

Abstract
Background
GNE myopathy is a rare genetic disease characterized by progressive muscle atrophy and weakness. It is caused by biallelic mutations in the GNE gene that encodes for the bifunctional enzyme, uridine diphosphate (UDP)‐N‐acetylglucosamine (GlcNAc) 2‐epimerase/N‐acetylmannosamine (ManNAc) kinase. Typical characteristics of GNE myopathy include progressive myopathy, first involving anterior tibialis muscle and sparing the quadriceps, and rimmed vacuoles on muscle biopsy. Identifying biallelic mutations by sequencing of the GNE gene confirms the diagnosis of GNE myopathy. In a subset of patients, diagnostic confirmation is challenged by the identification of mutations in only one allele, suggesting mutations in deep intronic regions or regulatory regions.
Methods
We performed targeted sequencing and copy number variant (CNV) analysis of GNE in two siblings who clinically presented with GNE myopathy. Further molecular and biochemical studies were done to characterize the effect of a previously uncharacterized GNE mutation.
Results
We report two siblings of Indian descent with characteristic features of GNE myopathy, including progressive skeletal muscle weakness initially involving the anterior tibialis, and rimmed vacuoles on muscle biopsy, in which a heterozygous mutation, p.Val727Met, was identified in both affected siblings, but no other deleterious variants in either coding region or exon–intron boundaries of the gene. Subsequent insertion/deletion analysis identified a novel 11.3‐kb deletion (Chr9 [GRCh37]: g.36257583_36268910del) encompassing the GNE promoter region, with breakpoints residing in Alu repeats. Gene expression analysis revealed reduced GNE mRNA and protein levels, confirming decreased expression of the deleted allele harboring the deletion.
Conclusions
We have identified GNE as one of the genes susceptible to Alu‐mediated recombination. Our findings suggest that the deletion may encompass the promoter or another region necessary for GNE expression. In patients with typical manifestations of GNE myopathy and a single GNE variant identified, copy number variant (CNV) analysis may be useful in arriving at the diagnosis.
Keywords: Alu‐SINE repeat, array‐CGH, copy number variant, genomic rearrangement, GNE isoforms, GNE myopathy, precision medicine, sialic acid
Introduction
GNE myopathy (MIM 605820) is an autosomal recessive muscle disease caused by biallelic mutations in the GNE gene (MIM 603824) that encodes for the bifunctional enzyme, uridine diphosphate (UDP)‐N‐acetylglucosamine (GlcNAc) 2‐epimerase/N‐acetylmannosamine (ManNAc) kinase (Eisenberg et al. 2001; Huizing et al. 2014). This enzyme catalyzes the first two committed steps in the N‐acetylneuraminic acid (Neu5Ac, sialic acid) biosynthetic pathway (Hinderlich et al. 1997; Keppler et al. 1999; Lucka et al. 1999). Sialic acids are negatively charged carbohydrate derivatives that are widely distributed across different tissues with distinctive roles in each cell type including cell proliferation, cell interaction, and immune defense (Varki and Varki 2007). Decreased sialic acid production in GNE myopathy is thought to cause hyposialylation of glycoproteins or glycolipids in muscle fibers (Keppler et al. 1999; Tajima et al. 2005; Malicdan et al. 2009; Huizing et al. 2014; Patzel et al. 2014).
The GNE enzymatic activities, localization, and gene expression are highly regulated (Lucka et al. 1999; Ghaderi et al. 2007; Reinke et al. 2009a). Eight human GNE (hGNE) isoforms are reported; hGNE1 is the major, ubiquitously expressed isoform, while isoforms hGNE2 to hGNE8 are differentially expressed and may act as tissue‐specific regulators of sialylation (Reinke et al. 2009a,b; Yardeni et al. 2011). Total hGNE mRNA is highest in liver and placenta, while skeletal muscle has low expression (Lucka et al. 1999; Yardeni et al. 2011).
