

Abstract
Huntington's disease (HD) is an inherited neurodegenerative disease caused by a polyglutamine expansion in the huntington protein (htt). The neuropathological hallmark of HD is the loss of neurons in the striatum and, to a lesser extent, in the cortex. Foxp1 is a member of the Forkhead family of transcription factors expressed selectively in the striatum and the cortex. In the brain, three major Foxp1 isoforms are expressed: isoform-A (∼90 kDa), isoform-D (∼70 kDa), and isoform-C (∼50 kDa). We find that expression of Foxp1 isoform-A and -D is selectively reduced in the striatum and cortex of R6/2 HD mice as well as in the striatum of HD patients. Furthermore, expression of mutant htt in neurons results in the downregulation of Foxp1. Elevating expression of isoform-A or -D protects cortical neurons from death caused by the expression of mutant htt. On the other hand, knockdown of Foxp1 promotes death in otherwise healthy neurons. Neuroprotection by Foxp1 is likely to be mediated by the transcriptional stimulation of the cell-cycle inhibitory protein p21Waf1/Cip1. Consistently, Foxp1 activates transcription of the p21Waf1/Cip1 gene promoter, and overexpression of Foxp1 in neurons results in the elevation of p21 expression. Moreover, knocking down of p21Waf1/Cip1 blocks the ability of Foxp1 to protect neurons from mut-Htt-induced neurotoxicity. We propose that the selective vulnerability of neurons of the striatum and cortex in HD is related to the loss of expression of Foxp1, a protein that is highly expressed in these neurons and required for their survival.
SIGNIFICANCE STATEMENT Although the mutant huntingtin gene is expressed widely, neurons of the striatum and cortex are selectively affected in Huntington's disease (HD). Our results suggest that this selectivity is attributable to the reduced expression of Foxp1, a protein expressed selectively in striatal and cortical neurons that plays a neuroprotective role in these cells. We show that protection by Foxp1 involves stimulation of the p21Waf1/Cip1 (Cdkn1a) gene. Although three major Foxp1 isoforms (A, C, and D) are expressed in the brain, only isoform-A has been studied in the nervous system. We show that isoform-D is also expressed selectively, neuroprotective and downregulated in HD mice and patients. Our results suggest that Foxp1 might be an attractive therapeutic target for HD.
Keywords: Foxp1, Huntington's disease, neurodegeneration, neuroprotection
Introduction
Huntington's disease (HD) is an inherited neurodegenerative disease caused by an abnormal expansion of a CAG repeat in the first exon of the huntingtin gene, resulting in a mutant protein with a polyglutamine expansion (Zuccato et al., 2010). Although mutant huntingtin (mut-Htt) is ubiquitously expressed in the brain, in both neurons and glial cells, neurodegeneration in HD is mostly restricted to medium spiny neurons (MSNs) of the striatum and, to a lesser degree, some neurons in layers II, V, and VI of the cortex (Zuccato et al., 2010). We propose that a key factor in the selective vulnerability of the striatum and cortex is the altered expression of a survival-promoting gene called Foxp1, which is expressed most highly in neurons of the striatum and, to a lower level, in the cortex (Desplats et al., 2006, 2008). Foxp1 is one of four members of the Foxp subfamily of Forkhead (FOX) proteins (Bowers and Konopka, 2012). Whereas Foxp1, Foxp2, and Foxp4 are expressed in the brain, Foxp3 is not (Tam et al., 2011; Le Fevre et al., 2013). Mutations of Foxp1 are linked to speech and language disorders, delays in motor development, autism, and mental retardation (Hamdan et al., 2010; Horn et al., 2010; Bowers and Konopka, 2012; Le Fevre et al., 2013; Bacon et al., 2014; Sollis et al., 2016). MRI scans have revealed abnormally enlarged ventricles in humans with Foxp1 mutations raising the possibility of neuronal loss (Pariani et al., 2009).
Conventional Foxp1 knock-out mice (Foxp1−/−) die at embryonic day 14.5 (E14.5) with cardiac abnormalities (Wang et al., 2004). Analyses of Foxp1+/− mice have revealed a role in motor neuron diversification, connectivity, and differentiation of stem cells into dopaminergic neurons (Dasen et al., 2008; Konstantoulas et al., 2010). Knockdown of Foxp1 in the developing cortex has revealed a role in regulating neuronal migration and morphogenesis (Li et al., 2015). These observations demonstrate the importance of Foxp1 in brain development. However, Foxp1 is expressed throughout adulthood, but little is known about its function in the adult brain. Within the striatum, Foxp1 is expressed in MSNs with no expression in striatal interneurons (Tamura et al., 2004). Brain-specific deletion of Foxp1 causes pronounced reduction in striatal volume (Bacon et al., 2014), which was suggested to be attributable to reduced proliferation of neuronal progenitors. Interestingly, the striatal reduction was not observed until 3 weeks of age. Since neurogenesis in the striatum is complete before this time, it is possible that degeneration is responsible for the smaller striatum.
The Foxp1 gene can be alternatively spliced to produce multiple isoforms, the largest of which is ∼90 kDa (also referred to as isoform-A). These isoforms are expressed tissue-specifically and may have distinct functions (Brown et al., 2008; Green et al., 2009; Santos et al., 2011). For example, ES-Foxp1, an isoform expressed only in embryonic stem cells and referred to as isoform-B, contributes to the maintenance of stem cell pluripotency (Gabut et al., 2011). Almost all published work on Foxp1 has focused on isoform-A. Although the brain expresses two other major isoforms of ∼50 and ∼70 kDa molecular weight (isoform-C and -D, respectively), the significance of these Foxp1 isoforms for neurons or the brain is not known.
We have investigated the role of Foxp1 in the regulation of neuronal survival. We describe that Foxp1 expression is reduced in the R6/2 mouse model of HD and in the striatum of HD patients. Forced expression of mut-Htt results in a downregulation of Foxp1 expression, whereas elevating expression of either isoform-A or -D inhibits mut-Htt neurotoxicity. To our knowledge, this is the first report demonstrating a protective role of Foxp1 in HD. Furthermore, this is the first study that investigates the function of the smaller isoforms of Foxp1 in the nervous system. We suggest that Foxp1 protects neurons by stimulating the expression of p21Waf1/Cip1, a cell-cycle regulatory protein with well documented neuroprotective effects.
Materials and Methods
Materials.
Unless stated otherwise, all tissue culture media were purchased from Invitrogen, and chemicals and reagents were purchased from Sigma-Aldrich. Poly-l-lysine for primary neuronal cultures was purchased from Trevigen. Antibodies used in this study are as follows: Foxp1 (from three sources: Cell Signaling Technologies, which recognizes Foxp1 amino acids adjacent to Leu584, catalog #D35D10, RRID:AB_10545755; Abcam, catalog #ab134055, RRID:AB_2632402; Bethyl Laboratories, which recognizes Foxp1 amino acids 627–677, catalog #A302-620A, RRID:AB_10555081), FLAG (Sigma-Aldrich, catalog #F1804, RRID:AB_262044), α-Tubulin (Sigma-Aldrich, catalog #T9026, RRID:AB_477593), ERK1/2 (Cell Signaling Technology, catalog #9102, RRID:AB_330744), GFP (Santa Cruz Biotechnology, mouse GFP, catalog #sc-9996a, RRID:AB_627695; and rabbit GFP, catalog #sc-8334, RRID:AB_641123), p21 (BD PharMingen, catalog #556430, RRID:AB_396414), and HRP-conjugated secondary antibodies (Pierce). Immunocytochemistry secondary antibodies Dylight 594 (catalog #115-585-146, RRID:AB_2338881) and Alexa Fluor 488 (catalog #115-545-146, RRID:AB_2307324), were obtained from Jackson ImmunoResearch Laboratories. Human caudate tissue samples from both male and female HD patients along with unaffected controls were obtained from NIH NeuroBioBank.
Expression plasmids.
FLAG-tagged Foxp1 plasmid pCMV10-mFoxp1 was a gift from Benjamin Blencowe (University of Toronto, Ontario, Canada) (Addgene plasmid 35170). GFP-tagged Foxp1 plasmids and Foxp1 isoform-D were cloned in our laboratory. GFP- and RFP-tagged Htt plasmids were a kind gift from Dr. Troy Littleton (Massachusetts Institute of Technology, Cambridge, MA). The p21Waf1/Cip1 promoter luciferase construct was a kind gift from Dr. Ming Zhang (Northwestern University Feinberg School of Medicine, Chicago, IL). FLAG-tagged p21Waf1/Cip1 was a gift from Mien-Chie Hung (The University of Texas MD Anderson Cancer Center, Houston, TX) (Addgene plasmid 16240).
Culturing, treatment, and transfection of cerebellar granule neurons.
