previously linked to elevated ALT levels and to the development of hepatic steatosis in the general population,(13) its association with elevated ALT after remission induction therapy suggested that asparaginase may influence blood ALT levels through dysregulation of hepatic lipids. We confirmed that PEGylated E. coli asparaginase caused hepatic steatosis in mice, as evidenced by a doubling in Oil-red O (ORO) staining in livers of mice treated with the drug compared to control (Figure S7).
Discussion
Patients with ALL are at increased risk for the development of toxicities, including hepatotoxicity, which is partly reflected by elevated serum aminotransferase levels.(3) Our objectives were to use a genome-wide approach to determine inherited variation associated with ALT elevation among a racially diverse set of pediatric patients with ALL at a uniform time point (the end of remission induction), adjusting for clinical and other covariates potentially associated with increased ALT levels. Although many ALL medications can contribute to elevations in serum ALT, our multivariate analysis suggested that elevated post-induction chemotherapy ALT levels were linked to asparaginase exposure (Figures 1A&B, Table 3). Patients on Total XVI (who all received PEGylated E. coli asparaginase) had higher ALT levels compared to the Total XV patients (who all received the less potent native E. coli asparaginase) (P = 0.041, Figure 1A, Table 3), despite the fact that patients on Total XV received pre-induction high-dose methotrexate whereas those on Total XVI did not (Table 1, Figure S9), suggesting an effect of asparaginase preparation on hepatotoxicity. Moreover, those who received additional asparaginase during induction had higher ALT levels compared to those that did not receive additional asparaginase (P = 2.7×10−4, Figure 1B, Table 3). Interestingly, serum bilirubin at end of induction did not differ by asparaginase exposure (data not shown). Adjusting for treatment, host-related covariates (e.g., age, BMI percentile) and ancestry, we found that the PNPLA3 rs738409 I148M variant associated with higher ALT levels. This is the identical genetic variant that has previously been linked to ALT elevation in the general population,(10) non-alcoholic fatty liver disease (NAFLD),(12, 14, 15) NAFLD severity,(16) and hepatic fibrosis(15) in genome-wide studies. The effect on NAFLD susceptibility and severity was confirmed by meta-analyses.(17) This variant was also associated with poor chronic hepatitis C treatment response,(18) hepatocellular carcinoma,(19) and insulin resistance.(20) Overall in the discovery cohort of 715 patients, the median increase in ALT was 12 IU/L, or 33.3% of the median of the major allele homozygous (C/C) group, for each minor allele of PNPLA3 (P = 0.032), compared to a 6.0% increase for each allele observed in the general population,(10) suggesting that the effect of the variant is amplified during ALL therapy, and consistent with larger effect sizes for pharmacogenetic traits.(21)
Because of hepatic infiltration of ALL at diagnosis and the prolonged use of potentially hepatotoxic chemotherapy, there is no optimal time to measure “baseline” ALT in ALL.(22) We did examine ALT levels at diagnosis within the SJCRH cohort and showed that serum ALT levels were lower at diagnosis than at end-of-induction (P < 2.2×10−16, paired t-test, Figure S8, Table 2), and that PNPLA3 rs738409 remains a significant risk factor (P = 4.4×10−6) for post-induction ALT elevation even after including diagnostic ALT as a covariate.
Patatin-like Phospholipase Domain Containing Protein 3 (PNPLA3 or adiponutrin) is a member of the family of patatin-like lipolytic enzymes involved in triacylglycerol metabolism and signaling.(23) It has been shown that substituting methionine for isoleucine at position 148 markedly compromised the catalytic velocity of PNPLA3.(24) This is reflected in knock-in mice studies suggesting that the PNPLA3 I148M variant leads to an increased risk of hepatotoxicity due to accumulation of PNPLA3 on lipid droplets in the liver, a reduction in triglyceride hydrolase activity, and accumulation of hepatic triglycerides.(25) Our finding that the top ranked genetic variation associated with drug-induced liver dysfunction after ALL therapy is identical to one of the top genetic risk factors for constitutive liver dysfunction is remarkable, and has implications for the field of pharmacogenetics and its use to predict drug toxicities.
