

Abstract
Proteins that bind small molecules (ligands) can be used as biosensors, signal modulators, and sequestering agents. When naturally occurring proteins for a particular target ligand are not available, artificial proteins can be computationally designed. We present a protocol based on Rosetta Ligand to redesign an existing protein pocket to bind a target ligand. Starting with a protein structure and the structure of the ligand, Rosetta can optimize both the placement of the ligand in the pocket and the identity and conformation of the surrounding sidechains, yielding proteins that bind the target compound.
Keywords: Computational design, Protein/small molecule interaction, Sequence optimization, Protein design, Ligand docking
1 Introduction
Proteins which bind to small molecules (i.e. ligands) are involved in many biological processes such as enzyme catalysis, receptor signaling, and metabolite transport. Designing these interactions can produce reagents which can serve as biosensors, in vivo diagnostics, signal modulators, molecular delivery devices, and sequestering agents [1 – 5]. Additionally, the computational design of proteins which bind small molecules serves as a critical test of our understanding of the principles that drive protein/ligand interactions.
While in vitro techniques for the optimization of protein/ligand interactions have shown success [6], these are limited in the number of sequence variants which can be screened, and often require at least a modest starting affinity which to further optimize [7]. Computational techniques allow searching larger regions of sequence space and permit design in protein scaffolds with no detectable intrinsic affinity for the target ligand. Computational and in vitro techniques are often complementary and starting activity achieved via computational design can often be improved via in vitro techniques ([8] and Chapter 9 of this volume). Although challenges remain, computational design of small molecule interactions have yielded success on a number of occasions [5, 9], and further attempts will refine our predictive ability to generate novel ligand binders.
The Rosetta macromolecular modeling software suite [10, 11] has proven to be a robust platform for protein design, having produced novel protein folds [12, 13], protein/DNA interactions [14], protein/peptide interactions [15], protein/protein interactions [16], and novel enzymes [17 – 19]. Technologies for designing protein/ligand interactions have also been developed and applied [4, 8, 20]. Design of ligand binding proteins using Rosetta approaches the problem in one of two ways. One method derives from enzyme design, where predefined key interactions to the ligand are emplaced onto a protein scaffold and the surrounding context is subsequently optimized around them [8]. The other derives from ligand docking, in which the interactions with a movable ligand are optimized comprehensively [4, 20]. Both approaches have proven successful in protein redesign, and features from both can be combined using the RosettaScripts system [21], tailoring the design protocol to particular design needs.
Here we present a protocol derived from RosettaLigand ligand docking [22 – 25], which designs a protein binding site around a given small molecule ligand (Fig. 1). After preparing the protein and ligand structures, the placement of the ligand in the binding pocket is optimized, followed by optimization of sidechain identity and conformation. This process is repeated iteratively, and the proposed designs are sorted and filtered by a number of relevant structural metrics, such as predicted affinity and hydrogen bonding. This design process should be considered as part of the integrated program of computational and experimental work, where proteins designed computationally are tested experimentally and the experimental results are used to inform subsequent rounds of computational design.
Fig. 1.
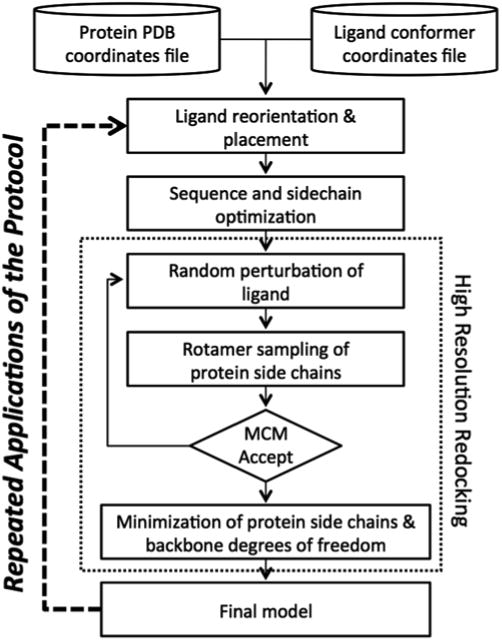
Flowchart of RosettaLigand design protocol. From the combined input coordinates of the protein and ligand, the position of the ligand is optimized. Next, residues in the protein/ligand interface are optimized for both identity and position. After several cycles of small molecule perturbation, sidechain rotamer sampling, Monte Carlo minimization with Metropolis (MCM) criterion, and a fi nal gradient-based minimization of the protein to resolve any clashes (“high resolution redocking”), the fi nal model is the output. Further optimization can occur by using the fi nal models of one round of design as the input models of the next round. Most variables in this protocol are user-defi ned, and will be varied to best fit the protein–ligand complex under study
2 Materials
A computer running a Unix-like operating system such as Linux or MacOS. Use of a multi-processor computational cluster is recommended for productions runs, although test runs and small production runs can be performed on conventional laptop and desktop systems.