GNE myopathy usually presents in early adulthood and progresses over decades (Huizing et al. 2014; Mori‐Yoshimura et al. 2014; Nishino et al. 2015). The anterior tibialis is typically the first affected muscle, resulting in foot drop and tripping. The disease progresses to involve other muscles of the lower and upper extremities, with relative sparing the quadriceps. Eventually patients require a wheelchair and assistance with activities of daily living (Huizing et al. 2014; Mori‐Yoshimura et al. 2014). The prevalence of GNE myopathy is 1:500 among Persian Jews (Eisenberg et al. 2001) and estimated 6/1,000,000 in the general population (Celeste et al. 2014). More than 150 causative GNE mutations have been reported for GNE myopathy (Celeste et al. 2014).
In this study, we describe two siblings of Indian descent who are compound heterozygous for a well‐described missense mutation and a novel 11.3‐kb deletion in the GNE gene. We discuss the effect of the deletion on GNE protein and hGNE gene expression, including effects on the different hGNE splice variants.
Subjects and Methods
Subjects
Both patients were evaluated under NIH protocol 11‐HG‐0218, “A Natural History of Patients with GNE Myopathy” (ClinicalTrials.gov Identifier NCT01417533). Open muscle biopsies were obtained under protocol 15‐HG‐0068, “An Open Label Phase 2 Study of ManNAc in Subjects with GNE Myopathy” (ClinicalTrials.gov Identifier NCT02346461). Both protocols were approved by the National Human Genome Research Institute Institutional Review Board. Patients provided written informed consent. Evaluations were performed at the NIH Clinical Center and included medical and family history, as well as comprehensive physical, biochemical, and imaging studies. Muscle imaging was performed using a single 3‐T whole‐body MRI system (Verio, Siemens Medical Systems, Erlangen, Germany).
DNA analysis
DNA was isolated from whole blood. Mutation analysis of the GNE coding exons (NM_001128227.2) was performed by Sanger sequencing (primer sequences available upon request). CNV analysis was performed on Prevention Genetics’ high‐density gene‐centric comparative genomic hybridization array (aCGH); a custom‐designed oligonucleotide array (720K probes; Oxford Gene Technology). Analysis was performed using Cytosure Interpretation Software (Oxford Gene Technology, Begbroke, UK). Primer sequences used for PCR confirmation and Sanger sequencing across the deletion are provided in Table S1.
RNA expression analysis
Total RNA was isolated from whole blood (PAX gene Blood RNA Kit; Ambion, Inc., Austin, TX, USA). First strand cDNA was synthesized with high‐capacity RNA‐to‐cDNA kit (Applied Biosystems). Quantitative real‐time PCR (qPCR) was performed on 100 ng/μL cDNA using power SYBR Green mix (Applied Biosystems, Foster City, CA, USA) and Bio‐Rad qPCR machine with standard qPCR parameters to analyze hGNE isoform expression compared to POLR2A, using the comparative CT method (Livak and Schmittgen 2001). For tissue‐specific expression studies, human multiple tissue cDNA panels (Clontech Laboratories, Mountain View, CA, USA) were amplified with isoform‐specific primers (primer sequences available upon request) and visualized on a 2% Agarose gel.
Immunoblotting
Epstein–Barr virus (EBV) transformed lymphoblasts from patients were grown in RPMI medium. Cell lysates were subjected to immunoblotting, using a 4–20% Tris‐Glycine gel (Life Technologies, Carlsbad, CA, USA) and nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). Membranes were blocked with Li‐Cor blocking buffer (Li‐Cor Biosciences, Lincoln, NE, USA), and incubated with goat polyclonal anti‐GLCNE (GLCGNE [T‐19]; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse monoclonal anti‐ACTB (anti‐β‐actin; Sigma‐Aldrich, St. Louis, MO, USA) antibodies followed by incubation with secondary antibodies (Li‐Cor Biosciences). Protein bands were visualized and quantified using Odyssey® imaging system (LI‐COR Biosciences).
