

The ionic mechanisms underlying CXCL10-induced neuronal activation and allergic itch are largely unexplored. This study revealed that CXCL10 evoked an ionic current mainly carried by Cl− channels. We suggest that Cl− channels are likely key molecular candidates responsible for the CXCL10-evoked neuronal activation and itch-like behaviors in a murine model of allergic contact dermatitis induced by the antigen squaric acid dibutylester. Cl− channels may emerge as a promising drug target for the treatment of allergic itch in which CXCL10/CXCR3 signaling may participate.
Keywords: CXCR3, CXCL10, itch, pain, Cl− channel, allergic contact dermatitis
Abstract
Persistent itch often accompanies allergic contact dermatitis (ACD), but the underlying mechanisms remain largely unexplored. We previously demonstrated that CXCL10/CXCR3 signaling activated a subpopulation of cutaneous primary sensory neurons and mediated itch response after contact hypersensitivity (CHS), a murine model of ACD, induced by squaric acid dibutylester. The purpose of this study was to determine the ionic mechanisms underlying CXCL10-induced neuronal activation and allergic itch. In whole cell recordings, CXCL10 triggered a current in dorsal root ganglion (DRG) neurons innervating the area of CHS. This current was modulated by intracellular Cl− and blocked by the general Cl− channel inhibitors. Moreover, increasing Ca2+ buffering capacity reduced this current. In addition, blockade of Cl− channels significantly suppressed CXCL10-induced Ca2+ response. In behavioral tests, injection of CXCL10 into CHS site exacerbated itch-related scratching behaviors. Moreover, the potentiating behavioral effects of CXCL10 were attenuated by either of two Cl− channel blockers. Thus we suggest that the Cl− channel acts as a downstream target mediating the excitatory and pruritic behavioral effects of CXCL10. Cl− channels may provide a promising therapeutic target for the treatment of allergic itch in which CXCL10/CXCR3 signaling may participate.
NEW & NOTEWORTHY The ionic mechanisms underlying CXCL10-induced neuronal activation and allergic itch are largely unexplored. This study revealed that CXCL10 evoked an ionic current mainly carried by Cl− channels. We suggest that Cl− channels are likely key molecular candidates responsible for the CXCL10-evoked neuronal activation and itch-like behaviors in a murine model of allergic contact dermatitis induced by the antigen squaric acid dibutylester. Cl− channels may emerge as a promising drug target for the treatment of allergic itch in which CXCL10/CXCR3 signaling may participate.
allergic contact dermatitis (ACD) is a common inflammatory skin disease initiated by T lymphocytes that are specific for an allergen (Grabbe and Schwarz 1998). Persistent itch (pruritus) and burning sensation are the major clinical sensory manifestations of ACD (Buddenkotte and Steinhoff 2010). Although the physiopathology of ACD is well studied, the pruritic mechanisms in ACD are largely unknown.
The C-X-C motif chemokine10 (CXCL10), also known as interferon-γ-inducible protein 10 (IP-10), is exclusively expressed in ACD but not irritant contact dermatitis reactions (Enk and Katz 1992; Flier et al. 1999). CXCL10 is predominantly produced by epidermal cells in the challenged skin of contact hypersensitivity (CHS; Flier et al. 1999; Goebeler et al. 2001; Tokuriki et al. 2002) and modulates innate and adaptive immune responses by specifically attracting T cells and dendritic cells bearing its receptor CXCR3 to the site of allergen reaction (Dufour et al. 2002). In addition to immune cells, both CXCL10 and CXCR3 are detected in primary sensory neurons (Bhangoo et al. 2007) and have been implicated in the maintenance of a chronic pain state under inflammatory pain or neuropathic pain conditions (Bhangoo et al. 2007; Fu et al. 2010; Strong et al. 2012). Our recent study revealed that CXCL10/CXCR3 signaling was upregulated in dorsal root ganglion (DRG) neurons after CHS. Moreover, CXCL10 may exert its pruritic effects by directly exciting primary sensory neurons through CXCR3 (Qu et al. 2015). However, the ionic mechanisms underlying the excitatory and pruritic effects of CXCL10 are largely unexplored.
Cl− channels, including Ca2+-activated Cl− channels (CaCCs), are present in primary sensory neurons and play an important role in the regulation of neuronal excitability (Boudes et al. 2009; Hartzell et al. 2005). Moreover, in peripheral sensory neurons, the higher expression of Na+-K+-Cl− cotransporter increases intracellular chloride concentration ([Cl−]i); therefore, the activation of CaCCs gives rise to the outward Cl− flow and cell depolarization. (Kamaleddin 2017; Mao et al. 2012). Accordingly, Cl− channels have been proposed to participate in somatosensory transduction. Indeed, anoctamin 1, one type of CaCCs, was identified to act as a heat sensor that mediates or amplifies thermal nociception (Cho et al. 2012). Certain pruritogens and algogens were shown to activate specific types of Cl− channels to elicit acute pruritic and nociceptive responses, respectively (Cho et al. 2012; Liu et al. 2010). In addition, some types of Cl− channels have been implicated in the maintenance of chronic neuropathic pain (Pineda-Farias et al. 2015). In murine microglia, Cl− was identified as a key downstream transduction channel in CXCL10/CXCR3 signaling (Rappert et al. 2002). Therefore, our purpose was to investigate the potential role of Cl− channels in mediating CXCL10-induced neuronal activation and allergic itch by using a mouse model of CHS induced by a hapten, squaric acid dibutylester (SADBE).