Rosetta. The Rosetta modeling package can be obtained from the RosettaCommons website (https://www.rosettacommons.org/software/license-and-download). Rosetta licenses are available free to academic users. Rosetta is provided as source code and must be compiled before use. See the Rosetta Documentation (https://www.rosettacommons.org/docs/latest/) for instructions on how to compile Rosetta. The protocol in this paper has been tested with Rosetta weekly release version 2015.12.57698.
A program to manipulate small molecules. OpenBabel [26] is a free software package which allows manipulation of many small molecule file formats. See http://openbabel.org/ for download and installation information. The protocol in this paper has been tested with OpenBabel version 2.3.1. Other small molecule manipulation programs can also be used.
A ligand conformer generation program. We recommend the BCL [27] which is freely available from http://meilerlab.org/index.php/bclcommons for academic use but does require an additional license to the Cambridge Structural Database [28] for conformer generation. The protocol in this paper has been tested with BCL version 3.2. Other conformer generation programs such as Omega [29], MOE [30], or RDKit [31] can also be used.
The structure of the target small molecule in a standard format such as SDF or SMILES (see Note 1).
The structure of the protein to be redesigned, in PDB format (see Notes 2 and 3).
3 Methods
Throughout the protocol ${ROSETTA} represents the directory in which Rosetta has been installed. File contents and commands to be run in the terminal are in italics. The use of a bash shell is assumed—users of other shells may need to modify the syntax of command lines.
3.1 Pre-relax the Protein Structure into the Rosetta Scoring Function [32]
Structure from non-Rosetta sources or structures from other Rosetta protocols can have minor structural variations resulting in energetic penalties which adversely affect the design process (see Notes 4 and 5).
${ROSETTA}/main/source/bin/relax.linuxgccrelease -ignore_ unrecognized_res -ignore_zero_occupancy_false -use_input_sc -flip_HNQ -no_optH false -relax:constrain_relax_to_start_coords -relax:coord_constrain_sidechains -relax:ramp_constraints false -s PDB.pdb
For convenience, rename the output structure.
mv PDB_0001.pdb PDB_relaxed.pdb
3.2 Prepare the Ligand
- Convert the small molecule to SDF format, including adding hydrogens as needed (see Note 6).obabel LIG.smi --gen3D -O LIG_3D.sdfobabel LIG_3D.sdf -p 7.4 -O LIG.sdf
- Generate a library of ligand conformers (see Notes 7 and 8).bcl.exe molecule: ConformerGenerator-top_models 100-ensemble_filenames LIG.sdf-conformers_single_file LIG_conf.sdf
- Convert the conformer library into a Rosetta-formatted “params file” (see Notes 9 and 10).${ROSETTA}/main/source/src/python/apps/public/molfile_to_params.py -n LIG -p LIG --conformers-in-one-file LIG_conf.sdf
This will produce three files: “LIG.params”, a Rosetta-readable description of the ligand; “LIG.pdb”, a selected ligand conformer; and “LIG_conformers.pdb”, the set of all conformers (see Note 11).
3.3 Place the Ligand into the Protein (See Notes 12 and 13)
Identify the location of desired interaction pockets. Visual inspection using programs like PyMol or Chimera [33] is normally the easiest method (see Note 14). Use the structure editing mode of PyMol to move the LIG.pdb file from step 3.2.3 into the starting conformation. Save the repositioned molecule with its new coordinates as a new file (LIG_positioned.pdb) (see Note 15).
If necessary, use a text editor to make the ligand be residue 1 on chain X (see Note 16).