Cerebellar granule neurons (CGNs) were cultured from 7- to 9-d-old Wistar rats and plated in Eagle's basal medium supplemented with 10% FBS, 25 mm KCl, 2 mm glutamine, and 0.2% gentamycin, as described previously (D'Mello et al., 1993). For 4- or 24-well dishes, cells were plated at a density of 1 × 106/well, and for 60 mm dishes, they were plated at a density of 10 × 106. The antimitotic agent cytosine arabinofuranoside was added 16–20 h later at a concentration of 10 μm. Treatment with high-potassium (HK) or low-potassium (LK) medium was performed 5 d after plating by switching the cultures to serum-free medium with 25 mm KCl (HK) or without KCl (LK) supplementation. Cell death was quantified by 4′6′-diamidino-2-phenylindole hydrochloride (DAPI) staining as described previously (Dastidar et al., 2011; Bardai et al., 2013). Cells with condensed or fragmented nuclei were scored as dead. Transfection of CGN cultures was performed 4–5 d after plating using the calcium phosphate method as described previously (Dastidar et al., 2011; Bardai et al., 2013). Unless mentioned otherwise, neurons were switched to HK or LK treatment 24 h after transfection and viability quantified 24 h later using immunocytochemistry to detect transfected cells. The proportion of transfected cells undergoing apoptosis was quantified by DAPI staining. The viability is normalized to that of the neurons transfected with GFP in the HK condition. Transfections for each experiment were performed in duplicates, and the experiments were repeated at least three times.
Culturing, treatment, and transfection of embryonic cortical neurons.
Cortical neurons were cultured from E17–E18 Wistar rat embryos as we have previously described (Dastidar et al., 2011; Bardai et al., 2013). The neurons were plated in Neurobasal medium with B27 supplement. Transfection was performed on day 5 of culture using the calcium phosphate method, and after 24 h of transfection, the neurons were subjected to apoptotic stimuli by adding 1 mm homocysteic acid (HCA), which induces oxidative stress (Ratan et al., 1994). After 16–18 h, the proportion of cells undergoing apoptosis was quantified by DAPI staining, and viability was normalized to that of the neurons transfected with GFP in the untreated condition. Transfections for each experiment were performed in duplicates, and the experiments were repeated at least three times.
Culturing of cortical glial cells.
The cortex was isolated from postnatal days 2–3 rat pups and placed in HBSS buffer. The tissue was then dissociated by the addition of trypsin, followed by mechanical dissociation. The trypsin was then neutralized by adding serum, and the cells were plated in DMEM with 10% FBS. The cell culture medium was replaced every 2 d, and the cells were subcultured when they reached 80% confluence. After two passages, the neurons were eliminated, and the cortical glial cells were used for the experiments.
Culture and transfection of cell lines.
Cell lines HEK293T (catalog #CRL-11268, RRID: CVCL_1926) and N2A (catalog #CCL-131, RRID: CVCL_0470) were obtained from ATCC. Each were cultured in DMEM supplemented with 10% FBS and transfected with Lipofectamine 2000 (Invitrogen) in Opti-MEM medium according to the manufacturer's instructions. Transfection efficiency was ∼70–80% in N2A cells and ∼80–90% in HEK293T cells.
Western blot analysis.
Cell culture and tissue samples were lysed, protein concentration was quantified, and Western blots were performed as described previously (Dastidar et al., 2011; Bardai et al., 2013). Typically, 50 μg of protein was used per lane and transferred following PAGE to PVDF membrane. After blocking with milk, membranes were incubated overnight in primary antibodies that were used at a 1:500 to 1:1000 dilution in TBST with 5% BSA. Appropriate secondary antibodies were used at 1:20,000 dilution in 5% milk in TBST for 1 h. Immunoreactivity was detected by enhanced chemiluminescence (GE Healthcare Bio-Sciences) according to the manufacturer's directions.
cDNA synthesis and RT-PCR.
RNA was extracted by lysing the cells in TRIzol (Invitrogen) and following the manufacturer's instructions. The RNA extracted was quantified, and 3 μg of RNA was used to generate cDNA using the Superscript First Strand Synthesis System (Invitrogen). GoTaq Green Master Mix (Promega) was used to perform PCR; initially, 1 μl of cDNA was used to check the expression levels of actin, and the quantity of cDNA used was adjusted according to actin normalization.
The following primers were used for amplification: Foxp1 MR For: 5′-GGACGATAGAAGCACAGCTCAATGT-3′ and Foxp1 MR Rev: 5′-ATGGGCACGTTGTATTTGTCTGAGT-3′; Foxp1 HUM For: 5′-GTTCCCGTGTCAGTGGCTAT-3′ and Foxp1 HUM Rev: 5′-TGTGATTGTTGCCTGTGGTT-3′; Actin For: 5′-GAGAGGGAAATCGTGCGTGAC-3′ and Actin Rev: 5′-CATCTGCTGGAAGGTGGACA-3′; c-Jun For: 5′-GATGGAAACGAC CTTCTACG-3′ and c-Jun Rev: 5′-GTTGAAGTTGCTGAGGTTGG-3′; p21 For: 5′-CCTGGTGATGTCCGACCTGTTCC-3′ and p21 Rev: 5′-GCGCTTGGAGTGATAGAAATCTG-3′.
shRNA-mediated knockdown.
Five shRNA constructs were obtained from Sigma-Aldrich to be used for knockdown analysis of Foxp1 [TRCN0000072003 (shA), TRCN0000072004 (shB), TRCN0000072005 (shC), TRCN0000072006 (shD), and TRCN0000321549 (shE)]. For knocking down the expression of p21Waf1/Cip1 shRNA constructs, TRCN0000042585 (sh1) and TRCN0000042587 (sh2), from Sigma-Aldrich, were used. The pLKO.1-TRC control vector that encodes an 18 bp nonhairpin-forming insert purchased from Addgene (catalog #10879) was used as control. The shRNAs were transfected into cortical neurons on day 4 in vitro and allowed to express for 72 h before quantifying the viability of transfected cells. The transfected cells were visualized by cotransfecting the shRNA with EGFP in a ratio of 6.5:1. The viability of these transfected cells was quantified by DAPI staining.
Site-directed mutagenesis.
Foxp1 mutant plasmids Foxp1ΔLZ [lacking the leucine zipper (LZ) region] and Foxp1ΔFH [lacking the Forkhead (FH) domain] were generated using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies) following the manufacturer's instructions. The GFP-tagged Foxp1 plasmid with the CMV promoter was used as the template. The plasmids were sequenced to confirm the mutations and expressed in HEK293 cells to confirm protein expression. The following primers were used to generate the mutants: Foxp1ΔLZ-376–397-For: 5′-GTAGAGTACAAATGCAGGTTCATGTGAAGTCTACAGAACC-3′; Foxp1ΔLZ-376–397-Rev: 5′-GGTTCTGTAGACTTCACATGAACCTGCATTTGTACTCTAC-3′; Foxp1ΔFH-493–583-For: 5′-GAATTTTATAAGAACGCGGAAGTTATTAAAAACATGCAGAGCAGCCAC-3′; Foxp1ΔFH-493–583-Rev: 5′-GTGGCTGCTCTGCATGTTTTTAATAACTTCCGCGTTCTTATAAAATTC-3′.
Adenovirus generation.
Adenovirus was generated using the ViraPower Adenovirus Expression kit from Invitrogen. The Foxp1 gene was cloned into the pDONR 221 shuttle vector, which was then transferred into the adenoviral vector pAd/CMV/V5-DEST. The gene-containing vector was then linearized with PacI and transfected into HEK293A cells for virus amplification. The crude lysate was then freeze thawed four times, followed by CsCl gradient ultracentrifugation to purify the virus. The approximate titer of the virus ranged from 1010 to 1011 pfu/ml. CGNs and cortical neuron cultures were infected with adenovirus on day 5 in vitro, and protein/gene expression changes were analyzed 36–48 h later. The transduction efficiency of ∼25–30% was observed using the adenovirus in neurons.
Bromodeoxyuridine assay.
Bromodeoxyuridine (BrdU) incorporation assays were performed as described previously (Majdzadeh et al., 2008; Mallick and D'Mello, 2014). Briefly, cell lines were transfected with the experimental plasmids by Lipofectamine 2000, and 22 h later, BrdU (200 μm) was added to the cell culture medium and incubated for 2 h. The cells were fixed with 4% paraformaldehyde, and immunocytochemistry was performed. The dividing cells (BrdU-positive cells) were identified using a monoclonal mouse antibody against BrdU (catalog #B8434, Sigma), whereas transfected cells were identified with GFP or FLAG antibodies. The proportion of transfected cells that were BrdU positive was quantified.
Luciferase assay for transcriptional activity.
A total of 3.75 μg p21Waf1/Cip1 luciferase reporter plasmid with firefly luciferase and 0.25 μg of renilla luciferase were transfected into N2A cells along with 4 μg of experimental plasmids. After 24 h, the cells were lysed with Passive Lysis Buffer, and the transcriptional assay was performed using the Dual-Luciferase reporter assay system (catalog #E1910, Promega). First, the substrate for firefly luciferase was added and the relative luminescence units (RLU) were measured in a luminometer, followed by the addition of Stop and Glow Reagent (which has a substrate for renilla luciferase), and then RLU were measured for renilla. The RLU values of firefly luciferase, which were normalized with renilla, were used to calculate the fold induction.