MATERIALS AND METHODS
Animals.
C57BL/6 male mice used in the study were 2-3 mo of age and weighed 20–30 g. All the experimental procedures were approved by the Institutional Animal Care and Use Committee of Yale University School of Medicine and were consistent with the guidelines provided by the National Institute of Health and the International Association for the Study of Pain.
Model of allergic contact dermatitis.
Allergic contact dermatitis (ACD) or contact hypersensitivity (CHS) was elicited by using the contact sensitizer squaric acid dibutylester (SADBE; Sigma, St. Louis, MO), as described previously (Fu et al. 2014; Qu et al. 2014). Mice were sensitized with 1% SADBE in acetone (25 μl) topically applied to the shaved abdomen once daily for 3 consecutive days. Five days later, mice were challenged with a topical application of 1% SADBE (25 μl) onto the right cheek (for behavioral testing) for 1 day or to the hairy skin of foot and the calf of hind leg (for electrophysiology and calcium imaging) once a day for 2 consecutive days. Separate groups of mice were challenged with the acetone alone and served as controls.
Retrograde labeling of cutaneous sensory neurons.
Rationale for using neurons from DRG rather than trigeminal ganglia (TG). We chose to study neurons from the DRG rather than TG because lumbar ganglia were used in our previous studies of the role of CXCL10/CXCR3 signaling in the mouse. We found that SADBE challenge to the skin of mouse cheek (cheek model) and calf (calf model) each induced analogous spontaneous itch- and pain-like behaviors directed to the skin of CHS (Qu et al. 2014). Moreover, CXCL10/CXCR3 signaling was involved in allergic itch associated with CHS in both cheek and calf models (Qu et al. 2015). Thus it is likely that CHS caused the similar biological changes of DRG and TG neurons. DRG neurons instead of TG neurons were chosen for in vitro experiments.
For in vitro studies, DRG cell bodies were identified as cutaneous and as having innervated the area of CHS (or vehicle treatment) by the presence of a retrogradely transported red fluorescent dye, DiI (Invitrogen). DiI (1.7 mg/ml in 1% DMSO) was injected subcutaneously at the SADBE application sites on the hairy skin of the calf (2 injections) and also on the dorsum of the foot (1 injection) of one hind leg of mice (10 µl per site) at least 1 wk before the first challenge with SADBE or acetone vehicle.
Cultures of dissociated DRG neurons.
At 24 h after the second challenge, L3–L5 lumbar DRGs, ipsilateral to either the acetone- or SADBE-treated skin, were harvested and placed in oxygenated complete saline solution (CSS) for cleaning and then mincing. The CSS consisted of (in mM) 137 NaCl, 5.3 KCl, 1 MgCl2, 3 CaCl2, 25 Sorbitol, and 10 HEPES, adjusted to pH 7.2 with NaOH. For 20 min the DRGs were digested with 0.35 U/ml Liberase TM (Roche Diagnostics, Indianapolis, IN) and then for 15 min with 25 U/ml Liberase TL (0.25 U/ml; Roche Diagnostics) and papain (30 U/ml; Worthington Biochemical, Lakewood, NJ) in CSS containing 0.5 mM EDTA at 37°C. The tissue was triturated with a fire-polished Pasteur pipette. The DRG neurons were suspended in DMEM medium containing 1 mg/ml trypsin inhibitor and 1 mg/ml bovine serum albumin (Sigma) and then plated onto poly-d-lysine/laminin-coated glass coverslips (BioCoat; BD Biosciences, Billerica, MA). The DMEM medium had equivalent amounts of DMEM and F-12 (GIBCO, Grand Island, MD) with 10% fetal calf serum (Sigma) and 1% penicillin-streptomycin (Invitrogen). The cells were maintained in 5% CO2 at 37°C in a humidified incubator and used within 16–24 h after plating.
Calcium imaging.