Results
Patients 1 and 2 are siblings from a nonconsanguineous family of Indian descent with no prior family history of neurological or muscle disease. Patient 1 is a female who presented at 25 years of age with ankle pain, tripping, and abnormal gait. When she was first evaluated at 27 years of age, she had bilateral foot drop, waddling gait, inability to stand on her toes or heels, or to stand up from a squat. Previous targeted sequencing for MYH2 (MIM 160740) and VCP (MIM 601023) genes did not reveal pathogenic variants. When she was evaluated at the NIH at 28 years of age, the disease had progressed to involve lower and upper extremity muscles. The patient used bilateral ankle foot orthotics and a cane for ambulation, had difficulty walking, climbing upstairs, standing from a squat, opening jars, and combing her hair. She denied problems with diplopia, facial weakness, chewing, swallowing, or sensation. On physical examination, she was unable to stand on her toes or walk on her heels, and had steppage gait. Otherwise she had a normal cranial nerve examination, no facial weakness, and normal coordination, sensation, and deep tendon reflexes. There was no respiratory or cardiac involvement. Electrocardiogram, echocardiogram, and pulmonary function tests, including supine to sitting, were normal. Creatine kinase was 167 U/L. Muscle biopsy showed characteristic findings of GNE myopathy, including marked fiber size variation, rimmed vacuoles, and absence of inflammation in both biceps brachii and gastrocnemius muscles; in addition, the gastrocnemius muscle showed marked fibrosis (Fig. 1). Muscle strength was evaluated by Quantitative Muscle Strength Assessment (QMA; Aeverl Medical, Gainesville, GA, USA). The results are expressed as percent of predicted strength for each muscle accounting for age, gender, height, and weight (The National Isometric Muscle Strength (NIMS) Database Consortium 1996). Ankle dorsiflexion strength was <1% of predicted bilaterally and knee extension >50% of predicted, showing a characteristic pattern of GNE myopathy with advanced ankle dorsiflexion weakness and relative sparing of the quadriceps. T1‐weighted MRI showed atrophy of hamstrings and lower leg muscles (Fig. 1A). Sequencing of the GNE gene revealed a single disease‐causing mutation c.2179G>A (p.Val727Met) in exon 13 (Fig. 2A). Deletion/duplication analysis of the GNE gene revealed a novel 11,328‐bp deletion [c.51+7981_52‐8189del] (or Chr9(GRCh37):g.36257583_36268910del (Fig. 2B), not previously reported nor found in other genomic databases. Primer sets around the suspected deletion breakpoints amplified fragments across the deletion (Table S1, Fig. 2B,C). Sanger sequencing revealed the exact deletion breakpoints (Fig. 2D), consistent with the expected sizes of the PCR fragments (Fig. 2B).
Figure 1.
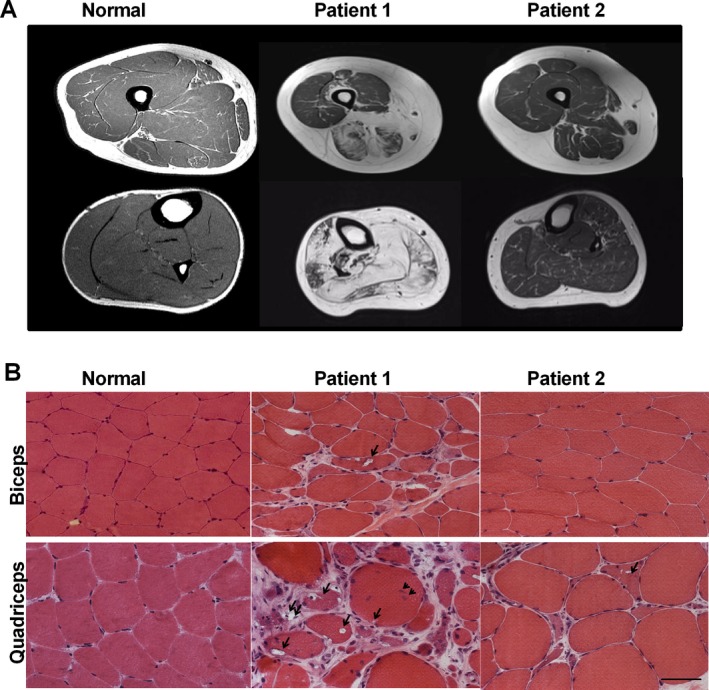
Muscle imaging and pathology. (A) T1‐weighted MRI of the thigh (upper panel) and lower leg (lower panel). Atrophy of hamstrings and lower leg muscles (Patient 1) and the proximal anterior tibialis (Patient 2) gave rise to fatty infiltration, apparent as white on the MRI. (B) Muscle biopsies of biceps brachii and lower extremity muscles (gastrocnemius medialis in Patient 1, anterior tibialis in Patient 2, quadriceps femoris in Normal) show characteristic findings of GNE myopathy, including rimmed vacuoles (arrows), fatty and fibrous tissue replacement (double arrows), marked variation in fiber size, and central nucleation (arrowhead). Note that the biceps muscle of Patient 2 appears normal, except for a mild variation in fiber size. Scale bar = 50 microns.