Using a structure viewing program, inspect and validate the placement of the ligand (LIG_positioned.pdb) in the binding pocket of the protein (PDB_relaxed.pdb) (see Note 17).
3.4 Run Rosetta Design
- Prepare a residue specification file. A Rosetta resfile allows specification of which residues should be designed and which should not. A good default is a resfile which permits design at all residues at the auto-detected interface (see Note 18).ALLAAAUTOstart1 × NATAA
-
Prepare a docking and design script (“design.xml”). The suggested protocol is based off of RosettaLigand docking using the RosettaScripts framework [22 – 25]. It will optimize the location of ligand in the binding pocket (low_res_dock), redesign the surrounding sidechains (design_interface), and refine the interactions in the designed context (high_res_dock). To avoid spurious mutations, a slight energetic bonus is given to the input residue at each position (favor_native).
<ROSETTASCRIPTS> <SCOREFXNS> <ligand_soft_rep weights=ligand_soft_rep/> <hard rep weights=ligandprime/> </SCOREFXNS> <TASKOPERATIONS> <DetectProteinLigandInterface name=design_ interface cut1=6.0 cut2=8.0 cut3=10.0 cut4=12.0 design=1 resfi le=“PDB.resfile”/> # see Note 19 </TASKOPERATIONS> <LIGAND_AREAS> <docking_sidechain chain=X cutoff=6.0 add_ nbr_radius=true all_atom_mode=true minimize_ ligand=10/> <final_sidechain chain=X cutoff=6.0 add_nbr radius=true all_atom_mode=true/> <final_backbone chain=X cutoff=7.0 add_ nbr_radius=false all_atom_mode=true Calpha_ restraints=0.3/> </LIGAND_AREAS> <INTERFACE_BUILDERS> <side_chain_for_docking ligand_areas=docking_ sidechain/> <side_chain_for_final ligand_areas=final_ sidechain/> <backbone ligand_areas=final_backbone extension_ window=3/> </INTERFACE_BUILDERS> <MOVEMAP_BUILDERS> <docking sc_interface=side_chain_for_docking minimize_water=true/> <final sc_interface=side_chain_for_final bb_ interface=backbone minimize_water=true/> </MOVEMAP_BUILDERS> <SCORINGGRIDS ligand_chain=X width=15> # see Note 20 <vdw grid_type=ClassicGrid weight=1.0/> </SCORINGGRIDS> <MOVERS> <FavorNativeResidue name=favor_native bonus= 1.00/> # see Notes 21 and 22 <Transform name=transform chain=X box_size= 5.0 move_distance=0.1 angle=5 cycles=500 repeats=1 temperature=5 rmsd=4.0/> # see Note 23 <HighResDocker name=high_res_docker cycles=6 repack_every_Nth=3 scorefxn=ligand_soft_rep movemap_builder=docking/> <PackRotamersMover name=designinterface score- fxn=hard_rep task_operations=design_inter- face/> <FinalMinimizer name=final scorefxn=hard_rep movemap_builder=final/> <InterfaceScoreCalculator name=add_scores chains=X scorefxn=hard_rep/> <ParsedProtocol name=low_res_dock> <Add mover_name=transform/> </ParsedProtocol> <ParsedProtocol name=high_res_dock> <Add mover_name=high_res_docker/> <Add mover_name=final/> </ParsedProtocol> </MOVERS> <PROTOCOLS> <Add mover_name=favor_native/> <Add mover_name=low_res_dock/> < Add mover_name=design_interface/> # see Note 24 <Add mover_name=high_res_dock/> <Add mover_name=add_scores/> </PROTOCOLS> </ROSETTASCRIPTS> - Prepare an options file (“design.options”). Rosetta options can be specified either on the command line or in a file. It is convenient to put options which do not change run-to-run (such as those controlling packing and scoring) into an options file rather than the command line.-ex1-ex2-linmem_ig 10-restore_pre_talaris_2013_behavior # see Note 25
- Run the design application (see Notes 26 and 27). This will produce a number of output PDB files (named according to the input file names, see Note 28) and a summary score file (“design_results.sc”).${ROSETTA}/main/source/bin/rosetta_scripts.linuxgccre-lease@design.options-parser:protocol design.xml -extra_ res_fa LIG.params -s “PDB_relaxed.pdb LIG_positioned.pdb” -nstruct <number of output models> -out:file:scorefile design_results.sc
3.5 Filter Designs
-
Most Rosetta protocols are stochastic in nature. The output structures produced will contain a mixture of good and bad structures. The large number of structures produced need to be filtered to a smaller number of structures taken on to the next step.