Calcium imaging was performed on cultured mouse DRG neurons, as described previously (Qu et al. 2011). Only small-diameter neurons (≤25 μm) were used that were labeled as cutaneous by the presence of DiI and innervated the chemically treated areas. DRG neurons were first loaded with 2 μM fura 2 acetoxymethyl ester (Invitrogen) in the dark for 45 min at 37°C and subsequently washed twice in a HEPES buffer containing (in mM) 145 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (adjusted to pH 7.4 with NaOH). DRG neurons were alternatively excited at 340 and 380 nm using a Polychrome V monochromator (TILL Photonics). Images were recorded at 2-s intervals at a room temperature of 20–22°C using a cooled charge-coupled device camera (Sensicam; PCO, Kelheim, Germany) that was controlled by a computer with Image Workbench 5.2 software (Indec Biosystems, Los Altos, CA). The ratio of 340- to 380-nm fluorescence intensity (R340/380) within a certain region of interest was used as a relative measure of the intracellular concentration of calcium ([Ca2+]i). At the end of the experiment, the viability of the neurons was confirmed by an increase in [Ca2+]i evoked by a 5-s application of 50 mM K+. Cells were considered to be responsive to a chemical if an increase in R340/380 was ≥15% above baseline (Wilson et al. 2011). Mouse recombinant CXCL10 (50 nM; R&D Systems), niflumic acid (NFA; 100 µM in 0.1% DMSO; Sigma), or 4,4′-diisothiocyanato-2,2′-stilbenedisulfonic acid (DIDS; 100 µM in 0.1% DMSO; Sigma) was added to HEPES buffer. Capsaicin (1 µM; 10 s) was applied at the end of recordings to identify capsaicin-sensitive nociceptors. All agents were then applied locally to the neuronal cell bodies through a micropipette with a tip diameter of 100 μm and connected to an 8-channel pressure-controlled drug application system (AutoMate Scientific, Berkeley, CA).
Electrophysiological recordings.
Whole cell recordings were made from small-diameter (≤25 μm), Dil-labeled DRG neurons, typically those that had been identified as responsive to CXCL10 using calcium imaging. Whole cell voltage-clamp experiments were performed at a room temperature of 20–22°C by means of a Multiclamp 700A amplifier and pClamp 9 software (Molecular Device, Sunnyvale, CA), as described previously (Qu et al. 2011, 2012). Signals were sampled at either 10 or 20 kHz and were filtered at 2 kHz. The patch pipettes were pulled from borosilicate glass capillaries with a P97 horizontal puller (Sutter Instruments, Novarto, CA). The patch pipettes, after being filled with internal solution, had a resistance of 3–4 MΩ, and their series resistance was routinely compensated at 60–80%. Only neurons with a resting membrane potential more negative than −40 mV were included in the study.
The DRG neurons were continuously perfused with HEPES buffer. The regular internal solution contained (in mM) 120 K+-gluconate, 30 NMDG-Cl−, 2 MgCl2·6H2O, 10 HEPES, 2 MgATP, 1 CaCl2·2H2O, and 11 EGTA 11, with pH adjusted to 7.2 using Tris base. The high-[Cl−]i internal solution contained (in mM) 140 NMDG-Cl, 30 K+-gluconate, 2 MgCl2·6H2O, 10 HEPES, 2 MgATP, 1 CaCl2·2H2O, and 11 EGTA, adjusted to pH 7.2. In the low-[Cl−]i internal solution, NMDG-Cl was decreased from 140 to 4 mM (Cho et al. 2012). Accordingly, K+-gluconate was increased from 30 to 136 mM. The internal solution with high Ca2+-buffering capacity was obtained by replacing 11 mM EGTA with 10 mM BAPTA in high-[Cl−]i internal solution.
Behavioral testing.
For the “cheek model,” either CXCL10 (2 µg/10 µl in PBS; R&D Systems) or its vehicle alone (10 µl PBS) was injected intradermally into the right cheek 24 h after the first SADBE challenge (when the skin was inflamed but in less fragile condition than after the second challenge). Behavioral responses were video recorded with a camcorder for 30 min starting after the injection. The video recording was played back offline in slow motion to assess the total number of site-directed scratching bouts with the hind paw and wiping with the forepaw for 30 min (Fu et al. 2014). In other tests, the effects of Cl− channel inhibitors on CXCL10-induced behavioral responses were tested. Either DIDS (50 nM/site, 10 µl; Sigma), NFA (50 nM/site, 10 µl; Sigma), or its vehicle alone (10 µl; 0.1 M NaHCO3 in PBS) was injected intradermally into the right cheek 1 h before the cheek injection of CXCL10. The dose of Cl− channel blockers was chosen on the basis of pilot tests and published dose-response findings (Liu et al. 2010). All behavioral tests were performed by the experimenters blinded to experimental conditions.
Statistical analysis.
Data are means ± SE. Student’s t-test was used to test the significance of differences between means between two groups. Comparisons for more than three groups were carried out using a one-way analysis of variance (ANOVA) followed by Bonferroni multiple-comparison corrections. Comparisons of proportions were made using Fisher’s exact test. The probability criterion for significant differences was P < 0.05. The type of statistical tests used for each comparison was indicated in the figure legends.
RESULTS
CXCL10 activated a Cl− conductance in cutaneous DRG neurons from CHS mice.