CRISPR knockdown.
CRISPR plasmids were constructed by cloning the target sequence of Foxp1 in vector PX458 (Addgene plasmid 48138), which has the cas9 gene in the backbone. The oligos for target sequences were melted in a hotplate, allowed to anneal at room temperature, and cloned into the sgRNA scaffold of PX458 digested with BbsI. The oligo sequences are as follows: Foxp1-CPR-1 For: 5′-CACCGTGACACTCGGTCCAACGGAG-3′; Foxp1-CPR-1 Rev: 5′-AAACCTCCGTTGGACCGAGTGTCAC-3′; Foxp1-CPR-2 For: 5′-CACCGCGAGAGCTGTCCATTGGTAG-3′; Foxp1-CPR-2 Rev: 5′-AAACCTACCAATGGACAGCTCTCGC-3′; Foxp1-CPR-3-For: 5′-CACCGTCTTCAGGTTCCCGTGTCAG-3′; Foxp1-CPR-3-Rev: 5′-AAACCTGACACGGGAACCTGAAGAC-3′.
The CRISPR plasmids were transfected into N2A cells by Lipofectamine 2000 to check for efficiency of knockdown by Western blotting and into primary neurons by the calcium phosphate method. Seventy-two hours later, immunocytochemistry was performed with the GFP antibody to detect the transfected cells, and the viability of the neurons was determined using DAPI as described previously.
Fly stocks and genetics.
Mammalian GFP-tagged full-length Foxp1 isoform-A and Foxp1 isoform-D were excised from mammalian expression vectors and subcloned into the Not1-Xba1 site of the modified fly upstream activation sequence (UAS) expression vector (Exelixis). The expression vectors containing the Foxp1 gene were then microinjected into flies by BestGene as part of a fee-for-service arrangement to obtain transgenic flies. The UAS-Foxp1A and UAS-Foxp1D flies were recombined with Appl-GAL4 to drive their expression in all neurons. The GMR-Htt Q120 flies were obtained from the Bloomington Stock Center (Indiana University Bloomington). All flies were maintained and crosses were set at room temperature (25°C) in standard Drosophila melanogaster medium.
Paraffin sections.
Paraffin sections for determining retinal degeneration were prepared as described by Bettencourt da Cruz et al. (2005). Briefly, whole flies were fixed in Carnoy's solution and dehydrated in an ethanol series, followed by incubation in methyl benzoate before embedding in paraffin. Sections were cut at 7 μm and imaged using the auto-fluorescence caused by the dispersed eye pigment.
Fast phototaxis.
Fast phototaxis assays were conducted in the dark using the countercurrent apparatus described by Benzer (1967) and a single light source. A detailed description of the experimental conditions has been published previously (Strauss and Heisenberg, 1993). Flies were starved for 4 h but had access to water before being tested. Five consecutive tests were performed in each experiment with a time allowance of 6 s to make a transition toward the light and into the next vial.
The R6/2 transgenic mouse model of HD.
Female C57BL/6J ovarian transplant mice hemizygous for exon 1 of the mutant Htt gene containing 120 ± 5 CAG repeats were bred with wild-type C57BL/6J males (The Jackson Laboratory), and the genotype of the progeny was analyzed by PCR. Transgenic mice (R6/2) and their wild-type gender-matched littermates were killed, and the brains were dissected. Cortex, striatum, cerebellum, and the rest of the brain/other brain parts were separated to be analyzed by Western blotting. Mice of both genders were used.
Hdh knock-in mouse model of HD.
Dissected brain tissue from Hdh knock-in mice with different polyQ lengths were a kind gift from Dr. Peter Detoff (University of Alabama at Birmingham, Birmingham, AL). The knock-in mice were as described by Kumar et al. (2016). The tissue was homogenized in RIPA buffer and analyzed by Western blotting.
Statistical analysis.
GraphPad Prism 5 software was used to generate the graphs in this study. For RT-PCR, Kodak 1D Software was used to quantify the intensity of the bands, which were normalized to that of housekeeping genes. For Western blots, ImageJ software was used to calculate the area and intensity of the bands. The intensity of the bands was then normalized with the loading control. Statistical analysis was performed by an unpaired two-tailed Student's t test for sample sizes <5 (n = 5). For larger sample sizes, the Mann–Whitney U test was used to calculate the significance level. For comparing more than two datasets, one-way ANOVA with post-test analysis was performed. The results are displayed as mean ± SD. For viability experiments, the transfections were performed in duplicate and the experiments were repeated at least three times, and >200 cells were counted for each condition. The p values <0.05 were considered as statistically significant, and asterisks in figure legends indicate the p values as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Animal usage.
All procedures conducted using animals were reviewed and approved by the Southern Methodist University Institutional Animal Care and Use Committee.
Results
Foxp1 is highly expressed in the cortex and striatum
To examine the expression of Foxp1 in the adult mouse brain, we performed Western blots. Foxp1-isoform-A (∼90 kDa) is expressed widely, but with highest expression in the striatum and cortex (Fig. 1). This is consistent with the results of previous studies (Desplats et al., 2006, 2008; Tang et al., 2012). In addition to isoform-A, expression of two other Foxp1 isoforms is clearly detectable in the brain. These have previously been designated as isoform-C (∼50 kDa) and isoform-D (∼70 kDa; Brown et al., 2008; Green et al., 2009; Santos et al., 2011). Isoform-D is also expressed selectively in the striatum and cortex and, in fact, more so than isoform-A (Fig. 1). Isoform-C is expressed at lower levels and in a pattern that is different from the two larger isoforms with expression in the hippocampus and other brain parts, but not cortex (Fig. 1).
Figure 1.

Foxp1 expression levels in different brain regions. Foxp1 protein expression levels in the mouse cortex (CTX), cerebellum (CBM), olfactory bulb (OB), hippocampus (HIP), striatum (STR), and other brain parts (OBP) at 12 months of age, analyzed using Foxp1 antibody (Bethyl Laboratories). α-Tubulin was used as a loading control.
Foxp1 expression is reduced in cortical neurons exposed to oxidative stress
Oxidative stress is an underlying feature in several neurodegenerative diseases, including HD (Johri and Beal, 2012; Schapira et al., 2014). Cortical neurons are particularly sensitive to oxidative stress and die when exposed to HCA (Ratan et al., 1994). Foxp1 mRNA and protein expression is reduced in HCA-treated neurons, and this occurs well before neuronal death is observed (Fig. 2A–D). The reduction in mRNA suggests that the downregulation occurs at the transcriptional level. Whereas Foxp1 expression was reduced, the expression of c-jun, commonly used as a marker of neuronal death, was increased by HCA (Fig. 2A). Although expressed in cultured CGNs at a low level, Foxp1 expression is not reduced during LK-induced death (Fig. 2E,F). This suggests that expression of Foxp1 during neuronal death is regulated cell-specifically.
Figure 2.

Foxp1 expression levels change in neurons under apoptotic stress. A, Foxp1 RNA expression analysis in cortical neurons subjected to apoptotic conditions. mRNA was prepared from cortical neurons treated with 1 mm HCA for 3 and 6 h. c-jun is used as a marker for apoptosis, and actin is used as a normalization control. B, Quantification of Foxp1 RNA expression levels normalized to the expression levels of β-actin. *p < 0.05 (n = 4). C, Foxp1 protein levels in cortical neurons under apoptotic stress from HCA treatment. Protein lysates were prepared at 3, 6, and 9 h of HCA treatment and subjected to Western blotting using Foxp1 antibody (Cell Signaling Technology). Erk1/2 is used as a loading control. D, Quantification of Foxp1 protein levels in cortical neurons treated with HCA. Foxp1 protein levels were normalized to the expression levels of Erk1/2. *p < 0.05 (n = 4). E, Foxp1 protein levels in CGNs subjected to apoptotic stress by LK treatment. Protein lysates were collected at 3 and 6 h of treatment and subjected to Western blotting using Foxp1 antibody (Cell Signaling Technology). Erk1/2 is used as a loading control. F, Quantification of Foxp1 protein expression levels normalized with expression levels of ERK. Not significant, n = 3. UN, Untreated.