We confirmed that asparaginase exposure in mice indeed led to an accumulation of hepatic lipids and neutral triglycerides (Figure S7). The results suggest that the mechanism of asparaginase-induced hepatotoxicity may be similar to that of hepatic steatosis, and may be attenuated by therapies that can mitigate hepatic triglyceride accumulation. These results may have particular implications for adults with ALL, in whom hepatotoxicity is common.(26) Interestingly, an early postmortem study from 31 pediatric ALL patients that received native E. coli asparaginase found that 87% had developed steatosis after exposure to asparaginase.(27)
We observed that ALT was higher (Table 3, Table S6) in those with ancestry indicating Hispanic status or whites than in blacks, as others have reported.(11) Differences in the risk allele frequency of the PNPLA3 rs738409 variant mirrored the differences in ALT among race groups (Figure 3B), although this variant did not reach statistical significance in blacks or Hispanics (Figures 3A&S3B), likely due to relatively low power because of small sample sizes for non-whites (with an effect size of 1.36 for the rs738409 variant, our power to detect a significant effect in separate GWASs of blacks and Hispanics was only 0.32 and 0.37, respectively). Although the PNPLA3 rs738409 variant accounted for some variability in ALT across race groups, race remained a significant risk factor for ALT even after accounting for PNPLA3 rs738409.
We found that some of the additional association of race with ALT might be due to other genetic variants. For example, black patients carrying the PIGV rs12748152 and GREB1 rs149940960 variants had lower ALT levels compared to non-carriers (Figure 3C&D, Table S3). PIGV rs12748152 is an expression quantitative trait locus (eQTL) for PIGV, ARID1A and ZDHHC18,(28, 29) and is also a QTL for cholesterol and triglycerides levels,(30) whereas GREB1 rs149940960 is in the 3′ untranslated region (3′-UTR) of GREB1, which regulates circulating levels of chemoattractant protein-1 (CCL2) and insulin-like growth factor binding protein 3 (IGFBF-3).(31) High levels of the latter are associated with adiposity and insulin resistance(32) while the former is a cytokine that recruits immune cells into damaged areas of the liver.(33) Reports suggest that infiltrating immune cells directly kill distressed hepatocytes overloaded with triglycerides. Preventing this infiltration with NF-κB inhibitors may be another possible avenue of intervention.(34) Although it has not been implicated in NAFLD, PIGV is involved in the biosynthesis of glycosylphosphatidylinositol. The PIGV rs12748152 variant explained 26.6% of the variability in ALT levels among black patients. However, there was no significant association in those with low West African ancestry (P = 0.27), and the variant was actually present at a lower frequency among blacks than other race groups (MAF = 2.0% in blacks, 13.3% in non-blacks). Limitations of these speculations lie in the small sample size of blacks in our cohort and low MAF of these genetic variants among them.
Although we think asparaginase was the most likely cause of post-induction ALT elevations in this study, we acknowledge that other elements of ALL induction therapy can also cause increases in ALT. Interestingly, high-dose methotrexate is well-known to cause acute elevations in ALT,(35) but this was unlikely to have contributed to the end-of-induction ALT (which was measured 42–46 days after a single dose of methotrexate for Total XV), and because high-dose methotrexate was used prior to induction only in Total XV, not Total XVI (Table 1, Figure S9), and yet ALT was higher in patients enrolled on Total XVI compared to Total XV (Figure 1A). Likewise, although thiopurines can cause elevations in ALT, they are more prominent with mercaptopurine (used in Total XV) than with thioguanine (used in Total XVI) (Table 1),(36, 37) and thus unlikely to explain the higher ALT on Total XVI compare to Total XV. The use of PEG-asparaginase at 3000 units/m2 on Total XVI results in much higher and more prolonged serum asparaginase concentrations than the doses of native E. coli asparaginase that we used on Total XV.(38) Most tellingly, in both Total XV and Total XVI, ALT was higher in the 25% of patients who required extra doses of asparaginase due to high MRD during induction therapy than it was in those who did not receive extra doses (Figure 1B).