Figure 2.
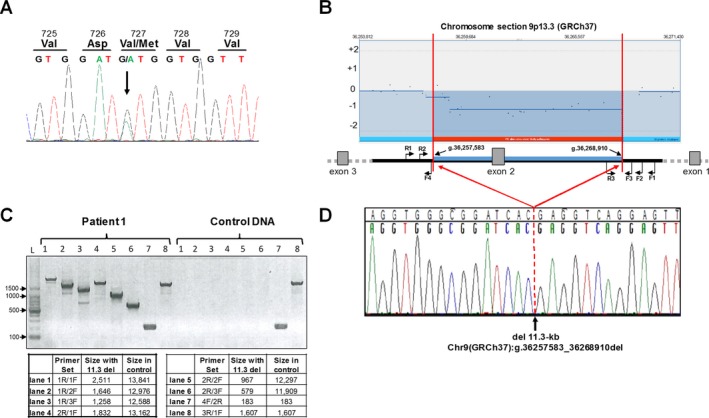
GNE mutation analysis. (A) Sanger sequencing of GNE exon 13 confirmed the heterozygous GNE mutation [NM_001128227.2:c.2179G>A;p.Val727Met] in both siblings (Patient 1 displayed). (B) Enlarged region of chromosome 9 (GRCh37) aCGH analysis that showed heterozygosity of a large (>10 kb) region in both patients. Primer locations for deletion analysis are indicated (not to scale, note that GNE gene is transcribed on reverse strand). (C) The size and breakpoints of the deletion were established by PCR analysis across the deletion; expected fragment sizes for each primer set are indicated. (D) Sanger sequencing across the deletion (Primer 1R; reverse sequence shown) determined the exact breakpoints and deletion size as: Chr9(GRCh37):g.36257583_36268910del (del 11,328‐bp).
Patient 2, the male brother of Patient 1, was diagnosed presymptomatically by genetic testing at the age of 24 years and harbors the same GNE gene variants as his affected sister. He was evaluated at 25 years of age at the NIH. At that time, he reported difficulty running and occasional ankle pain. On physical examination, he was unable to walk on his heels and walked with difficulty on his toes, but no other abnormalities could be identified. There were no cardiac or respiratory abnormalities identified by electrocardiogram, echocardiogram, or pulmonary function tests. Creatine kinase was 136 U/L. Tibialis anterior biopsy showed fiber size variation and scattered fibers with rimmed vacuoles; biceps muscles showed mild variation in fiber size (Fig. 1). Muscle strength by QMA showed mild weakness of different muscle groups and was significant for decreased ankle dorsiflexion strength of 25% of predicted bilaterally, T1‐weighted MRI showed atrophy of the proximal anterior tibialis (Fig. 1A), which confirmed the physical examination findings.
Because of the characteristic presentation of anterior tibialis weakness presenting in young adults, relative sparing of the quadriceps, family cosegregation with autosomal recessive inheritance (data not shown), and presence of one heterozygous disease‐associated GNE variant, we evaluated the pathogenicity of the 11,328‐bp deletion.