Foxp1 expression is reduced in the striatum of HD mice and patients
Since Foxp1 is selectively expressed in the striatum and cortex, the same brain regions that are selectively affected in HD, we looked at the expression of Foxp1 in the brains of R6/2 mice, a widely used transgenic mouse model of HD (Bates et al., 1997). The expression of isoform-A and -D were dramatically reduced in the striatum and cortex (Fig. 3A). We and others have previously described that motor deficits in R6/2 mice can be observed at ∼7 weeks and, thus, Foxp1 downregulation in the R6/2 brain occurs before motor deficits are obvious. The downregulation was selective for the striatum and cortex because no reduction was observed in the cerebellum or other brain parts (Fig. 3C,D). Foxp1 expression in the striatum of R6/2 was comparable to wild-type mice at 2 weeks of age (Fig. 3B), arguing against a developmental effect of expression and indicating that the reduction in Foxp1 may trigger disease pathogenesis. In support of this conclusion, Foxp1 expression levels were also decreased in striatum of Hdh knock-in mice (Fig. 3F) but not to a significant extent in the cortex and cerebellum (Fig. 3E,G). The reduction in Foxp1 protein levels in the striatum of these mice correlated with the increase in length of the polyglutamine expansion, which previous studies have shown correlates with disease severity (Fig. 3H; Kumar et al., 2016). In addition to our own studies, microarray analyses conducted by other groups also list Foxp1 as one of the genes that is significantly downregulated in various mouse HD models (Table 1). We extended our analyses to lysates from the caudate of human HD patients and found a dramatic decrease in expression of Foxp1 mRNA levels, both in males (Fig. 4A) and females (Fig. 4B) compared with age- and gender-matched normal individuals (see Table 2 for sample details). In Western blots, Foxp1 isoform-A and isoform-D were decreased in lysates of HD patients compared with normal controls, whereas expression of isoform-C appeared to be increased (Fig. 4C,D). Reduced Foxp1 expression has also been described by other laboratories in R6/1 mice, a related HD model (Desplats et al., 2006; Tang et al., 2012). And although expression of Foxp1 protein has not been studied previously in HD patients, reduction of Foxp1 mRNA levels in the HD caudate has been described by another laboratory (Desplats et al., 2006; Tang et al., 2012). It is noteworthy that Foxp1 expression is not altered in the striatum or substantia nigra of 6-hydroxy dopamine-administered rats, a commonly used model of Parkinson's disease (Tang et al., 2012). Together with the absence of a downregulation in LK-treated CGNs, this observation suggests that Foxp1 downregulation is both cell type and disease selective.
Figure 3.

Foxp1 expression levels in Huntington's disease models. A, Western blotting analysis of Foxp1 in various brain regions of wild-type (WT) and R6/2 transgenic littermates of mice at different ages using Foxp1 antibody (Bethyl Laboratories). B, C, Western blotting analysis of Foxp1 in various brain regions of WT and R6/2 mice at 2 weeks of age (B) and at 12 weeks of age (C) using Foxp1 antibody (Cell Signaling Technology). D, Quantification of protein expression levels of Foxp1 (isoform-A plus isoform-D) in different brain regions of WT and R6/2 mice at 12 weeks of age. Erk1/2 was used as a normalization control. ***p < 0.001 compared with expression levels in WT mice (n = 3). E–G, Western blotting analysis of Foxp1 expression levels in cortex (E), striatum (F), and cerebellum (G) of knock-in mice having various lengths of polyglutamine expansion in the Huntingtin gene using Foxp1 antibody (Abcam). Tubulin is used as a loading control. H, Quantification of Foxp1 protein levels in various brain regions of Htt knock-in mice normalized with expression levels of Tubulin. **p < 0.01; ***p < 0.001 (n = 3). CTX, Cortex; CBM, cerebellum; STR, striatum; OBP, other brain parts.
Table 1.
Decreased Foxp1 mRNA levels reported in published datasets from human HD caudate and HD mouse models
| Model | Age/grade | Region | Gene ID | Log2 FC | p value | Reference |
|---|---|---|---|---|---|---|
| Human | Grades 0–4 | Caudate | FOXP1 | −0.578 | 2.57E-10 | Hodges et al. (2006) |
| R6/1 | 6 months | Striatum | Foxp1 | −1.152 | 1.70E-03 | Desplats et al. (2006) |
| R6/2-Q150 | 12 weeks | Striatum | Foxp1 | −0.775 | 3.94E-06 | Kuhn et al. (2007) |
| R6/2-Q150 | 12 weeks | Striatum | Foxp1 | −0.304 | 3.80E-03 | Tang et al. (2011) |
| HdhQ92 | 18 months | Striatum | Foxp1 | −1.737 | 1.02E-02 | Kuhn et al. (2007) |
| Yac128 | 12 months | Striatum | Foxp1 | −0.403 | 4.31E-02 | Kuhn et al. (2007) |
| CHL2 (HdhQ150) | 22 months | Striatum | Foxp1 | −0.979 | 7.55E-06 | Kuhn et al. (2007) |
| Het-KI-Q111 | 10 months | Striatum | Foxp1 | −0.223 | 5.10E-04 | Langfelder et al. (2016) |
| Het-KI-Q140 | 10 months | Striatum | Foxp1 | −0.341 | 1.06E-07 | Langfelder et al. (2016) |
| Het-KI-Q175 | 10 months | Striatum | Foxp1 | −0.325 | 4.09E-07 | Langfelder et al. (2016) |
FC, Fold change.
Figure 4.

FOXP1 mRNA and protein expression in caudate of HD patients. A, B, RNA extracted from caudate of age-matched normal male individuals and HD patients (A) or normal female individuals and HD patients (B) was subjected to RT-PCR analysis using primers amplifying regions common to all the isoforms of FOXP1. β-ACTIN was used as the normalization control. C, D, Foxp1 protein expression levels in normal male individuals and HD patients (C) or normal female individuals and HD patients (D) analyzed by Western blotting using Foxp1 antibody (Abcam). α-TUBULIN was used as the loading control, and a downregulation of isoform-A and -D and upregulation of isoform-C was observed. E, F, Quantification of Foxp1 RNA expression levels (E) and protein expression levels (F) in caudate of normal individuals and HD patients. G, Western blotting analysis of Foxp1 protein levels using Foxp1 antibody (Abcam) in N2A neuroblastoma cells and in cortical neurons (CN), after infection with GFP or Q138 adenovirus. H, Foxp1 isoform-A is shown, and the expression level in cortical neurons is quantified.
Table 2.
Summary of neuropathology and clinical diagnosis of human caudate tissue from NeuroBioBank
| NeuroBioBank sample ID | Age (years) | Gender | Postmortem interval | HD Vonsattel grade | Neuropathology | Other pathology |
|---|---|---|---|---|---|---|
| 13082 | 41 | Male | 27 h | n.a. | Normal | n.d. |
| 4340 | 47 | Male | 12.5 h | n.a. | Normal | Esophageal cancer with metastases to liver |
| 4615 | 49 | Male | 15 h | n.a. | Normal | Colon cancer with metastases to liver |
| 13058 | 50 | Male | 8 h | n.a. | Normal | n.d. |
| 13069 | 53 | Male | 25 h | n.a. | Normal | n.d. |
| 4827 | 55 | Male | 11.4 h | n.a. | Normal | Schizophrenia, Tourette's syndrome |
| 13106 | 59 | Male | 5 h | n.a. | Normal | n.d. |
| HCtZU_16_08 | 62 | Male | 29 h | n.a. | Normal | n.d. |
| 2983 | 30 | Male | 2 h | NG | HD | Early Huntington's disease |
| 2706 | 43 | Male | 17 h | 1–2 | HD | Dementia |
| 4518 | 49 | Male | 12.7 h | 3 | HD | Dementia, ataxia |
| 2858 | 50 | Male | 12 h | 2 | HD | n.d. |
| 3573 | 51 | Male | 32 h | 3 | HD | n.d. |
| 4254 | 53 | Male | 9.3 h | 3 | HD | Depression, dementia, pneumonia |
| 3744 | 55 | Male | 19 h | 1–2 | HD | n.d. |
| 2488 | 59 | Male | 9.5 h | 4 | HD | n.d. |
| 5293 | 41 | Female | 11.5 h | n.a. | Normal | Liver failure, pneumonia |
| 4836 | 52 | Female | 15.1 h | n.a. | Normal | Depression, suicide, ethanol abuse |
| 3175 | 54 | Female | 21.5 h | n.a. | Normal | Pancytopenia, diabetes type I, hypothyroidism |
| 4715 | 55 | Female | 4 h | n.a. | Normal | n.d. |
| 3558 | 59 | Female | 19.5 h | n.a. | Normal | Lymphoma, non-Hodgkins |
| HBJO_16_01 | 67 | Female | 21 h | n.a. | Normal | Asperger's disorder, opiod abuse |
| HCtYD_16_01 | 68 | Female | 19 h | n.a. | Normal | n.d. |
| HCtZZC_16_01 | 82 | Female | 14 h | n.a. | Normal | n.d. |
| 3383 | 40 | Female | 13 h | 1 | HD | n.d. |
| 2669 | 44 | Female | 11 h | NG | HD | Advanced Huntington's disease |
| HBGY_16_01 | 48 | Female | 32 h | NG | HD | Dementia, depression, malnutrition |
| 4072 | 50 | Female | 16.3 h | 2 | HD | Breast cancer |
| HBLF_16_01 | 62 | Female | 14 h | NG | HD | n.d. |
| HBCS_16_01 | 68 | Female | 43 h | NG | HD | n.d. |
| HBDY_16_01 | 73 | Female | 15 h | NG | HD | n.d. |
| HBFO_16_01 | 80 | Female | 38 h | NG | HD | n.d. |
n.d., Not determined; n.a., not applicable; NG, No grade.