To determine the ionic mechanisms underlying the CXCL10-induced membrane depolarization, we performed whole cell recordings on the cultured cutaneous DRG neurons from CHS mice before and after the application of CXCL10. Bath application of CXCL10 (50 nM) for 2 min induced an inward current (ICXCL10) with a peak amplitude of 68.3 ± 8.7 pA (n = 9) when the DRG neurons were held at −60 mV (Fig. 1A). Because Cl− channels have been associated with the activation of ionic currents by CXCL10 in murine microglia (Rappert et al. 2002), we next asked whether the ICXCL10 recorded in DRG neurons was mediated by a Cl− channel. The directly measured normal [Cl−]i in DRG neurons was more than 30 mM (Rocha-González et al. 2008). Thus we set the Cl− concentration in control internal solution at 36 mM. When the concentration of Cl− was increased from 36 to 146 mM in the internal solution, the peak of the ICXCL10 was significantly potentiated (Fig. 1, B and D). In contrast, lowering the concentration of Cl− from 36 to 10 mM in the internal solution dramatically reduced the ICXCL10 (Fig. 1, C and D), suggesting that this current is likely mediated by a Cl− channel. Because the peak amplitude of the ICXCL10 was larger under the high-[Cl−]i condition, the high-[Cl−]i (146 mM) internal solution was chosen throughout the following experiments to facilitate the recordings of this current.
Fig. 1.
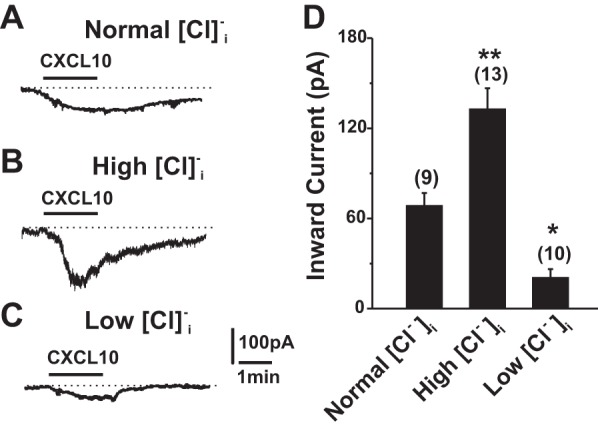
CXCL10-induced currents in DRG neurons after CHS were associated with the activation of a Cl− conductance. The neurons were held at the membrane potential of −60 mV. A–C: representative traces of inward currents (ICXCL10) induced by CXCL10 (50 nM; 2 min) recorded with the internal solution containing concentrations of Cl− that were normal (36 mM; A), high (146 mM; B), or low (10 mM; C). D: increasing [Cl−]i dramatically enhanced the amplitude of ICXCL10, whereas lowering [Cl−]i significantly attenuated this current. *P < 0.05; **P < 0.01 vs. normal [Cl−]i, one-way ANOVA with Bonferroni posttest.
To further determine the potential involvement of Cl− channels, we examined the effects of DIDS and NFA, the broad-spectrum Cl− channel antagonists (Malekova et al. 2007), on the ICXCL10. Pretreatment with DIDS (100 μM) or NFA (100 μM) for 3 min almost abolished the ICXCL10 (Fig. 2, A–C, E), indicating that the current induced by CXCL10 was likely due to the opening of the Cl− channels.
Fig. 2.
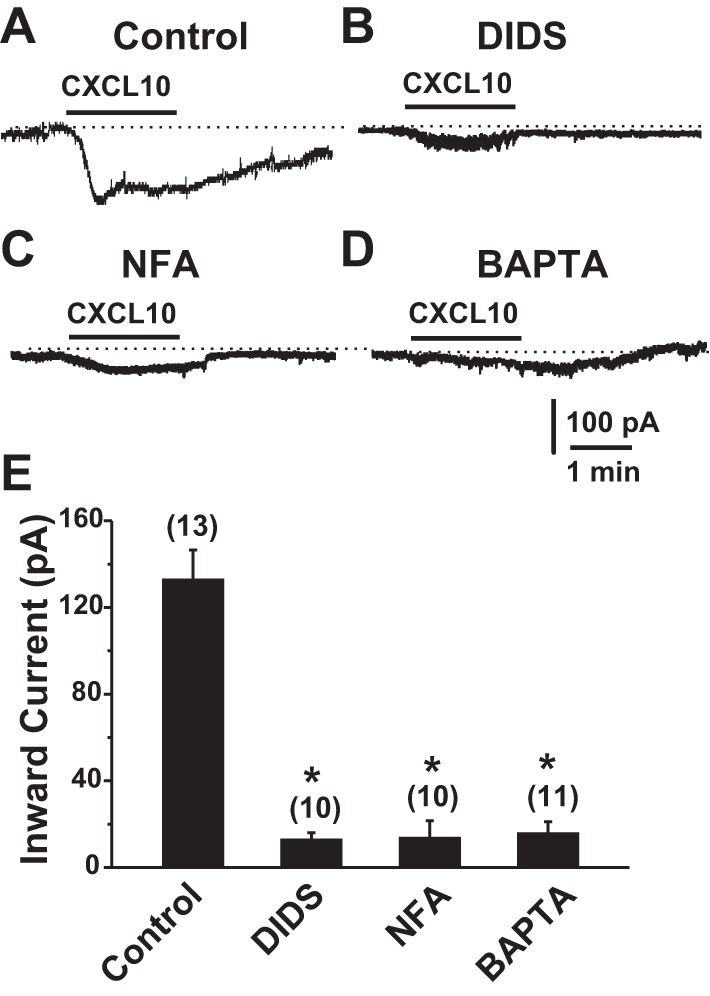
Effects of Cl− channel antagonists and intracellular Ca2+ on CXCL10-induced currents in DRG neurons innervating CHS skin. A and B: sample traces of the ICXCL10 recorded in the absence (A) and presence of the Cl− channel blockers DIDS (100 µM; B) or NFA (100 µM; C) applied to the bath or in the presence of 10 mM BAPTA in the pipette solution (D). The high-[Cl−]i pipette solution was used. E: pretreatment with DIDS or NFA for 3 min significantly reduced the peak amplitude of ICXCL10. Replacement of 11 mM EGTA with 10 mM BAPTA in the pipette solution almost abolished this inward current. The numbers of DRG neurons tested are given in parentheses. *P < 0.05 vs. control, one-way ANOVA with Bonferroni posttest.
CXCL10-induced Cl− current was modulated by intracellular Ca2+ in DRG neurons.