GenBank describes five of the eight human GNE splice variants (Yardeni et al. 2011): hGNE1 (NM_005746), hGNE2 (NM_001128227), hGNE3 (NM_001190388), hGNE4 (NM_001190383), and hGNE5 (NM_001190384) (Fig. 3A). The 11.3 kb deleted area in our patients is located in the deep intronic (intron 1) region for two transcripts (hGNE2 and hGNE3) and in the 5′ UTR and promoter region of three others (hGNE1, hGNE4, and hGNE5) (Fig. 3A). Online tools predicted a splicing defect (www.interactive-biosoftware.com/alamut-visual) due to the deep intronic 11.3 kb deletion in hGNE2 and hGNE3. We measured expression of hGNE2 and hGNE3, with transcript‐specific primers by PCR in whole blood (lymphoblast) cDNA, which showed no amplification products in controls (Fig. S1, EBV) nor in cDNA from both patients (not shown); this indicated that these hGNE transcripts are not expressed in blood lymphoblasts. PCR on multiple tissue cDNA panels showed expression of hGNE2 in kidney, liver, and heart; and hGNE3 in kidney, liver, and muscle (Fig. S1), similar to previous results (Yardeni et al. 2011), thereby validating our primer choice for these splice variants.
Figure 3.
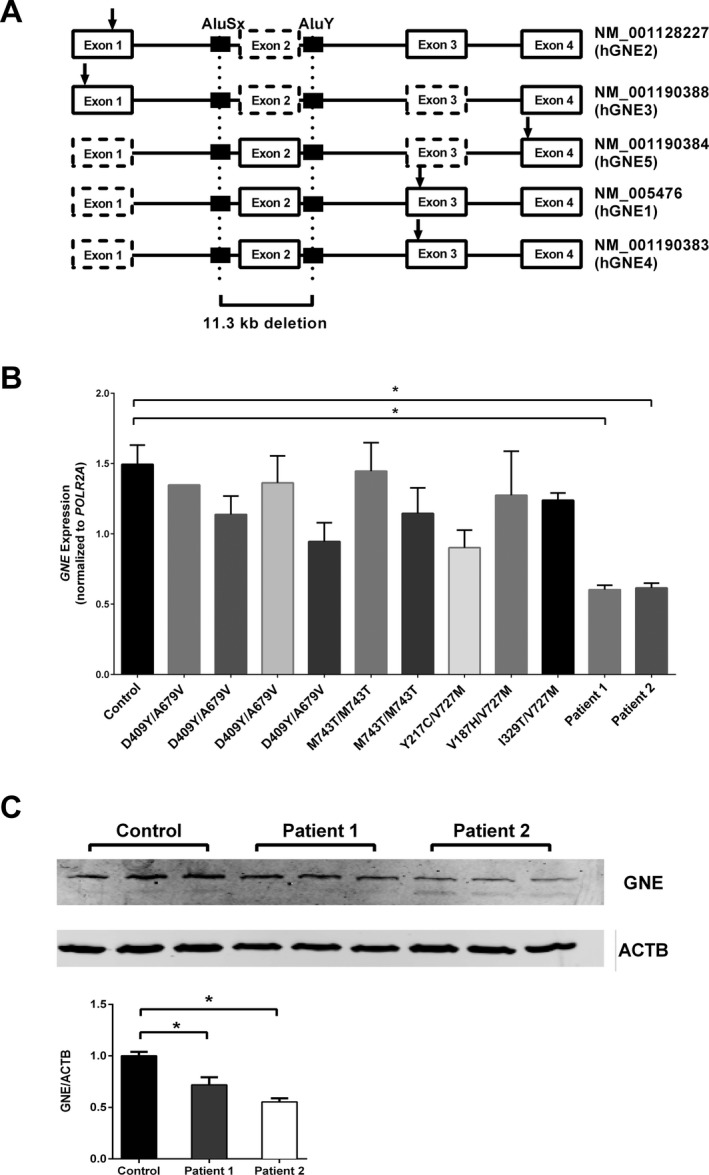
GNE gene and protein expression. (A) Display of the 5′ terminal region of the five GNE splice variants listed in GenBank. Exons represented by dotted boxes are absent in respective transcripts. The 11.3 kb deletion breakpoints (vertical dotted lines) within Alu‐repeat regions (black boxes) and the start codon (ATG) position (arrow) of each isoform is shown. The deletion is located deeply intronic for two isoforms (hGNE2 and hGNE3) and in the 5′ UTR and promoter region of others (hGNE1,hGNE4, and hGNE5). (B) Blood GNE mRNA levels normalized with POLR2A. Patients 1 and 2, harboring the 11.3 kb GNE deletion in combination with a missense, have reduced GNE expression, as compared to other GNE myopathy patients whose biallelic missense mutations in GNE are mentioned. *P < 0.05. (C) Measurement of GNE protein expression in lymphoblastoid cell lines from control and Patients 1 and 2. Expression levels were normalized with ACTB, and expressed as a ratio with control (lower bar graph). *P < 0.05.