To examine whether we could recapitulate the downregulation of Foxp1 expression seen in mouse models (Fig. 3D,H) and human HD patients (Fig. 4E,F) in a cell culture model of HD, we expressed mut-Htt in primary cortical neurons and Neuro2A cells, a mouse striatal cell line. As observed in HD mice and patients, substantial downregulation of Foxp1 was observed in mut-Htt-expressing neurons as well as N2A cells (Fig. 4G,H).
Foxp1 is needed for neuronal survival
The downregulation of Foxp1 isoform-A expression in both oxidatively stressed cortical neurons and degenerating regions of the R6/2 and human HD brain suggests that elevated expression of Foxp1 isoform-A is necessary for the full survival capacity of neurons. To investigate this possibility, we tested five commercially available shRNAs against Foxp1 and identified three that reduced Foxp1 protein levels significantly, designated as shA, shD, and shE (Fig. 5A). Expression of these three shRNAs induced cell death in otherwise healthy cortical neurons (Fig. 5B). In contrast, another shRNA, shC, which was incapable of knocking down Foxp1 expression, and shB, which only binds to Foxp1 isoform-A but not to the other isoforms, had no effect on neuronal survival (Fig. 5B). Similarly, when Foxp1 expression was knocked down using CRISPR/Cas9 technology using two separate guide RNA constructs, CPR-1 and CPR-2, the survival of cortical neurons was reduced (Fig. 5C,D). The knockdown of Foxp1 was confirmed in cortical neurons by immunocytochemistry using endogenous Foxp1 antibodies (Fig. 5E).
Figure 5.
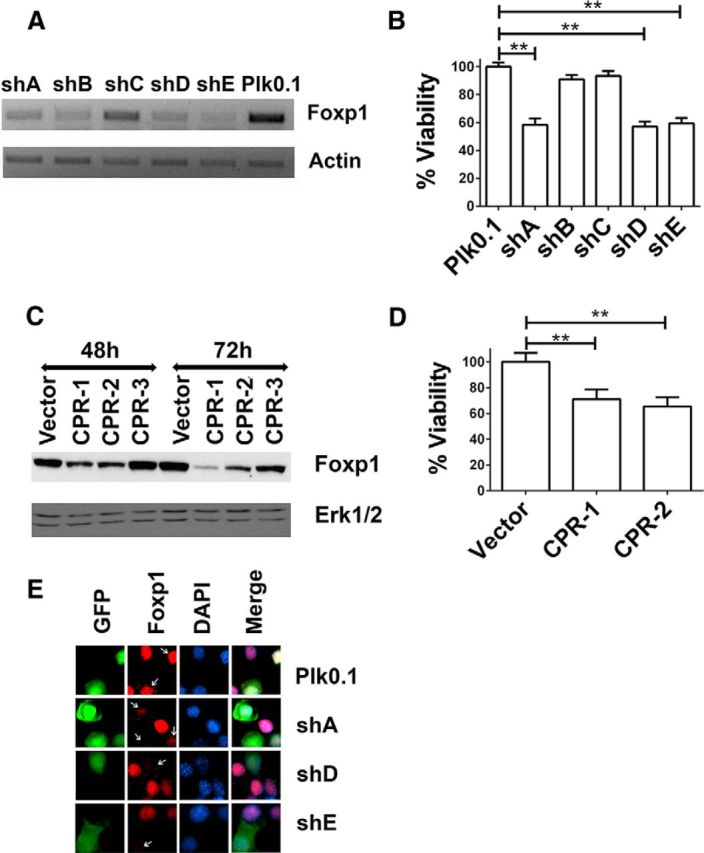
Suppression of Foxp1 expression induces apoptosis in healthy neurons. A, RNA was collected from cortical neurons infected with lentivirus generated from shRNA constructs (shA–shE) to knock down Foxp1 expression or by lentivirus Plk0.1 as control. β-Actin is used as the normalization control. B, Cortical neurons were cotransfected with Plk0.1 (control) or shRNA plasmids and GFP in a ratio of 6.5:1 for 72 h. Immunocytochemistry was performed using a GFP antibody to identify the transfected cells, and viability was determined by DAPI staining. **p < 0.01 (n = 3), compared with the viability of cells transfected with Plk0.1. Data are expressed as mean ± SD. C, CRISPR plasmids CPR-1, CPR-2, and CPR-3 and a vector control (PX-458) were transfected into N2A cells for 48 and 72 h. Protein lysates were collected and analyzed by Western blotting using Foxp1 antibody (Cell Signaling Technology). Erk1/2 is used as the loading control. D, Cortical neurons were transfected with PX-458 (vector) or CRISPR plasmids CPR-1 and CPR-2 for 72 h, immunocytochemistry was performed using a GFP antibody, and viability was quantified by DAPI staining. Viability was normalized to cortical neurons transfected with PX-458. The data are represented as mean ± SD. **p < 0.01, compared with neurons transfected with PX-458 (n = 3). E, Cortical neurons were cotransfected with Plk0.1 or shRNA plasmids along with GFP in a ratio of 6.5:1, and 72 h later, immunocytochemistry was performed to detect the endogenous levels of Foxp1 protein using Foxp1 antibody (Cell Signaling Technology) and to determine the efficiency of the shRNA in knocking down Foxp1.
Elevating Foxp1 levels protects against mut-Htt neurotoxicity
Whereas reduced Foxp1 expression results in loss of neuronal viability, restoring elevated levels of Foxp1 isoform-A by ectopic expression protected cortical neurons from HCA-induced death (Fig. 6A) and against mut-Htt neurotoxicity (Fig. 6B). The two domains essential for Foxp1 transcriptional activity are the LZ and the FH domains. We, therefore, generated mutant forms of Foxp1 lacking these two domains, Foxp1ΔLZ and Foxp1ΔFH, and tested their ability to protect against mut-Htt neurotoxicity. When ectopically expressed in neurons, Foxp1ΔLZ maintains a nuclear localization like wild-type Foxp1 whereas Foxp1ΔFH, which lacks the nuclear localization signal, shows a cytoplasmic localization (Fig. 6C). Neither of these mutant constructs could protect against mut-Htt neurotoxicity (Fig. 6B).
Figure 6.

Overexpression of Foxp1 protects neurons from apoptosis. A, Cortical neurons were transfected with GFP or Foxp1 for 24 h and treated with medium with or without 1 mm HCA for another 24 h. Viability of transfected neurons was quantified by DAPI staining, and the viability was normalized to GFP-transfected cortical neurons in medium without HCA. The data are represented as mean ± SD. **p < 0.01, compared with neurons transfected with GFP and treated with HCA (n = 4). UN, Untreated. B, Cortical neurons were transfected with either GFP alone or GFP, Foxp1A, Foxp1D, Foxp1ΔLZ, and Foxp1ΔFH along with Q138-RFP. After 24 h of transfection, the cells were fixed and immunocytochemistry was performed with GFP and RFP antibodies, and the viability of transfected cells was quantified by DAPI staining. Viability was normalized to neurons transfected with GFP alone. The data are represented as mean± SD. ***p < 0.001, compared with GFP and Q138-cotransfected cells. C, N2A cells were cotransfected with GFP or Foxp1 plasmids along with Q138-RFP, and immunocytochemistry was performed to determine the intracellular localization of the proteins. D, CGNs were transfected with GFP or Foxp1 plasmids for 24 h, and the medium was changed to HK or LK medium for another 24 h. Immunocytochemistry was performed with GFP/FLAG antibody to identify the transfected cells, and viability was determined by DAPI staining. Viability was normalized to cells transfected with GFP in HK medium. The data are represented as mean ± SD. ***p < 0.001, compared with neurons transfected with GFP and treated with HK medium (n = 6).
As shown in Figure 2, E and F, Foxp1 is expressed relatively weakly in CGNs, and its expression is not reduced in these neurons when cell death is induced by LK treatment. Yet, the overexpression of Foxp1 completely prevents LK-induced death of CGNs (Fig. 6D). This indicates that Foxp1 is a bona fide neuroprotective protein.
Virtually nothing is known about the functional significance of the smaller isoforms of Foxp1 in the nervous system. Because Foxp1 isoform-D is also downregulated in the striatum of patients and R/2 mice, we investigated whether it was also capable of protecting neurons. As shown in Figure 6B, Foxp1 isoform-D also protected cortical neurons against mut-Htt toxicity.