The association of PNPLA3 rs738409 with ALT after induction therapy was replicated in the COG AALL0232 protocol. The replication is somewhat remarkable in that there were several important factors that differed between the discovery and replication cohorts. First, patients on AALL0232 were older than those in the SJCRH discovery cohort (Table 2), and, as expected, exhibited higher ALT grades. Second, discrete ALT values were not available for the AALL0232 cohort, only CTCAE grades; and very few cases of grade 1 and 2 ALT elevation were recorded. Thus, we used linear regression in the discovery cohort but used case (grade > 2)-control (grade ≤ 2) logistic regression for the replication cohort (Figure S6). Finally, BMI was not available for patients in AALL0232. Despite these differences, the effect of PNPLA3 rs738409 variant was still replicated.
In summary, we have identified that elevated post-induction chemotherapy ALT levels were likely due primarily to asparaginase, and we found that the same PNPLA3 I148M variant that was previously shown to be the most important determinant of NAFLD in a general population of adults is associated with drug-induced increases in serum ALT. Together, our genetic data from patients with ALL and our preclinical data in mice suggest that hepatotoxicity caused by ALL remission-induction chemotherapy may be mediated by induction of fatty liver and that the toxicity can be modulated by inherited germline polymorphisms that affect hepatic triglyceride homeostasis.
Materials and Methods
Patients
The discovery cohort consisted of patients enrolled on SJCRH protocols Total XV(39) (n = 373, ClinicalTrials.gov identifier: NCT00137111) and Total XVI(40) (n = 342, ClinicalTrials.gov identifier: NCT00549848) for newly diagnosed ALL. The replication cohort consisted of children with newly diagnosed ALL who were treated on the COG AALL0232 protocol (N = 2,285, ClinicalTrials.gov identifier: NCT00075725) for high-risk B-precursor ALL. Patients included in the genetic association analyses represented 85% (n = 715 of 840) of patients in the SJCRH discovery cohort, and 80% (n = 2285 of 2868) of participants in the COG replication cohort (Figure S1).
Informed consent from the parents or guardians or patients, and assent from the patients, where appropriate, were obtained according to guidelines used at the Institutional Review Boards of the participating institutions. The COG AALL0232 protocol was also approved by the National Cancer Institute.
For the SJCRH cohort, ALT values were graded using CTCAE version 3.0.(41) ALT values between 1.0 ~ 2.5 fold of normal upper limit were assigned as grade 1, between 2.5 ~5.0 fold as grade 2, between 5.0 ~20.0 fold as grade 3, and > 20.0 fold as grade 4. Patient BMI at the time of ALT sample collection was calculated and BMI percentile was determined as described previously with obesity defined as ≥ 95th percentile.(42) For the COG AALL0232 cohort, ALT values were graded using the CTCAE version 4.0. ALT values between 1.0 ~ 3.0 fold of normal upper limit were assigned as grade 1, between 3.0 ~5.0 fold as grade 2, between 5.0 ~20.0 fold as grade 3, and > 20.0 fold as grade 4.