The 11.3 kb deletion most likely affects the three other hGNE splice variants (hGNE1, hGNE4, and hGNE5) by altering mRNA expression, stability, and/or translation. We designed a primer pair that specifically amplified these three transcripts together and measured GNE mRNA expression in blood by quantitative PCR. There was a 40–50% reduction in hGNE expression in both affected siblings (Fig. 3B), when compared to control and other patients with biallelic mutations in GNE. Immunoblotting showed a significant decrease (55–70%) in the amount of GNE protein expressed in lymphoblastoid cells of both patients compared to control (Fig. 3C), likely resulting from the decreased mRNA expression.
Discussion
In this study, we describe two siblings of Indian descent with characteristic clinical and histopathological manifestations of GNE myopathy with compound heterozygous GNE mutations. One mutation is a missense variant, p.Val727Met in the GNE‐kinase domain, a common mutation in patients of Indian ancestry with GNE myopathy (Nalini et al. 2013; Celeste et al. 2014). The other mutation is a novel 11.3‐kb deletion located in the 5′ UTR and promoter region of hGNE. The site of the deletion is deeply intronic to be detected by conventional Sanger sequencing. Therefore, genomic rearrangement (copy number variant [CNV]) analysis should be considered when a single mutation is identified in patients with autosomal recessive inheritance and characteristic manifestations of GNE myopathy.
Indeed, while preparing this report, a study reported 9 novel CNVs in 13 Asian GNE myopathy patients with a previously identified monoallelic GNE mutation. The CNVs ranged from 0.5 kb to >40 kb, and most appeared to include GNE exon 2 and were flanked by Alu repeats. The study identified GNE as an Alu‐rich gene, and particularly exon 2 as a hotspot of genomic GNE rearrangements (Zhu et al. 2017). The 11.3 kb deletion in our siblings (annotated del g.13,132–24,459 on NG_008246.1 for direct comparison), was not identified in this previous study (nor were either of the breakpoints), but did include exon 2 and was indeed flanked by Alu repeats of the SINE/Alu family; AluSx on the 5′ end and AluY on the 3′ end of the deletion (Fig. 3A), which likely underlie the rearrangement/deletion mechanism. This novel 11.3 kb deletion should be checked in patients of Indian descent in which no or only one GNE variant was identified, such as the 4 Indian patients with monoallelic mutations reported by Nalini et al. (2013).
We demonstrated that the 11.3 kb deletion affects expression of isoforms hGNE1, hGNE5, and hGNE4, likely because of degradation due to nonsense‐mediated decay or aberrant expression due to promoter mutation of the deleted allele. The hGNE1 transcript is the major GNE isoform, as it is expressed in most tissues tested and its protein contains the highest epimerase and kinase activity (Reinke et al. 2009b; Yardeni et al. 2011). The hGNE1 transcript contains exon 2 in its noncoding 5′ UTR, and a deletion of this exon may explain decreased mRNA transcription in our patients (Fig. 3B).
Interestingly, while the hGNE mRNA expression is ~50% reduced in our patients’ lymphoblastoid cells, the amount of residual GNE protein in these cells is only moderately reduced (55–70% of normal) (Fig. 3C). This might be due to the cumulative translation of missense mutant alleles and/or expression of other unknown isoforms. Of note, biallelic GNE missense mutations in other patients with GNE myopathy appeared to not affect GNE mRNA expression (Fig. 3B), which was not previously reported. It is difficult to predict the effect of the patients’ 11.3 kb deletion and p.Val727Met substitution on GNE enzyme activity, since we could not completely assess the protein expression of all GNE isoforms due to lack of specific antibodies and/or appropriate patients’ tissue; an additional complication is the fact that GNE homodimerizes to oligomeric structures with different enzyme activities (Ghaderi et al. 2007; Reinke et al. 2009b).