Because [Ca2+]i was increased after exposure to CXCL10 (Qu et al. 2015) and certain types of Cl− channels are activated by intracellular Ca2+ (Duran et al. 2010; Hartzell et al. 2005), we next tested whether intracellular Ca2+ modulated Cl− channels induced by CXCL10. When the intracellular Ca2+-buffering capacity was enhanced by replacement of EGTA in the internal solution with the fast Ca2+ chelator BAPTA (10 mM), the peak of the ICXCL10 was significantly attenuated (Fig. 2, D and E), suggesting that CXCL10-induced Cl− current was sensitized or regulated by intracellular Ca2+.
Cl− channels contributed to CXCL10-induced neuronal activation in CHS mice.
CXCL10 was shown to activate cutaneous DRG neurons from CHS mice (Qu et al. 2015). We next asked whether Cl− channels were involved in CXCL10-induced neuronal activation. In the presence of vehicle (0.1% DMSO), 42.1% (40 of 95) of cutaneous DRG neurons from CHS mice responded to CXCL10. Of all CXCL10-responsive neurons in CHS mice, 47.5% (19 of 40) were capsaicin insensitive, consistent with our published findings (Qu et al. 2015). Preincubation with a nonselective Cl− channel blocker, NFA (100 μM), for 3 min significantly reduced the percentage of CXCL10-responsive neurons in CHS mice (Fig. 3). Of all the remaining CXCL10-responsive cells, 42.9% (9 of 21) were capsaicin insensitive. These findings suggested that Cl− channels may be required for the excitatory effects of CXCL10 in primary sensory neurons.
Fig. 3.

Effects of Cl− channel blockade on CXCL10-evoked Ca2+ responses in cutaneous DRG neurons after CHS. A and B: representative traces of a CXCL10-evoked Ca2+ response in the presence of vehicle (0.1% DMSO; A) and the Cl− blocker NFA (100 µM) in the vehicle (B). C: preincubation with NFA for 3 min, compared with its vehicle, significantly suppressed the percentage of CXCL10-responsive neurons. Numbers of responsive neurons divided by total number tested responding and/or tested are given in parentheses. Cap, capsaicin. *P < 0.05 vs. vehicle, Fisher's exact test.
Cl− channel was involved in CXCL10-mediated itch-like behaviors in CHS mice.
Our recent study showed that CXCL10 injection into the cheek enhanced itch-related scratching behaviors in CHS but not in naive mice (Qu et al. 2015). CXCL10 did not evoke pain-like wiping behaviors in either CHS or naive mice (Qu et al. 2015). Because Cl− channels were identified as mediating the excitatory neuronal effects of CXCL10 in vitro, we next tested whether the potentiating effect of CXCL10 on scratching behavior in CHS mice was mediated through Cl− channels by using the cheek model. At 24 h after the first challenge, intradermal injection of CXCL10 (2 µg/10 µl) into the cheek of CHS mice significantly increased the number of scratching bouts compared with the injection of vehicle (PBS) (Fig. 4A). There were no significant differences in the number of site-directed pain-like wiping behaviors between CXCL10 and vehicle alone (data not shown). Local intradermal injection of either of the Cl− channel blockers DIDS (50 nM/site, 10 µl) or NFA (50 nM/site; 10 µl), but not of their vehicles (0.1 M NaHCO3 in saline; 10 µl), significantly reduced CXCL10-evoked scratching response in CHS mice (Fig. 4B), indicating that Cl− channel contributes to CXCL10-elicted pruritic responses in the settings of skin inflammation.