A rule of thumb is that filtering should remove unlikely solutions, rather than selecting the single “best” result. Successful designs are typically good across a range of relevant metrics, rather than being the best structure on a single metric (see Note 29).
The metrics to use can vary based on the desired properties of the final design. Good standard metrics include the predicted interaction energy of the ligand, the stability score of the complex as a whole, the presence of any clashes [34], shape complementarity of the protein/ligand interface [35], the interface area, the energy density of the interface (binding energy per unit of interface area), and the number of unsatisfied hydrogen bonds formed on binding.
- Prepare a file (“metric_thresholds.txt”) specifying thresholds to use in filtering the outputs of the design runs. IMPORTANT: The exact values of the thresholds need to be tuned for your particular system (see Note 30).req total_score value < -1010 # measure of protein stabilityreq if_X_fa_rep value < 1.0# measure of ligand clashesreq ligand_is_touching_X value > 0.5# 1.0 if ligand is in pocketoutput sortmin interface_delta_X# binding energy
- Filter on initial metrics from the docking run. This will produce a file (“filtered_pdbs.txt”) containing a list of output PDBs which pass the metric cutoffs.perl ${ROSETTA}/main/source/src/apps/public/enzdes/DesignSelect.pl -d <(grep SCORE design_results.sc) -c metric_thresholds.txt -tag_column last > filtered_designs.sc awk ‘{print $NF “.pdb”}’ filtered_designs.sc> filtered_pdbs.txt
- Calculate additional metrics (see Note 31). Rosetta's InterfaceAnalyzer [36] calculates a number of additional metrics. These can take time to evaluate, though, so are best run on only a pre- filtered set of structures. After the metrics are generated, the structures can be filtered as in steps 3.5.1 and 3.5.2. This will produce a score file (“design_interfaces.sc”) containing the calculated metric values for the selected PDBs.${ROSETTA}/main/source/bin/InterfaceAnalyzer. linuxgccrelease -interface A_X -compute_packstat -pack_ separated -score:weights ligandprime -no_nstruct_label -out:file:score_only design_interfaces.sc -l filtered_ pdbs.txt -extra_res_fa LIG.params
- Filter on additional metrics. The commands are similar to those used in step 3.5.2, but against the design_interfaces.sc score file, and with a new threshold file.perl ${ROSETTA}/main/source/src/apps/public/enzdes/DesignSelect.pl -d <(grep SCORE design_results.sc) -c metric_thresholds.txt -tag_column last > filtered_ designs.scawk ‘{print $NF “.pdb”}’ filtered_designs.sc> filtered_pdbs.txt
Example contents of metric_thresholds2.txt:
req packstat value > 0.55 # packing metric; 0-1 higher better
req sc_value value > 0.45# shape complementarity; 0-1 higher better
req delta_unsatHbonds value < 1.5# unsatisfied hydrogen bonds on binding
req dG_separated/dSASAx100 value < -0.5 # binding energy per contact area
output sortmin dG_separated# binding energy
3.6 Manually Inspect Selected Sequences
While automated procedures are continually improving and can substitute to a limited extent [37], there is still no substitute for expert human knowledge in evaluating designs. Visual inspection of interfaces by a domain expert can capture system-specific requirements that are difficult to encode into an automated filter (see Note 32).
3.7 Reapply the Design Protocol, Starting at Step 3.4
Improved results can be obtained by repeating the design protocol on the output structures from previous rounds of design. The number of design rounds depends on your system and how quickly it converges, but 3–5 rounds of design, each starting from the filtered structures of the previous one, is typical (see Note 33).
3.8 Extract Protein Sequences from the Final Selected Designs into FASTA Format
${ROSETTA}/main/source/src/python/apps/public/pdb2fasta.py $(cat final_filtered_pdbs.txt) > selected_ sequences.fasta
3.9 Iteration of Design
Only rarely will the initial design from a computational protocol give exactly the desired results. Often it is necessary to perform iterative cycles of design and experiment, using information learned from experiment to alter the design process (Fig. 2).