The 11.3 kb deletion most likely spares the hGNE2 and hGNE3 isoforms, since these transcripts do not contain exon 2 (Fig. 3A). Therefore, it is possible that accumulation of substrates due to reduced hGNE1, hGNE5, and hGNE4 expression promotes overexpression of hGNE2 and hGNE3 as a compensatory mechanism. Unfortunately, expression in tissues could not be evaluated due to limited patient tissue samples. hGNE2 and hGNE3 were not expressed in control blood lymphoblasts (EBV, Fig. S1). Both transcripts were expressed only in selected tissues including liver and kidney, consistent with earlier findings (Yardeni et al. 2011). We also detected hGNE2 expression in heart and hGNE3 in skeletal muscle (Fig. S1), in contrast with a previous report (Yardeni et al. 2011).
GNE myopathy is a slowly progressive muscle disease. There have been reports of variability in the disease, even among siblings (Ro et al. 2005). However, both patients had onset of symptoms at 25 years of age. Patient 1 is older than her brother, and therefore, at the time of evaluation, she was at a more advanced stage of progression than her brother. At follow‐up visits, both patients have shown a similar rate of progression.
In conclusion, this study emphasizes considering genomic rearrangement/deep intronic and CNV analysis in the GNE gene, with emphasis on the exon 2 containing region, in patients with characteristic features of GNE myopathy, in which no biallelic pathologic GNE variants could be identified. Such analysis could improve patient diagnosis, since biallelic disease‐causing mutations in the GNE gene ultimately confirm the diagnosis. A delayed or incomplete diagnosis not only causes emotional hardship for the patient but also delays proper management of the disease and may influence eligibility to enroll in clinical trials and/or response to therapy. Furthermore, our findings suggest that the area encompassed by the deletion harbors a region that plays a critical role in GNE expression that should be further investigated.
Conflict of Interest
None declared.
Supporting information
Figure S1 (A) PCR amplification of tissue‐specific cDNA from selected human tissues (human multiple tissue cDNA panels, Clontech Laboratories) or EBV cells (Epstein–Barr virus [EBV] transformed human lymphoblasts) with primers specifically amplifying hGNE2 (114 bp), hGNE3 (109 bp), or a C‐terminal common region expressed in all GNE transcripts (392 bp).
Table S1 Primer sequences on chromosome 9p13.3 (GRCh37) used for Fig. 2B*.
Acknowledgments
We thank the patients and their families for participating in this study. This research was supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Center for Advancing Translational Sciences, the National Institute of Neurological Disorders and Stroke and the Common Fund, and the Office of the Director National Institutes of Health, Bethesda, MD, USA.
References
- The National Isometric Muscle Strength (NIMS) Database Consortium 1996. Muscular weakness assessment: use of normal isometric strength data. Arch. Phys. Med. Rehabil. 77:1251–1255. [DOI] [PubMed] [Google Scholar]
- Celeste, F. V. , Vilboux T., Ciccone C., de Dios J. K., Malicdan M. C., Leoyklang P., et al. 2014. Mutation update for GNE gene variants associated with GNE myopathy. Hum. Mutat. 35:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg, I. , Avidan N., Potikha T., Hochner H., Chen M., Olender T., et al. 2001. The UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat. Genet. 29:83–87. [DOI] [PubMed] [Google Scholar]
- Ghaderi, D. , Strauss H. M., Reinke S., Cirak S., Reutter W., Lucka L., Hinderlich S.. 2007. Evidence for dynamic interplay of different oligomeric states of UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase by biophysical methods. J. Mol. Biol. 369:746–758. [DOI] [PubMed] [Google Scholar]
- Hinderlich, S. , Stäsche R., Zeitler R., Reutter W.. 1997. A bifunctional enzyme catalyzes the first two steps in N‐acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase. J. Biol. Chem. 272:24313–24318. [DOI] [PubMed] [Google Scholar]