Foxp1 stimulates p21Waf1/Cip1 expression and inhibits cell-cycle progression
Tang et al. (2012) conducted an RNA-Seq analysis to identify genes regulated by Foxp1 isoform-A overexpression in cultured striatal neurons. One of the genes identified in that screen was CDKN1A, which encodes the cell-cycle inhibitory protein p21Waf1/Cip1 (Tang et al., 2012). Interestingly, a separate RNA-Seq study of the striatum of Foxp1+/− mice also identified p21Waf1/Cip1 as a gene that was positively regulated by Foxp1 (Araujo et al., 2015). We and several other laboratories have previously described neuroprotective effects of p21Waf1/Cip1, including protection against mut-Htt toxicity (Poluha et al., 1996; Tomita et al., 2006; Harms et al., 2007; Langley et al., 2008; Ma et al., 2013; Mallick and D'Mello, 2014). We, therefore, investigated whether Foxp1 exerts its neuroprotective effect through stimulation of p21Waf1/Cip1. Overexpression of Foxp1 isoform-A in striatally derived N2A cells led to an increase in p21Waf1/Cip1 protein expression (Fig. 7A). In contrast, Foxp1 mutants Foxp1ΔLZ and Foxp1ΔFH failed to stimulate p21Waf1/Cip1. Like Foxp1 isoform-A, expression of Foxp1 isoform-D also increased p21Waf1/Cip1 expression, whereas Foxp1 isoform-C had no effect (Fig. 7A). Foxp1 stimulated expression of p21Waf1/Cip1 in cortical neurons both under normal and apoptotic conditions (Fig. 7B). Similar results were obtained in CGNs upon overexpression (Fig. 7C). If stimulation of p21Waf1/Cip1 is necessary for neuroprotection by Foxp1, then knockdown of p21Waf1/Cip1 would be expected to abolish the ability of Foxp1 to protect. Indeed, knocking down p21Waf1/Cip1 using two separate shRNA constructs blocked the ability of Foxp1 to protect neurons from cell death (Fig. 7D). To examine whether p21Waf1/Cip1 was a direct target of Foxp1, we performed transcriptional assays in N2A cells by coexpressing Foxp1 plasmids with a p21Waf1/Cip1 promoter-Luciferase reporter plasmid. Both Foxp1 isoform-A and Foxp1 isoform-D stimulated the activity of the p21Waf1/Cip1 promoter (Fig. 7E).
Figure 7.

Foxp1 induces the expression of p21. A, N2A cells were transfected with GFP or Foxp1 plasmids for 24 h. The protein lysates were subjected to Western blotting with p21 antibody. Erk1/2 was used as the loading control. B, Adenovirus for GFP or Foxp1 was used to infect cortical neurons, and 48 h later, the neurons were maintained in either normal survival condition or HCA-induced apoptotic condition for 3 h, and protein lysates were collected and subjected to Western blotting with a p21 antibody. Erk1/2 was used as a loading control. C, CGNs were infected with GFP or Foxp1 adenovirus, and 48 h later, the protein lysates were collected and subjected to Western blotting with p21 antibody. Erk1/2 was used as a loading control. UN, Untreated. D, p21 shRNA plasmids were cotransfected with GFP or Foxp1-GFP plasmids in cortical neurons for 72 h, and the viability was accessed by immunocytochemistry and DAPI staining. Viability is normalized to cortical neurons transfected with Plk0.1 along with GFP. The data are represented as mean ± SD (n = 3). E, N2A cells were transfected with GFP or Foxp1 plasmids along with a p21 promoter luciferase construct for 24 h. Renilla was used as a transfection control. Promoter activity is expressed as a ratio of firefly luciferase to renilla luciferase. Fold change represents the ratio of p21 promoter activity in experimental transfected cells to p21 promoter activity in GFP-transfected cells. **p < 0.01; *p < 0.05, Foxp1 fold induction compared with the fold induction in GFP-transfected cells.
A large body of evidence suggests that aberrant re-entry into the cell cycle underlies cell loss in neurodegenerative diseases (Becker and Bonni, 2004; Greene et al., 2007). Since Foxp1 stimulates p21Waf1/Cip1 expression and given that p21Waf1/Cip1 is a strong cell-cycle inhibitor, it was likely that Foxp1 has an inhibitory effect on the cell-cycle machinery. Indeed, BrdU-incorporation assays indicate that Foxp1 does inhibit cell-cycle progression in N2A cells (Fig. 8A,B) and in rat primary cortical astrocytes (Fig. 8C).
Figure 8.

Foxp1 inhibits cell-cycle progression. A, N2A cells were transfected with plasmids expressing GFP, Foxp1, or p21Cip1/Waf1, which serves as a positive control for cell-cycle inhibition. BrdU was added to the culture medium 22 h later for 2 h. Immunocytochemistry was performed with GFP or FLAG antibodies to label the transfected cells and BrdU antibodies to label the dividing cells. B, C, BrdU assay in N2A cells (B) and in cortical glial cells (C). Percentage of BrdU-positive cells was calculated as BrdU-positive cells per transfected cells. **p < 0.01 (n = 3), Foxp1-transfected cells compared with GFP-transfected N2A cells.
Foxp1 is protective in a Drosophila model of HD
To extend our results on Foxp1-dependent neuroprotection to an in vivo model, we used a Drosophila HD model. In these flies, the coding region for an N-terminal Htt fragment (amino acids 1–170) containing 120 CAG repeats is fused to the GMR promoter, resulting in expression in the eye (Jackson et al., 1998). Although the eye develops normally in these flies, they develop severe photoreceptor degeneration as they age (Jackson et al., 1998). We, therefore, analyzed the degenerative phenotype in 6- and 10-d-old GMR-Htt Q120 flies with and without Foxp1 expression. For this analysis, we generated transgenic flies that express UAS constructs of either Foxp1 isoform-D or Foxp1 isoform-A, the two isoforms we found to be reduced in R6/2 mice and HD patients. These UAS constructs can be readily induced by crossing the flies with flies carrying a promoter-GAL4 construct (Brand and Perrimon, 1993). However, inducing their expression in the retina with the strong GMR-GAL4 promoter construct resulted in defects in the development of the eye, rendering them unsuitable for our analysis of age-related degeneration. As an alternative we used Appl-GAL4, which induces expression in all neurons (including photoreceptors), but at much lower levels. Indeed, expressing Foxp1 isoform-A or Foxp1 isoform-D with this driver had no detectable effect on the development of the eye (data not shown). Intriguingly, expression of either isoform with Appl-GAL4 suppressed the degenerative eye phenotype in GMR-Htt Q120 flies. Wild-type flies show a regular pattern of ommatidia in the eye that contains eight photoreceptors (each with its own light-sensitive rhabdomere), but because photoreceptors R7 and R8 are positioned above each other, only seven of the photoreceptors/rhabdomeres are detectable in cross sections (fly was 6 d old; Fig. 9A). In contrast, 6-d-old GMR-Htt Q120 control flies (carrying the Appl-GAL4 promoter construct) showed a highly disorganized retina, with reduced numbers of rhabdomeres (Fig. 9B). When we induced UAS-Foxp1 isoform-A (via Appl-GAL4) in these flies, their retinae appeared less disrupted and more rhabdomeres were present (Fig. 9C), and a similar result was obtained with UAS-Foxp1 isoform-D (data not shown). To quantify this phenotype, we counted the number of rhabdomeres per ommatidia in each line. Wild-type flies showed an average of 6.6 rhabdomeres (Fig. 9D), close to the expected seven (the difference is most likely attributable to a rhabdomere occasionally not being discernable). GMR-Htt Q120 flies with only the Appl-GAL4 driver showed a significantly reduced number of rhabdomeres, with an average of 2.5 per ommatidia. Expression of Foxp1 isoform-A or Foxp1 isoform-D increased this number to 4.2 and 4.4, confirming a protective function for both isoforms. When we examined flies that only expressed the Foxp1 constructs, we found no effect on rhabdomere numbers with Foxp1-isoform-D, but Foxp1-isoform-A did show a slight reduction to 6.3 rhabdomeres per ommatidia, which was significantly different compared with wild type (Fig. 9D). Although this construct induced a weak degenerative phenotype, it nevertheless reduced the retinal degeneration caused by mutant Htt, showing that both isoforms are protective in combination with Htt Q120. A similar protective effect was detected in 10-d-old flies (data not shown) but was more difficult to quantify because of the eye becoming progressively more disrupted with age. Finally, to determine whether the increased survival of photoreceptor cells also improved visual function, we performed phototaxis assays, which measure behavioral responses to a light source. As expected from the severe retinal degeneration seen in 10-d-old GMR-Htt Q120 flies (with Appl-GAL4), they showed a significantly reduced performance in this assay (Fig. 9E). In contrast, expressing Foxp1 isoform-A or Foxp1 isoform-D with Appl-GAL4 in GMR-Htt Q120 flies dramatically increased their performance in this assay. Remarkably, the Foxp1-isoform-D-expressing flies actually performed as well as wild-type flies, revealing a dramatic functional improvement. Inducing Foxp1-isoform-A or Foxp1-isoform-D in the wild-type background did not cause detectable changes in phototaxis behavior.
Figure 9.