Fig. 2.
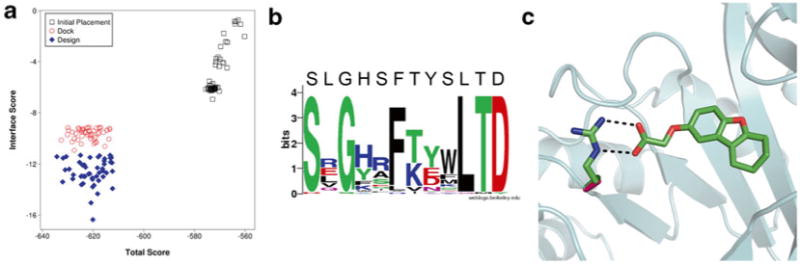
Protein/ligand interface design with RosettaLigand. (a) Comparison in improvements in Interface Score and Total Score for top models from an initial placement, docking without sequence design, and docking with design. (b) Sequence logo of mutation sites among the top models from a round of interface design [43]. For most positions, the consensus sequence resembles the native sequence. Amino acids with sidechains that directly interact with the ligand show a high prevalence to mutation as seen in the positions with decreased consensus. (c) Example of a typical mutation introduced by RosettaLigand. The protein structure is represented in cartoon (cyan). The native alanine (pink) is mutated to an arginine residue (green) to match ionic interactions with the negatively charged ligand (green). Image generated in PyMol [44]
4 Notes
While Rosetta can ignore chain breaks and missing loops far from the binding site, the structure of the protein should be complete in the region of ligand binding. If the binding pocket is missing residues, remodel these with a comparative modeling protocol, using the starting structure as a template.
Acceptable formats depend on the capabilities of your small molecule handling program. OpenBabel can be used to convert most small molecule representations, including SMILES and InChI, into the sdf format needed by Rosetta.
High resolution experimental structures determined in complex with a closely related ligand are most desirable, but not required. Experimental structures of the unliganded protein and even homology models can be used [38, 39].
The option “-relax:coord_constrain_sidechains” should be omitted if the starting conformation of the sidechains are from modeling rather than experimental results.
Rosetta applications encode the compilation conditions in their filename. Applications may have names which end with *.linuxgccrelease, *.macosclangrelease, *.linuxiccrelease, etc. Use whichever ending is produced for your system. Applications ending in “debug” have additional error checking which slows down production runs.
It is important to add hydrogens for the physiological conditions under which you wish to design. At neutral pH, for example, amines should be protonated and carboxylates deprotonated. The “-p” option of OpenBabel uses heuristic rules to reprotonate molecules for a given pH value. Apolar hydrogens should also be present.
Visually examine the produced conformers and manually remove any which are folded back on themselves or are otherwise unsuitable for being the target design conformation.
It is unnecessary to sample hydrogen positions during rotamer generation, although any ring flip or relevant heavy atom isomeric changes should be sampled.
molfile_to_params.py can take a number of options—run with the “-h” option for details. The most important ones are: “-n”, which allows you to specify a three letter code to use with the PDB file reading and writing, permitting you to mix multiple ligands; “-p”, which specifies output file naming; “--recharge”, which is used to specify the net charge on the ligand if not correctly autodetected; and “--nbr_atom”, which allows you to specify a neighbor atom (see Note 10)
Specifying the neighbor atom is important for ligands with offset “cores”. The neighbor atom is the atom which is superimposed when conformers are exchanged. By default the neighbor atom is the “most central” atom. If you have a ligand with a core that should be stable when changing conformers, you should specify an atom in that core as the neighbor atom.
LIG.params expects LIG_conformers.pdb to be in the same directory, so keep them together when moving files to a new directory. If you change the name of the files, you will need to adjust the value of the PDB_ROTAMERS line in the LIG. params file.
Rosetta expects the atom names to match those generated in the molfile_to_params.py step. Even if you have a starting structure with the ligand correctly placed, you should align the molfile_to_params.py generated structure into the pocket so that atom naming is correct.