Fig. 4.
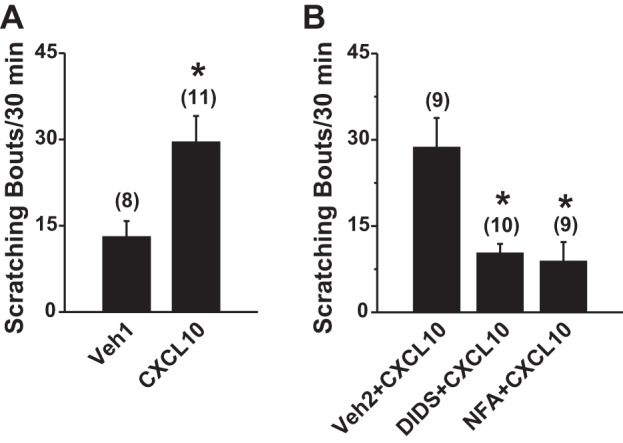
Effects of Cl− channel blockade on CXCL10-mediated itch-like behavior in CHS mice. The number of bouts of site-directed scratching with the hindlimb was quantified for 30 min immediately after the injection. A: at 24 h after the first challenge with SADBE (CHS), intradermal. injection of CXCL10 (2 µg/10 µl in PBS vehicle) into the SADBE-challenged cheek significantly increased the site-directed itch-related scratching compared with injection of PBS vehicle alone (Veh1). The number of animals tested is given in parentheses. *P < 0.05 vs. vehicle, unpaired t-tests. B: the CXCL10-evoked scratching in the SADBE-challenged cheek was significantly attenuated by preinjection, intradermally 1 h before, with the general Cl− channel blockers, either DIDS (50 nM/site; 10 µl) or NFA (50 nM/site; 10 µl), compared with prior intradermal injection of the vehicle (Veh2; 0.1 M NaHCO3 in PBS). The number of animals tested is given in parentheses. *P < 0.01 vs. vehicle, one-way ANOVA with Bonferroni posttest.
DISCUSSION
In this study, we have demonstrated that CXCL10 evokes an ionic current mainly carried by Cl− channels. We suggest that Cl− channels are likely key molecular candidates responsible for the CXCL10-evoked neuronal activation and itch-like behaviors in a murine model of ACD induced by the antigen SADBE.
In our previous study we found that cutaneous primary sensory neurons innervating the CHS skin became more excitable (Qu et al. 2014). Moreover, our recent findings revealed that upregulated CXCL10/CXCR3 signaling within DRG may contribute to neuronal hyperexcitability in the context of skin inflammation (Qu et al. 2015). The present study provided direct evidence to support the hypothesis that the Cl− channel might represent an ionic mechanism mediating CXCL10-induced membrane depolarization in DRG neurons under the CHS condition. In this study, we observed that an increase in [Cl−]i potentiated CXCL10-induced inward current, whereas a reduction in [Cl−]i nearly abolished it. Furthermore, this current was inhibited by general Cl− channel antagonists. The contribution of Cl− currents to CXCL10/CXCR3 signaling was also revealed in murine microglia (Rappert et al. 2002). Peripheral sensory neurons, compared with neurons in the central nervous system, have a greater activity of cation-Cl− cotransporters and thus normally maintain higher [Cl−]i levels (30-50 mM; Mao et al. 2012). In addition, inflammatory mediators may cause further Cl− accumulation in the sensory neurons under inflammatory conditions (Funk et al. 2008). Therefore, the equilibrium potential for Cl− is normally far more positive (−22 to −35 mV) than the resting membrane potential in primary sensory neurons (−60 to −55 mV; Mao et al. 2012; Rocha-González et al. 2008). Thus, the activation of Cl− conductance is thought to lead to the membrane depolarization and neuronal excitation in primary sensory neurons (Cho et al. 2012; Liu et al. 2010). In addition, our study showed that blockade of Cl− channels reduced the CXCL10-evoked Ca2+ response, suggesting that Cl− channel-induced depolarization is likely pro-excitatory in DRG neurons. Further studies are required to identify the molecular identity of CXCL10-activated Cl− channels.