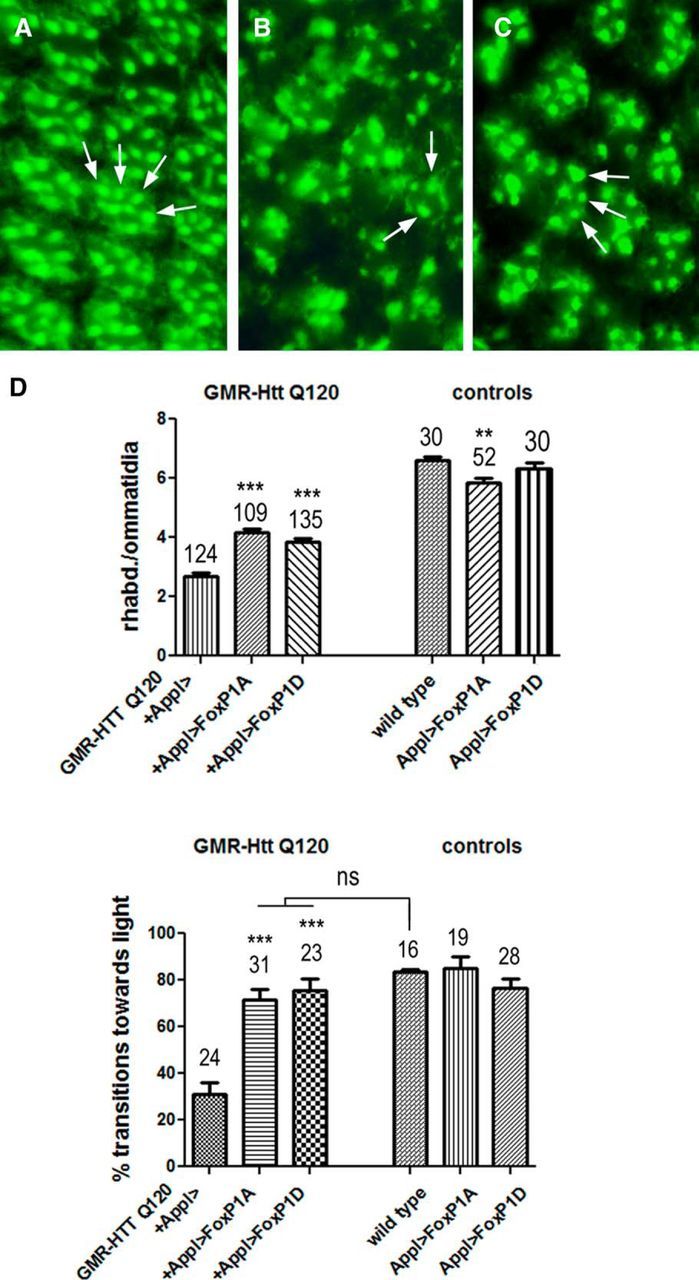
Foxp1 reduces photoreceptor loss in 6-d-old Htt Q120-expressing flies. A–C, Section through the retina of a 6-d-old wild-type fly (A), 6-d-old GMR-Htt Q120 fly (B), and 6-d-old GMR-Htt Q120, Appl-GAL4>UAS-Foxp1A fly (C). Some rhabdomeres are indicated by arrows. D, Counting the number of rhabdomeres per ommatidia revealed a significant increase from 2.5 rhabdomeres in GMR-Htt Q120 to 4.2 when Foxp1A was induced and 4.4 when Foxp1D was induced. The number of ommatidia analyzed is indicated (4–6 ommatidia were counted per retina and at least 5 flies were used). **p < 0.01; ***p < 0.001. E, The performance in the fast phototaxis assay is reduced in 10-d-old GMR-Htt Q120 flies compared with age-matched wild type, and this is also significantly improved by the expression of Foxp1A or Foxp1D. The number of flies tested is indicated. *p < 0.05; ***p < 0.001. All flies were females.
As a first step to determine whether the protective effects of Foxp1 were specific for Htt, we also determined the effects of Foxp1 isoform-A and Foxp1 isoform-D on amyloid β (Aβ)-induced degeneration and behavioral deficits but could not detect significant effects (data not shown). These studies confirm that both of these Foxp1 isoforms have a neuroprotective effect in vivo, and they indicate that Foxp1 specifically interacts with Htt, supporting the hypothesis that downregulation of Foxp1 is a key event in the pathology of HD.
Discussion
A defining feature of neurodegenerative diseases is the abnormal loss of neurons for which there is no cure or treatment strategy that can slow down the relentless death of neurons. A number of genes have been identified whose mutations cause familial forms of many of these diseases. An intriguing but yet unexplained feature about neurodegenerative diseases is the selective nature of neuronal loss. Specifically, although the mutant genes responsible for the various diseases are generally expressed widely in the brain, only specific brain regions and neuronal populations are affected in each disease. One possibility that may explain this selective vulnerability is that the mutant gene may affect, either directly or through other biochemical or metabolic changes, the expression of genes that are selectively expressed in the vulnerable brain regions and that regulate the survival of neurons in these regions. Reduced expression of such region- or cell-specific neuroprotective genes, or increased expression of region- or cell-specific neurotoxic genes, would thus explain the selective vulnerability of specific brain regions. Focusing on HD, we suggest that Foxp1 is one such gene. In support of this possibility, and as previously described (Desplats et al., 2006, 2008; Tang et al., 2012), we find that Foxp1 is expressed most highly in the striatum and to a lower level in the cortex of HD mice. Within the striatum, Foxp1 is expressed only in MSNs, which are the neuronal population that degenerates in HD (Desplats et al., 2006, 2008; Tang et al., 2012). Expression of Foxp1 is selectively reduced in the striatum and cortex of R6/2 HD mice and even more dramatically in the caudate of human HD patients. Forcibly knocking down Foxp1 induces death in otherwise healthy cortical neurons, supporting the idea that Foxp1 is necessary for their survival. In support of our hypothesis that the loss of specific neuronal populations in neurodegenerative diseases is attributable to an effect of the disease gene on molecules selectively expressed in those cell populations, we found that overexpression of mut-Htt is sufficient to reduce Foxp1 expression. Indeed, re-establishment of elevated levels of Foxp1 protects cultured cortical neurons from mut-Htt neurotoxicity. Furthermore, elevated Foxp1 protects against neuronal loss and improves behavioral performance in a Drosophila model of HD. It deserves mention that whereas neurons of the striatum and cortex selectively degenerate, other brain regions and neuronal types are also known to be affected in HD. Further investigation is needed to determine whether Foxp1 is expressed in these other neuronal types and whether its expression is reduced in these cells in HD. It is quite possible that although Foxp1 protects cortical and striatal neurons, the survival of other neuronal populations that are affected in HD may be safe-guarded by molecules other than members of the Foxp family or by other proteins all together.
The importance of Foxp for neuronal function is also supported by the finding that a neuronal knockdown of endogenous Foxp in flies causes defects in motor coordination (Lawton et al., 2014). However, whether this may be attributable to neuronal death was not investigated. Together with the protective function of Foxp1 in flies, this suggests that the neuroprotective function and the molecular pathways that mediate this function are conserved between Drosophila and mammals. An intriguing aspect of Foxp1-mediated neuroprotection is that although it can protect cultured cortical neurons and CGNs against HCA and LK-induced death, and can protect retinal neurons in Drosophila against mut-Htt toxicity, it cannot protect retinal neurons against Aβ toxicity. This suggests that Foxp1 cannot protect neurons in all circumstances. Certain neurotoxic stimuli may activate molecular mechanisms or a form of cell death that Foxp1 is unable to protect against. In the case of Aβ, it is possible that Aβ induces molecular alterations that Foxp1 cannot prevent or block the effects of.
Although it is known that the Foxp1 gene encodes multiple isoforms as a result of alternative splicing, the functional significance of the smaller isoforms of Foxp1 are poorly understood. Interestingly, isoform-D is even more selectively expressed in the striatum and cortex than Foxp1 isoform-A and is more robustly downregulated in R6/2 mice than the full-length form. That this isoform is also neuroprotective is shown by our findings that ectopic expression of isoform-D also protects neurons and flies from mut-Htt toxicity. Although further investigation is necessary, it is possible that downregulation of isoform-D is even more relevant to HD pathogenesis than downregulation of isoform-A. In contrast to these two other isoforms, isoform-C has a different pattern of expression and its levels are increased in the human HD striatum.
Our results point to p21Waf1/Cip1 as one target of Foxp1's neuroprotective action because elevating Foxp1 expression results in a stimulation of p21Waf1/Cip1 expression. Several other laboratories have described neuroprotective effects for p21Waf1/Cip1, including in the context of HD (Poluha et al., 1996; Tomita et al., 2006; Harms et al., 2007; Langley et al., 2008; Ma et al., 2013; Mallick and D'Mello, 2014). It is possible that once stimulated by Foxp1, p21Waf1/Cip1 acts by inhibiting cell-cycle progression. Aberrant cell-cycle progression has been described as a contributing factor in a variety of neurodegenerative diseases, including HD (Becker and Bonni, 2004; Greene et al., 2007; Liu et al., 2015). Whereas our results point to p21Waf1/Cip1 as a contributing factor, it is likely that there are other Foxp1 targets that contribute to its neuroprotective activity in the striatum and cortex. And it is also likely that there are other alterations besides reduced Foxp1 levels that combined result in HD. Indeed, although mutations in Foxp1 are associated with autism, speech/language deficiency, motor deficiencies, and mental retardation, they do not result in striatal degeneration or other major pathological features of HD. It may be noted, however, that mutations in other genes, whose reduced functions are widely believed to contribute to HD pathogenesis and considered as therapeutic targets, do not produce HD characteristics either. For example, mutations of BDNF, a molecule whose reduced activity and function are widely believed to contribute to HD, are associated with bulimia, anorexia, and congenital hypoventilation (Weese-Mayer et al., 2002; Ribases et al., 2004, 2005).