Other methods of placing the ligand in the pocket are also possible. Notably, Tinberg et al. [8] used RosettaMatch [40] both to place the ligand in an appropriate scaffold and to place key interactions in the scaffold.
Other pocket detection algorithms can also be used (see Chapter 1 of this volume and [41] for a review).
If you have a particularly large pocket, or multiple potential pockets, save separate ligand structures at different positions and perform multiple design runs. For a large number of locations, the StartFrom mover in RosettaScripts can be used to randomly place the ligand at multiple specified locations in a single run.
Being chain X residue 1 should be the default for molfile_to_ params.py produced structures. Chain identity is important as the protocol can be used to design for ligand binding in the presence of cofactors or multiple ligands. For fixed-location cofactors, simply change the PDB chain of the cofactor to something other than X, add the cofactor to the input protein structure, and add the cofactors' params file to the -extra_res_ fa command line option. For designing to multiple movable ligands, including explicit waters, see Lemmon et al. [42].
To refine the initial starting position of the ligand in the protein, you can do a few “design” runs as in step 3.4, but with design turned off. Change the value of the design option in the DetectProteinLigandInterface tag to zero. A good starting structure will likely have good total scores and good interface energy from these runs, but will unlikely result in ideal interactions. Pay more attention to the position and orientation of the ligand than to the energetics of this initial placement docking run.
The exact resfile to use will depend on system-specific knowledge of the protein structure and desired interactions. Relevant commands are ALLAA (allow design to all amino acids), PIKAA (allow design to only specified amino acids) NATAA (disallow design but permit sidechain movement), and NATRO (disallow sidechain movement). The AUTO specification allows the DetectProteinLigandInterface task operation to remove design and sidechain movement from residues which are “too far” from the ligand.
Change the name of the resfile in the XML script to match the full path and filename of the resfile you are using. The cut values decide how to treat residues with the AUTO specification. All AUTO residues with a C-beta atom within cut1 Angstroms of the ligand will be designed, as will all residues within cut2 which are pointing toward the ligand. The logic in selecting sidechains is similar for cut3 and cut4, respectively, but with sidechain flexibility rather than design. Anything outside of the cut shells will be ignored during the design phase, but may be moved during other phases.
The grid width must be large enough to accommodate the ligand. For longer ligands, increase the value to at least the maximum extended length of the ligand plus twice the value of box_size in the Transform mover.
Allison et al. [20] found that a value of 1.0 for the FavorNativeSequence bonus worked best over their benchmark set. Depending on your particular requirements, though, you may wish to adjust this value. Do a few test runs with different values of the bonus and examine the number of mutations which result. If there are more mutations than desired, increase the bonus. If fewer than expected, decrease the bonus.
More complicated native favoring schemes can be devised by using FavorSequenceProfile instead of FavorNativeSequence. For example, you can add weights according to BLOSUM62 relatedness scores, or even use a BLAST-formatted position-specific scoring matrix (PSSM) to weight the bonus based on the distribution of sequences seen in homologous proteins.
The value of box_size sets the maximum rigid body displacement of the ligand from the starting position. The value of rmsd sets the maximum allowed root mean squared deviation from the starting position. Set these to smaller values if you wish to keep the designed ligand closer to the starting conformation, and to larger values if you want to permit more movement. These are limits for the active sampling stage of the protocol only. Additional movement may occur during other stages of the protocol.
The provided protocol only does one round of design and minimization. Additional rounds may be desired for further refinement. Simply replicate the low_res_dock, design_interface, and high_res_dock lines in the PROTOCOLS section to add additional rounds of design and optimization. Alternatively, the EnzRepackMinimize mover may be used for finer control of cycles of design and minimization (although it does not incorporate any rigid body sampling).
Refinement of the Rosetta scorefunction for design of protein/ligand interfaces is an area of current active research. The provided protocol uses the standard ligand docking scorefunction which was optimized prior to the scorefunction changes in 2013, and thus requires an option to revert certain changes. Decent design performance has also been seen with the “enzdes” scorefunction (which also requires the -restore_pre_talaris_2013 option) and the standard “talaris2013” scorefunction.