Our recent data showed that CXCL10 evoked a Ca2+ influx from the extracellular space in DRG neurons (Qu et al. 2015). In the present study, we found that the ICXCL10 was modulated by intracellular Ca2+. Thus members of the CaCC family might contribute to CXCL10-activated Cl− currents. One hypothesis is that CaCC is activated secondarily to the CXCL10-induced Ca2+ increase, causing membrane depolarization and a further Ca2+ influx from extracellular space. However, our findings do not seem to support this possibility, because CXCL10-evoked Ca2+ responses were completely inhibited by the Cl− channel antagonist NFA. We suggest that CXCL10 binds to neuronal CXCR3 and activates a Cl− conductance, which results in membrane depolarization and subsequent activation of voltage-gated Ca2+ channels. The CXCL10-evoked increase in [Ca2+]i is probably due to an influx of Ca2+ through voltage-gated Ca2+ channels. The elevated [Ca2+]i may in turn enhance the activity of the Cl− channels. However, the cellular signaling whereby CXCR3 is coupled to Cl− channels in DRG neurons remains to be explored.
The upregulated excitatory neuronal CXCL10/CXCR3 signaling has been implicated in the chronic pain state in animal models of inflammatory pain (Bhangoo et al. 2007). Recently, we discovered that CXCL10, which is a nonpruritogenic chemokine in native mice, became a potent pruritogen that evoked itch-like behavior in ACD (Qu et al. 2015). In present study, we found that Cl− channel antagonists greatly inhibited the CXCL10-elicted itch behavior in the mice with CHS, suggesting a potential role of Cl− channels for CXCL10-evoked itch under the condition of skin inflammation. Indeed, Cl− channels have been involved in acute nociception and itch induced by several algogens and pruritogens, including bradykinin, capsaicin, endothelin 1, and histamine (Cho et al. 2012; Deba and Bessac 2015; Liu et al. 2010). Moreover, some types of Cl− channels, including anoctamin 1, are able to detect nociceptive thermal stimuli and possibly mediate thermal nociception (Cho et al. 2012). In addition, Cl− channels have been implicated in the maintenance of a chronic state of inflammatory and neuropathic pain (García et al. 2014; Pineda-Farias et al. 2015). However, the contribution of Cl− channels to spontaneous itch associated with CHS awaits further investigation. Because CXCR3 is widely expressed in immune cells, we cannot rule out a possible role of such nonneuronal cells in the pruritic effect of CXCL10 and the antipruritic effects of Cl− channel blockers in addition to the role of the sensory neurons.
In conclusion, we have demonstrated, for the first time to our knowledge, that Cl− channels mediate CXCL10-induced neuronal excitation and allergic itch under the CHS condition. We suggest that blocking Cl− channels may represent a therapeutic approach to treat the sensory symptoms of inflammatory disease where the CXCL10/CXCR3 axis may participate.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS047399 and NS014624 (to R. H. LaMotte). L. Qu is the recipient of a fellowship from the Canadian Institutes of Health Research and the Johns Hopkins Blaustein Pain Research Grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.Q. and R.H.L. conceived and designed research; L.Q., K.F., and S.G.S. performed experiments; L.Q., K.F., and S.G.S. analyzed data; L.Q. and S.G.S. interpreted results of experiments; L.Q. prepared figures; L.Q. drafted manuscript; L.Q. and R.H.L. edited and revised manuscript; L.Q. and R.H.L. approved final version of manuscript.
REFERENCES
- Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain 3: 38, 2007. doi: 10.1186/1744-8069-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudes M, Sar C, Menigoz A, Hilaire C, Péquignot MO, Kozlenkov A, Marmorstein A, Carroll P, Valmier J, Scamps F. Best1 is a gene regulated by nerve injury and required for Ca2+-activated Cl− current expression in axotomized sensory neurons. J Neurosci 29: 10063–10071, 2009. doi: 10.1523/JNEUROSCI.1312-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddenkotte J, Steinhoff M. Pathophysiology and therapy of pruritus in allergic and atopic diseases. Allergy 65: 805–821, 2010. doi: 10.1111/j.1398-9995.2010.01995.x. [DOI] [PubMed] [Google Scholar]
- Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, Back SK, Na HS, Harfe BD, Wang F, Raouf R, Wood JN, Oh U. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci 15: 1015–1021, 2012. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- Deba F, Bessac BF. Anoctamin-1 Cl− channels in nociception: activation by an N-aroylaminothiazole and capsaicin and inhibition by T16A[inh]-A01. Mol Pain 11: 55, 2015. doi: 10.1186/s12990-015-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 168: 3195–3204, 2002. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol 72: 95–121, 2010. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enk AH, Katz SI. Early molecular events in the induction phase of contact sensitivity. Proc Natl Acad Sci USA 89: 1398–1402, 1992. doi: 10.1073/pnas.89.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier J, Boorsma DM, Bruynzeel DP, Van Beek PJ, Stoof TJ, Scheper RJ, Willemze R, Tensen CP. The CXCR3 activating chemokines IP-10, Mig, and IP-9 are expressed in allergic but not in irritant patch test reactions. J Invest Dermatol 113: 574–578, 1999. doi: 10.1046/j.1523-1747.1999.00730.x. [DOI] [PubMed] [Google Scholar]