Treatment
During induction therapy, patients enrolled on the SJCRH Total XV or Total XVI protocols received asparaginase, prednisone, vincristine, daunorubicin, cyclophosphamide, cytarabine, and thiopurine (Table 1, Figure S9).(39, 40) Total XV patients received native E. coli asparaginase (Elspar) at 10,000 units/m2 IM, three times weekly for six doses and Total XVI patients received a single dose of PEGylated E. coli asparaginase (Oncaspar) at 3,000 units/m2 IV. Patients on Total XV with day 19 MRD ≥ 5% received three additional doses of native E. coli asparaginase over a week’s period, and patients on Total XVI with day 15 MRD ≥ 1% received an additional dose of PEGylated E. coli asparaginase (Table 1, Figure S9). Post-induction samples for ALT and total bilirubin measurement were drawn on day 46 (Total XV) and on day 42 (Total XVI) from the start of remission induction therapy, regardless of clinical suspicion of hepatotoxicity (Figure S9). ALT and total bilirubin were also measured at diagnosis (pre-induction therapy), before the patients were exposed to methotrexate or asparaginase (Figure S9).
During induction therapy, patients enrolled on COG AALL0232 received vincristine, daunorubicin, dexamethasone or prednisone, and PEGylated E. coli asparaginase.(43) Patients with day 29 MRD between 1% and 25% received extended induction consisting of additional doses of vincristine, daunorubicin, dexamethasone or prednisone, and PEGylated E. coli asparaginase. During consolidation, patients received cytarabine, vincristine, cyclophosphamide, mercaptopurine, and PEGylated E. coli asparaginase (Table S7). Measurement of ALT levels was a required observation on COG AALL0232 on day 1 of induction and at the start of consolidation and was performed at other times at the discretion of the treating physician. Sites were required to report all grade 3 and higher ALT elevation.
Genotyping
Germline DNA was genotyped using the Exome-24 BeadChip (Illumina, San Diego, CA) (689 patients in SJCRH cohort and 2,382 in AALL0232 trial) and either the Affymetrix Genome-Wide Human SNP Array 6.0 or the Affymetrix Human Mapping 500K Array Set (839 in SJCRH cohort and 2,666 for AALL0232 trial), as described previously.(44) MaCH-Admix (http://www.unc.edu/~yunmli/MaCH-Admix) was used to impute the remaining untyped SNPs using the 1000 Genomes Project (http://www.1000genomes.org) as described elsewhere.(45, 46) Germline DNA from 552 patients from the SJCRH cohort was also subjected to whole exome sequencing. For this, genomic libraries were prepared using the NimbleGen SeqCap EZ Exome Enrichment Kit v2.0 (Roche NimbleGen, Madison, WI) and sequencing was performed using the HiSeq 2000 Sequencing System (Illumina, San Diego, CA). The Hardy-Weinberg equilibrium (HWE) test was performed using PLINK on SNPs with a MAF ≥ 1% and among patients of European ancestry. SNPs with genotyping call rates < 95% and SNPs that were not in HWE (P values < 0.001) were excluded from the association analysis (Figure S1).
Genetic Ancestry
The genetic ancestry of patients was determined using STRUCTURE (http://pritchardlab.stanford.edu/structure.html) as previously described,(47) and percent ancestry was treated as a continuous variable or used to assign patients to race groups: whites were defined as having > 90% Northern European ancestry, blacks as having > 70% West African ancestry, Hispanics as having Native American ancestry > 10% and greater than their African ancestry percentage, Asians as having > 90% East Asian ancestry, and others as those whose ancestry was outside the above boundaries. In the SJCRH cohort, patients of Asian ancestry were grouped with others due to their small sample size.
Preclinical In vivo Asparaginase-induced Hepatotoxicity Studies
Male Balb/cJ mice (4-week-old) (Jackson Laboratories, Bar Harbor, ME) received 1,500 IU/kg of PEGylated E. coli asparaginase by intraperitoneal injection twice weekly for 6 weeks. Livers were collected at the end of the experiment and snap frozen in liquid nitrogen. Frozen livers were immersed in OCT media and 4 μm sections were stained with Oil-red O. The staining intensity was quantified using the Aperio Image analysis system (Aperio, Vista, CA). Three 1 mm2 square regions for each sample were analyzed and the number of strongly positive pixels in the regions was used to compare experimental and control samples. All experiments involving the use of mice were approved by the Institutional Animal Care and Use Committee (IACUC) of St. Jude Children’s Research Hospital. Mice were housed in an American Association of Laboratory Animal Care-accredited facility. The study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH).