- Huizing, M. , Malicdan M. C., Krasnewich D., Manoli I., Carrillo‐Carrasco N.. 2014. GNE myopathy. McGraw‐Hill, New York. [Google Scholar]
- Keppler, O. T. , Hinderlich S., Langner J., Schwartz‐Albiez R., Reutter W., Pawlita M.. 1999. UDP‐GlcNAc 2‐epimerase: a regulator of cell surface sialylation. Science 284:1372–1376. [DOI] [PubMed] [Google Scholar]
- Livak, K. J. , and Schmittgen T. D.. 2001. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lucka, L. , Krause M., Danker K., Reutter W., Horstkorte R.. 1999. Primary structure and expression analysis of human UDP‐N‐acetyl‐glucosamine‐2‐epimerase/N‐acetylmannosamine kinase, the bifunctional enzyme in neuraminic acid biosynthesis. FEBS Lett. 454:341–344. [DOI] [PubMed] [Google Scholar]
- Malicdan, M. C. , Noguchi S., Hayashi Y. K., Nonaka I., Nishino I.. 2009. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV‐hIBM mouse model. Nat. Med 15:690–695. [DOI] [PubMed] [Google Scholar]
- Mori‐Yoshimura, M. , Oya Y., Yajima H., Yonemoto N., Kobayashi Y., Hayashi Y. K., et al. 2014. GNE myopathy: a prospective natural history study of disease progression. Neuromuscul. Disord. 24:380–386. [DOI] [PubMed] [Google Scholar]
- Nalini, A. , Gayathri N., Nishino I., Hayashi Y. K. et al. 2013. GNE myopathy in India. Neurol India 61:371–374. [DOI] [PubMed] [Google Scholar]
- Nishino, I. , Carrillo‐Carrasco N., and Argov Z.. 2015. GNE myopathy: current update and future therapy. J. Neurol. Neurosurg. Psychiatry 86:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzel, K. A. , Yardeni T., Le Poec‐Celic E., Leoyklang P., Dorward H., Alonzi D. S., et al. 2014. Non‐specific accumulation of glycosphingolipids in GNE myopathy. J. Inherit. Metab. Dis. 37:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke, S. O. , Lehmer G., Hinderlich S., Reutter W.. 2009a. Regulation and pathophysiological implications of UDP‐GlcNAc 2‐epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol. Chem. 390:591–599. [DOI] [PubMed] [Google Scholar]
- Reinke, S. O. , Eidenschink C., Jay C. M., Hinderlich S.. 2009b. Biochemical characterization of human and murine isoforms of UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase (GNE). Glycoconj. J. 26:415–422. [DOI] [PubMed] [Google Scholar]
- Ro, L. S. , Lee‐Chen G. J., Wu Y. R., Lee M., Hsu P. Y., Chen C. M.. 2005. Phenotypic variability in a Chinese family with rimmed vacuolar distal myopathy. J. Neurol. Neurosurg. Psychiatry 76:752–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, Y. , Uyama E., Go S., Sato C., Tao N., Kotani M., et al. 2005. Distal myopathy with rimmed vacuoles: impaired O‐glycan formation in muscular glycoproteins. Am. J. Pathol. 166:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki, N. M. , and Varki A.. 2007. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab. Invest. 87:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni, T. , Choekyi T., Jacobs K., Ciccone C., Patzel K., Anikster Y., et al. 2011. Identification, tissue distribution, and molecular modeling of novel human isoforms of the key enzyme in sialic acid synthesis, UDP‐GlcNAc 2‐epimerase/ManNAc kinase. Biochemistry 50:8914–8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W. , Mitsuhashi S., Yonekawa T., Noguchi S., Huei J. C., Nalini A., et al. 2017. Missing genetic variations in GNE myopathy: rearrangement hotspots encompassing 5'UTR and founder allele. J. Hum. Genet. 62:159–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) PCR amplification of tissue‐specific cDNA from selected human tissues (human multiple tissue cDNA panels, Clontech Laboratories) or EBV cells (Epstein–Barr virus [EBV] transformed human lymphoblasts) with primers specifically amplifying hGNE2 (114 bp), hGNE3 (109 bp), or a C‐terminal common region expressed in all GNE transcripts (392 bp).
Table S1 Primer sequences on chromosome 9p13.3 (GRCh37) used for Fig. 2B*.