Use of a computational cluster is recommended for large production runs. Talk to your local cluster administrator for instructions on how to launch jobs on your particular cluster system. The design runs are “trivially parallel” and can either be manually split or run with an MPI-compiled version. If splitting manually, change the value of the -nstruct option to reduce the number of structures produced by each job, and use the options -out:file:prefix or -out:file:suffix to uniquely label each run. The MPI version of rosetta_scripts can automatically handle distributing structures to multiple CPUs, but requires Rosetta to be compiled and launched in cluster-specific ways. See the Rosetta documentation for details.
The Rosetta option “-s” takes a list of PDBs to use as input for the run. The residues from multiple PDBs can be combined into a single structure by enclosing the filenames in quotes on the command line. Multiple filenames not enclosed in quotes will be treated as independent starting structures.
The number of output models needed (the value passed to -nstruct) will depend on the size of the protein pocket and the extent of remodeling needed. Normally, 1000–5000 models is a good sized run for a single starting structure and a single protocol variant. At a certain point, you will reach “convergence” and the additional models will not show appreciable metric improvement or sequence differences. If you have additional computational resources, it is often better to run multiple smaller runs (100–1000 models) with slightly varying protocols (different starting location, number of rounds, extent of optimization, native bonus, etc.), rather than have a larger number of structures from the identical protocol.
Relevant metrics can be determined by using “positive controls”. That is, run the design protocol on known protein– ligand interactions which resemble your desired interactions. By examining how the known ligand–protein complexes behave under the Rosetta protocol, you can identify features which are useful for distinguishing native-like interactions from non-native interactions. Likewise, “negative controls”, where the design protocol is run without design (see Note 17) can be useful for establishing baseline metric values and cutoffs.
The thresholds to use are system-specific. A good rule of thumb is to discard at least a tenth to a quarter by each relevant metric. More important metrics can receive stricter thresholds. You may wish to plot the distribution of scores to see if there is a natural threshold to set the cut at. You will likely need to do several test runs to adjust the thresholds to levels which give the reasonable numbers of output sequences. “Negative controls” (the protocol run with design disabled, see Note 17) can also be used to determine thresholds.
Other system-specific metric values are available through the RosettaScripts interface as “Filters”. Adding “confidence = 0” in the filter definition tag will turn off the filtering behavior and will instead just report the calculated metric for the final structure in the final score file. Many custom metrics, such as specific atom–atom distances, can be constructed in this fashion. See the Rosetta documentation for details.
Certain automated protocol can ease this post-analysis. For example, Rosetta can sometimes produce mutations which have only a minor influence on binding energy. While the native bonus (see Notes 21 and 22) mitigates this somewhat, explicitly considering mutation-by-mutation reversions can further reduce the number of such “spurious” mutations seen. Nivon et al. [37] presents such a protocol.
In subsequent rounds, you will likely want to decrease the aggressiveness of the low resolution sampling stage (the box_ size and rmsd values of the Transform mover in step 3.4.2) as the ligand settles into a preferred binding orientation. As the output structure contains both the protein and ligand, the quotes on the values passed to the “-s” option (see step 3.4.4 and Note 27) are no longer needed. Instead, you may wish to use the “-l” option, which takes the name of a text file containing one input PDB per line. Each input PDB will each produce “-nstruct” models. Reduce this value such that the total number of unfiltered output structures in each round is approximately the same.
Acknowledgments
This work was supported through NIH (R01 GM099842, R01 DK097376, R01 GM073151) and NSF (CHE 1305874). RM is further partially supported by grant from the RosettaCommons.