Data Analysis
Univariate and multivariate linear regression was performed using a general linear model with R statistical software (version 2.13.2) to identify clinical risk factors associated with post-induction ALT levels as a continuous variable. For the COG AALL0232 replication cohort, associations between clinical risk factors and the frequency of grade > 2 hepatotoxicity were determined using Fisher’s exact test in R. The genome-wide association analysis between SNP genotypes and ALT was performed in PLINK 1.9 (https://www.cog-genomics.org/plink2) using linear or logistic regression as appropriate, adjusting for significant covariates and assuming an additive genetic model.(48) Within each race group defined above (whites, blacks, and Hispanics), the first five principal components were calculated using GCTA version 1.26.0 (http://cnsgenomics.com/software/gcta/) with genotype data acquired from the Affymetrix arrays. Gene-level association was done using the sequence kernel association test (SKAT) as implemented by PLINK/SEQ (https://atgu.mgh.harvard.edu/plinkseq/). Linkage disequilibrium based variant pruning was performed using PLINK. To assess the proportion of variability in ALT accounted for by specific genomic variants, a general linear model was used.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
Hepatotoxicity is a frequent complication leading to treatment interruptions, and several drugs used in ALL remission induction are associated with the development of hepatotoxicity, including asparaginase. There are no prior genome-wide association studies for hepatotoxicity in patients with ALL.
What question did this study address?
This study evaluated patient- and treatment- related variables associated with hepatotoxicity in two ancestrally diverse groups of pediatric ALL patients, using ALT levels measured at the end of remission induction as an indicator. The study identified genetic variants that contribute to the risk of hepatotoxicity on a genomic level.
What this study adds to our knowledge?
PNPLA3 rs738409 reached genome-wide significance as a risk factor of hepatotoxicity in patients that completed ALL remission induction. Asparaginase, especially in its PEGylated preparation, is likely a major contributor to higher ALT levels during this treatment phase.
How this might change clinical pharmacology or translational science?
The study accentuated asparaginase hepatotoxicity during ALL induction, and identified a pharmacogenetic risk factor in this setting, PNPLA3 rs738409, which overlaps with that for non-alcoholic fatty liver disease.
Acknowledgments
This study was supported by grants GM 1152579, CA 21765, CA 156449, CA 142665, CA 36401, U10 CA98543 (COG Chair’s grant), U10 CA98413 (COG Statistical Center), and U24 CA114766 (COG Specimen Banking) from the National Cancer Institute, and by the American Lebanese Syrian Associated Charities. S.P.H is the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics at the Children’s Hospital of Philadelphia.
Footnotes
Conflict of Interest
The authors have no conflicts of interest to disclose.
Authorship Contribution
M.V.R., Y.L., C.A.F., C.S., W.Y., C.C., C.P., N.K., C.L., L.R., S.E.K., L.J.J., E.C.L., N.W., W.L.C., M.L.L., E.A.R., S.P.H., M.D., J.J.Y., C.G.M., J.Z., W.E.E., S.J., and C-H.P. wrote the manuscript; M.V.R., Y.L., and C.A.F. designed the research; Y.L. and C.A.F. performed the research; M.V.R., Y.L., C.A.F., C.S., W.Y., and C.C. analyzed the data.