- Fu ES, Zhang YP, Sagen J, Candiotti KA, Morton PD, Liebl DJ, Bethea JR, Brambilla R. Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury. Pain 148: 509–518, 2010. doi: 10.1016/j.pain.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K, Qu L, Shimada SG, Nie H, LaMotte RH. Enhanced scratching elicited by a pruritogen and an algogen in a mouse model of contact hypersensitivity. Neurosci Lett 579: 190–194, 2014. doi: 10.1016/j.neulet.2014.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk K, Woitecki A, Franjic-Würtz C, Gensch T, Möhrlen F, Frings S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain 4: 32, 2008. doi: 10.1186/1744-8069-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García G, Martínez-Rojas VA, Rocha-González HI, Granados-Soto V, Murbartián J. Evidence for the participation of Ca2+-activated chloride channels in formalin-induced acute and chronic nociception. Brain Res 1579: 35–44, 2014. doi: 10.1016/j.brainres.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Goebeler M, Trautmann A, Voss A, Bröcker EV, Toksoy A, Gillitzer R. Differential and sequential expression of multiple chemokines during elicitation of allergic contact hypersensitivity. Am J Pathol 158: 431–440, 2001. doi: 10.1016/S0002-9440(10)63986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabbe S, Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol Today 19: 37–44, 1998. doi: 10.1016/S0167-5699(97)01186-9. [DOI] [PubMed] [Google Scholar]
- Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol 67: 719–758, 2005. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- Kamaleddin MA. Molecular, biophysical, and pharmacological properties of calcium-activated chloride channels. J Cell Physiol. First published May 15, 2017; doi: 10.1002/jcp.25823. [DOI] [PubMed] [Google Scholar]
- Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl− channels. J Clin Invest 120: 1240–1252, 2010. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malekova L, Tomaskova J, Novakova M, Stefanik P, Kopacek J, Lakatos B, Pastorekova S, Krizanova O, Breier A, Ondrias K. Inhibitory effect of DIDS, NPPB, and phloretin on intracellular chloride channels. Pflugers Arch 455: 349–357, 2007. doi: 10.1007/s00424-007-0300-9. [DOI] [PubMed] [Google Scholar]
- Mao S, Garzon-Muvdi T, Di Fulvio M, Chen Y, Delpire E, Alvarez FJ, Alvarez-Leefmans FJ. Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol 108: 834–852, 2012. doi: 10.1152/jn.00970.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Farias JB, Barragán-Iglesias P, Loeza-Alcocer E, Torres-López JE, Rocha-González HI, Pérez-Severiano F, Delgado-Lezama R, Granados-Soto V. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol Pain 11: 41, 2015. doi: 10.1186/s12990-015-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Fan N, Ma C, Wang T, Han L, Fu K, Wang Y, Shimada SG, Dong X, LaMotte RH. Enhanced excitability of MRGPRA3- and MRGPRD-positive nociceptors in a model of inflammatory itch and pain. Brain 137: 1039–1050, 2014. doi: 10.1093/brain/awu007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Fu K, Yang J, Shimada SG, LaMotte RH. CXCR3 chemokine receptor signaling mediates itch in experimental allergic contact dermatitis. Pain 156: 1737–1746, 2015. doi: 10.1097/j.pain.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Li Y, Pan X, Zhang P, LaMotte RH, Ma C. Transient receptor potential canonical 3 (TRPC3) is required for IgG immune complex-induced excitation of the rat dorsal root ganglion neurons. J Neurosci 32: 9554–9562, 2012. doi: 10.1523/JNEUROSCI.6355-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Zhang P, LaMotte RH, Ma C. Neuronal Fc-gamma receptor I mediated excitatory effects of IgG immune complex on rat dorsal root ganglion neurons. Brain Behav Immun 25: 1399–1407, 2011. doi: 10.1016/j.bbi.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl− current and chemotaxis in murine microglia. J Immunol 168: 3221–3226, 2002. doi: 10.4049/jimmunol.168.7.3221. [DOI] [PubMed] [Google Scholar]
- Rocha-González HI, Mao S, Alvarez-Leefmans FJ. Na+, K+, 2Cl− cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J Neurophysiol 100: 169–184, 2008. doi: 10.1152/jn.01007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong JA, Xie W, Coyle DE, Zhang JM. Microarray analysis of rat sensory ganglia after local inflammation implicates novel cytokines in pain. PLoS One 7: e40779, 2012. doi: 10.1371/journal.pone.0040779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki A, Seo N, Ito T, Kumakiri M, Takigawa M, Tokura Y. Dominant expression of CXCR3 is associated with induced expression of IP-10 at hapten-challenged sites of murine contact hypersensitivity: a possible role for interferon-gamma-producing CD8+ T cells in IP-10 expression. J Dermatol Sci 28: 234–241, 2002. doi: 10.1016/S0923-1811(01)00172-4. [DOI] [PubMed] [Google Scholar]
- Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, Bautista DM. TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci 14: 595–602, 2011. doi: 10.1038/nn.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]