References
- 1.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15(15):4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baeumner AJ. Biosensors for environmental pollutants and food contaminants. Anal Bioanal Chem. 2003;377(3):434–445. doi: 10.1007/s00216-003-2158-9. [DOI] [PubMed] [Google Scholar]
- 4.Morin A, Kaufmann KW, Fortenberry C, Harp JM, Mizoue LS, Meiler J. Computational design of an endo-1,4-beta-xylanase ligand binding site. Protein Eng Des Sel. 2011;24(6):503–516. doi: 10.1093/protein/gzr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morin A, Meiler J, Mizoue LS. Computational design of protein-ligand interfaces: potential in therapeutic development. Trends Biotechnol. 2011;29(4):159–166. doi: 10.1016/j.tibtech.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 7.Nannemann DP, Birmingham WR, Scism RA, Bachmann BO. Assessing directed evolution methods for the generation of biosynthetic enzymes with potential in drug biosynthesis. Future Med Chem. 2011;3(7):809–819. doi: 10.4155/fmc.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, Baker D. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501(7466):212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldmeier K, Hocker B. Computational protein design of ligand binding and catalysis. Curr Opin Chem Biol. 2013;17(6):929–933. doi: 10.1016/j.cbpa.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Progress in modeling of protein structures and interactions. Science. 2005;310(5748):638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- 11.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object- oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic level accuracy. Science. 2003;302(5649):1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 13.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D. Principles for designing ideal protein structures. Nature. 2012;491(7423):222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, Baker D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010;38(16):5601–5608. doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sammond DW, Bosch DE, Butterfoss GL, Purbeck C, Machius M, Siderovski DP, Kuhlman B. Computational design of the sequence and structure of a protein-binding peptide. J Am Chem Soc. 2011;133(12):4190–4192. doi: 10.1021/ja110296z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332(6031):816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319(5868):1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453(7192):190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 19.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329(5989):309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allison B, Combs S, DeLuca S, Lemmon G, Mizoue L, Meiler J. Computational design of protein-small molecule interfaces. J Struct Biol. 2014;185(2):193–202. doi: 10.1016/j.jsb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. RosettaScripts: a scripting language interface to the rosetta macromolecular modeling suite. PLoS One. 2011;6(6):20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meiler J, Baker D. ROSETTALIGAND: protein- small molecule docking with full side-chain flexibility. Proteins. 2006;65(3):538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 23.Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385(2):381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon G, Meiler J. Rosetta Ligand docking with flexible XML protocols. Methods Mol Biol. 2012;819:143–155. doi: 10.1007/978-1-61779-465-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLuca S, Khar K, Meiler J. Fully Flexible Docking of Medium Sized Ligand Libraries with RosettaLigand. PLoS One. 2015;10(7):e0132508. doi: 10.1371/journal.pone.0132508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kothiwale S, Mendenhall JL, Meiler J. BCL∷Conf: small molecule conformational sampling using a knowledge based rotamer library. J Cheminform. 2015;7:47. doi: 10.1186/s13321-015-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58(Pt 3 Pt 1):380–388. doi: 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- 29.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50(4):572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labute P. LowModeMD--implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J Chem Inf Model. 2010;50(5):792–800. doi: 10.1021/ci900508k. [DOI] [PubMed] [Google Scholar]
- 31.Ebejer JP, Morris GM, Deane CM. Freely available conformer generation methods: how good are they? J Chem Inf Model. 2012;52(5):1146–1158. doi: 10.1021/ci2004658. [DOI] [PubMed] [Google Scholar]
- 32.Nivon LG, Moretti R, Baker D. A Pareto- optimal refinement method for protein design scaffolds. PLoS One. 2013;8(4):e59004. doi: 10.1371/journal.pone.0059004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Sheffler W, Baker D. RosettaHoles: rapid assessment of protein core packing for structure prediction, refinement, design, and validation. Protein Sci. 2009;18(1):229–239. doi: 10.1002/pro.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234(4):946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 36.Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22(1):74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nivon LG, Bjelic S, King C, Baker D. Automating human intuition for protein design. Proteins. 2014;82(5):858–866. doi: 10.1002/prot.24463. [DOI] [PubMed] [Google Scholar]
- 38.Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc. 2013;8(7):1277–1298. doi: 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High- resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Rothlisberger D, Baker D. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15(12):2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henrich S, Salo-Ahen OM, Huang B, Rippmann FF, Cruciani G, Wade RC. Computational approaches to identifying and characterizing protein binding sites for ligand design. J Mol Recognit. 2010;23(2):209–219. doi: 10.1002/jmr.984. [DOI] [PubMed] [Google Scholar]
- 42.Lemmon G, Meiler J. Towards ligand docking including explicit interface water molecules. PLoS One. 2013;8(6):e67536. doi: 10.1371/journal.pone.0067536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLano WL. The PyMOL Molecular Graphics System 1.0. DeLano Scientific LLC; Palo Alto, CA, USA: 2007. edn. [Google Scholar]