References
- 1.Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(27):2938–48. doi: 10.1200/JCO.2014.59.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91–01. Blood. 2001;97(5):1211–8. doi: 10.1182/blood.v97.5.1211. [DOI] [PubMed] [Google Scholar]
- 3.Schmiegelow K, Pulczynska M. Prognostic significance of hepatotoxicity during maintenance chemotherapy for childhood acute lymphoblastic leukaemia. British journal of cancer. 1990;61(5):767–72. doi: 10.1038/bjc.1990.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adam de Beaumais T, Fakhoury M, Medard Y, Azougagh S, Zhang D, Yakouben K, et al. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. British journal of clinical pharmacology. 2011;71(4):575–84. doi: 10.1111/j.1365-2125.2010.03867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ladas EJ, Kroll DJ, Oberlies NH, Cheng B, Ndao DH, Rheingold SR, et al. A randomized, controlled, double-blind, pilot study of milk thistle for the treatment of hepatotoxicity in childhood acute lymphoblastic leukemia (ALL) Cancer. 2010;116(2):506–13. doi: 10.1002/cncr.24723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrow AC, Buchanan GR, Zwiener RJ, Bowman WP, Winick NJ. Serum aminotransferase elevation during and following treatment of childhood acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15(4):1560–6. doi: 10.1200/JCO.1997.15.4.1560. [DOI] [PubMed] [Google Scholar]
- 7.Cooper SL, Brown PA. Treatment of pediatric acute lymphoblastic leukemia. Pediatric clinics of North America. 2015;62(1):61–73. doi: 10.1016/j.pcl.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stock W, Douer D, DeAngelo DJ, Arellano M, Advani A, Damon L, et al. Prevention and management of asparaginase/pegasparaginase-associated toxicities in adults and older adolescents: recommendations of an expert panel. Leukemia & lymphoma. 2011;52(12):2237–53. doi: 10.3109/10428194.2011.596963. [DOI] [PubMed] [Google Scholar]
- 9.Gokbuget N, Hoelzer D. High-dose methotrexate in the treatment of adult acute lymphoblastic leukemia. Annals of hematology. 1996;72(4):194–201. doi: 10.1007/s002770050160. [DOI] [PubMed] [Google Scholar]
- 10.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nature genetics. 2011;43(11):1131–8. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2013;11(9):1183–90. e2. doi: 10.1016/j.cgh.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature genetics. 2008;40(12):1461–5. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS genetics. 2011;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Human genetics. 2013;132(7):783–92. doi: 10.1007/s00439-013-1294-3. [DOI] [PubMed] [Google Scholar]
- 16.Kawaguchi T, Sumida Y, Umemura A, Matsuo K, Takahashi M, Takamura T, et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PloS one. 2012;7(6):e38322. doi: 10.1371/journal.pone.0038322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883–94. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 18.Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology. 2011;53(3):791–9. doi: 10.1002/hep.24123. [DOI] [PubMed] [Google Scholar]
- 19.Corradini SG, Burza MA, Molinaro A, Romeo S. Patatin-like phospholipase domain containing 3 sequence variant and hepatocellular carcinoma. Hepatology. 2011;53(5):1776. doi: 10.1002/hep.24244. author reply 7. [DOI] [PubMed] [Google Scholar]
- 20.Wang CW, Lin HY, Shin SJ, Yu ML, Lin ZY, Dai CY, et al. The PNPLA3 I148M polymorphism is associated with insulin resistance and nonalcoholic fatty liver disease in a normoglycaemic population. Liver international : official journal of the International Association for the Study of the Liver. 2011;31(9):1326–31. doi: 10.1111/j.1478-3231.2011.02526.x. [DOI] [PubMed] [Google Scholar]
- 21.Maranville JC, Cox NJ. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. The pharmacogenomics journal. 2015 doi: 10.1038/tpj.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segal I, Rassekh SR, Bond MC, Senger C, Schreiber RA. Abnormal liver transaminases and conjugated hyperbilirubinemia at presentation of acute lymphoblastic leukemia. Pediatric blood & cancer. 2010;55(3):434–9. doi: 10.1002/pbc.22549. [DOI] [PubMed] [Google Scholar]
- 23.Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. Journal of lipid research. 2009;50(Suppl):S63–8. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. The Journal of biological chemistry. 2011;286(43):37085–93. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61(1):108–18. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aldoss I, Douer D, Behrendt CE, Chaudhary P, Mohrbacher A, Vrona J, et al. Toxicity profile of repeated doses of PEG-asparaginase incorporated into a pediatric-type regimen for adult acute lymphoblastic leukemia. European journal of haematology. 2015 doi: 10.1111/ejh.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pratt CB, Johnson WW. Duration and severity of fatty metamorphosis of the liver following L-asparaginase therapy. Cancer. 1971;28(2):361–4. doi: 10.1002/1097-0142(197108)28:2<361::aid-cncr2820280215>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 28.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45(10):1238–43. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45(6):580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Global Lipids Genetics C, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, et al. Discovery and refinement of loci associated with lipid levels. Nature genetics. 2013;45(11):1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PloS one. 2012;7(12):e51954. doi: 10.1371/journal.pone.0051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Runchey SS, Boyko EJ, Ioannou GN, Utzschneider KM. Relationship between serum circulating insulin-like growth factor-1 and liver fat in the United States. Journal of gastroenterology and hepatology. 2014;29(3):589–96. doi: 10.1111/jgh.12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Degre D, Lemmers A, Gustot T, Ouziel R, Trepo E, Demetter P, et al. Hepatic expression of CCL2 in alcoholic liver disease is associated with disease severity and neutrophil infiltrates. Clinical and experimental immunology. 2012;169(3):302–10. doi: 10.1111/j.1365-2249.2012.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51(6):1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber BL, Tanyer G, Poplack DG, Reaman GH, Feusner JH, Miser JS, et al. Transient acute hepatotoxicity of high-dose methotrexate therapy during childhood. NCIMonogr. 1987:207–12. [PubMed] [Google Scholar]
- 36.Lennard L, Davies HA, Lilleyman JS. Is 6-thioguanine more appropriate than 6-mercaptopurine for children with acute lymphoblastic leukaemia? British journal of cancer. 1993;68(1):186–90. doi: 10.1038/bjc.1993.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petit E, Langouet S, Akhdar H, Nicolas-Nicolaz C, Guillouzo A, Morel F. Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicology in vitro : an international journal published in association with BIBRA. 2008;22(3):632–42. doi: 10.1016/j.tiv.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL) Oncologist. 2007;12(8):991–8. doi: 10.1634/theoncologist.12-8-991. [DOI] [PubMed] [Google Scholar]
- 39.Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–41. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pui CHCD, Sandlund JT, Bhojwani D, Evans WE, Relling MV, Jeha S. Treatment of childhood acute lymphoblastic leukemia without cranial irradiation. Annals of hematology. 2011;90(Suppl 1):S61–3. [Google Scholar]
- 41.National Cancer Institute. Cancer Therapy Evaluation Program, Common Toxicity Criteria Version 3.0. 2006 Aug; Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
- 42.Inaba H, Surprise HC, Pounds S, Cao X, Howard SC, Ringwald-Smith K, et al. Effect of body mass index on the outcome of children with acute myeloid leukemia. Cancer. 2012;118(23):5989–96. doi: 10.1002/cncr.27640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsen EC, Devidas M, Chen S, Salzer WL, Raetz EA, Loh ML, et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016 doi: 10.1200/JCO.2015.62.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nature genetics. 2009;41(9):1001–5. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu EY, Li M, Wang W, Li Y. MaCH-admix: genotype imputation for admixed populations. Genetic epidemiology. 2013;37(1):25–37. doi: 10.1002/gepi.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karol SE, Yang W, Van Driest SL, Chang TY, Kaste S, Bowton E, et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2015;126(15):1770–6. doi: 10.1182/blood-2015-05-643601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang JJ, Cheng C, Devidas M, Cao X, Fan Y, Campana D, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nature genetics. 2011;43(3):237–41. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.