How does the presence of mut-Htt lead to a reduction in Foxp1 expression? It is possible that this reduction is mediated by HDAC3. We have previously described that HDAC3 is required for mut-Htt neurotoxicity (Bardai and D'Mello, 2011; Bardai et al., 2013). Other laboratories have described beneficial effects of HDAC3 inhibitors in mouse models of HD (Thomas et al., 2008; Jia et al., 2012). Although expressed in the healthy brain and required for proper brain development, HDAC3 promotes neuronal death when phosphorylated by GSK3β, whose increased activity has been noted in HD and other neurodegenerative diseases (Wei et al., 2001; Carmichael et al., 2002; Rangone et al., 2005). Other events that contribute to the transformation of HDAC3 to a neurotoxic protein include disassociation from wild-type Htt and association with HDAC1 (Bardai et al., 2013).
In summary, we describe for the first time that Foxp1, a gene expressed selectively in the striatum and cortex, is neuroprotective. Using both cultured neurons and an in vivo model, we present evidence indicating that the downregulation of Foxp1 in HD contributes to neurodegeneration. We show that a smaller isoform of Foxp1 is also neuroprotective and likely to be involved in HD pathogenesis. To our knowledge, this is the first attempt to investigate the functions of the smaller isoforms of Foxp1 in the nervous system. Finally, we suggest that Foxp1 preserves neuronal viability by stimulating the expression of p21Waf1/Cip1. We propose that elevating Foxp1 expression in vulnerable populations either pharmacologically or delivering it using viruses or other biological means could represent at attractive therapeutic approach for HD.
Footnotes
This work was supported by NIH Grant NS040408 to S.R.D. We thank Lulu Wang for technical assistance. We are grateful to Troy Littleton (Massachusetts Institute of Technology) and Dimitri Krainc (Northwestern University) for providing Htt and mutant-Htt plasmids. We thank Peter Detloff (University of Alabama at Birmingham) for the knock-in mouse samples. We also thank Ming Zhang (Northwestern University) for providing the p21Waf1/Cip1 promoter luciferase construct.
References
- Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, Rieger MA, Huang HC, Rajkovich K, Loerwald KW, Dekker JD, Tucker HO, Dougherty JD, Gibson JR, Konopka G (2015) FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 29:2081–2096. 10.1101/gad.267989.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, Rappold GA (2014) Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry 20:632–639. 10.1038/mp.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, D'Mello SR (2011) Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J Neurosci 31:1746–1751. 10.1523/JNEUROSCI.5704-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Verma P, Smith C, Rawat V, Wang L, D'Mello SR (2013) Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci 33:11833–11838. 10.1523/JNEUROSCI.5831-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP, Mangiarini L, Mahal A, Davies SW (1997) Transgenic models of Huntington's disease. Hum Mol Genet 6:1633–1637. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A (2004) Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol 72:1–25. [DOI] [PubMed] [Google Scholar]
- Benzer S. (1967) Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc Natl Acad Sci U S A 58:1112–1119. 10.1073/pnas.58.3.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt da Cruz A, Schwärzel M, Schulze S, Niyyati M, Heisenberg M, Kretzschmar D (2005) Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol Biol Cell 16:2433–2442. 10.1091/mbc.E04-11-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JM, Konopka G (2012) The role of the FOXP family of transcription factors in ASD. Dis Markers 33:251–260. 10.1155/2012/456787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415. [DOI] [PubMed] [Google Scholar]
- Brown PJ, Ashe SL, Leich E, Burek C, Barrans S, Fenton JA, Jack AS, Pulford K, Rosenwald A, Banham AH (2008) Potentially oncogenic B-cell activation-induced smaller isoforms of FOXP1 are highly expressed in the activated B cell-like subtype of DLBCL. Blood 111:2816–2824. 10.1182/blood-2007-09-115113 [DOI] [PubMed] [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC (2002) Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. J Biol Chem 277:33791–33798. 10.1074/jbc.M204861200 [DOI] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM (2008) Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell 134:304–316. 10.1016/j.cell.2008.06.019 [DOI] [PubMed] [Google Scholar]
- Dastidar SG, Landrieu PM, D'Mello SR (2011) FoxG1 promotes the survival of postmitotic neurons. J Neurosci 31:402–413. 10.1523/JNEUROSCI.2897-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats PA, Kass KE, Gilmartin T, Stanwood GD, Woodward EL, Head SR, Sutcliffe JG, Thomas EA (2006) Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J Neurochem 96:743–757. 10.1111/j.1471-4159.2005.03588.x [DOI] [PubMed] [Google Scholar]
- Desplats PA, Lambert JR, Thomas EA (2008) Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington's disease. Neurobiol Dis 31:298–308. 10.1016/j.nbd.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A 90:10989–10993. 10.1073/pnas.90.23.10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O'Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, Nedelec S, Wichterle H, Woltjen K, Hughes TR, Zandstra PW, Nagy A, Wrana JL, Blencowe BJ (2011) An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147:132–146. 10.1016/j.cell.2011.08.023 [DOI] [PubMed] [Google Scholar]
- Green MR, Gandhi MK, Courtney MJ, Marlton P, Griffiths L (2009) Relative abundance of full-length and truncated FOXP1 isoforms is associated with differential NFkappaB activity in follicular lymphoma. Leuk Res 33:1699–1702. 10.1016/j.leukres.2009.05.004 [DOI] [PubMed] [Google Scholar]
- Greene LA, Liu DX, Troy CM, Biswas SC (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta 1772:392–401. 10.1016/j.bbadis.2006.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs MO, Joober R, Lafrenière RG, Lacaille JC, Mottron L, Drapeau P, Beauchamp MH, Phillips MS, Fombonne E, Rouleau GA, Michaud JL (2010) De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet 87:671–678. 10.1016/j.ajhg.2010.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hubner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hörtnagl H (2007) Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci 27:4562–4571. 10.1523/JNEUROSCI.5110-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, et al. (2006) Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 15:965–977. 10.1093/hmg/ddl013 [DOI] [PubMed] [Google Scholar]
- Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S, Hempel M, Wagenstaller J, Gawthrope A, Monaco AP, et al. (2010) Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat 31:E1851–E1860. 10.1002/humu.21362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL (1998) Polyglutamine-expanded human Huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21:633–642. 10.1016/s0896-6273(00)80573-5 [DOI] [PubMed] [Google Scholar]
- Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, Sangrey GR, Darnell SB, Plasterer H, Sadri-Vakili G, Gottesfeld JM, Thompson LM, Rusche JR, Marsh JL, Thomas EA (2012) Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol Dis 46:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Beal MF (2012) Antioxidants in Huntington's disease. Biochim Biophys Acta 1822:664–674. 10.1016/j.bbadis.2011.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantoulas CJ, Parmar M, Li M (2010) FoxP1 promotes midbrain identity in embryonic stem cell-derived dopamine neurons by regulating Pitx3. J Neurochem 113:836–847. 10.1111/j.1471-4159.2010.06650.x [DOI] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RL, et al. (2007) Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 16:1845–1861. 10.1093/hmg/ddm133 [DOI] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Tallaksen-Greene S, Crowley MR, Crossman DK, Morton AJ, Van Groen T, Kadish I, Albin RL, Lesort M, Detloff PJ (2016) Allelic series of Huntington's disease knock-in mice reveals expression discorrelates. Hum Mol Genet 25:1619–1636. 10.1093/hmg/ddw040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, et al. (2016) Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci 19:623–633. 10.1038/nn.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR (2008) Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci 28:163–176. 10.1523/JNEUROSCI.3200-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton KJ, Wassmer TL, Deitcher DL (2014) Conserved role of Drosophila melanogaster FoxP in motor coordination and courtship song. Behav Brain Res 268:213–221. 10.1016/j.bbr.2014.04.009 [DOI] [PubMed] [Google Scholar]
- Le Fevre AK, Taylor S, Malek NH, Horn D, Carr CW, Abdul-Rahman OA, O'Donnell S, Burgess T, Shaw M, Gecz J, Bain N, Fagan K, Hunter MF (2013) FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A 161A:3166–3175. 10.1002/ajmg.a.36174 [DOI] [PubMed] [Google Scholar]
- Li X, Xiao J, Frohlich H, Tu X, Li L, Xu Y, Cao H, Qu J, Rappold GA, Chen JG (2015) Foxp1 regulates cortical radial migration and neuronal morphogenesis in developing cerebral cortex. PLoS One 10:e0127671. 10.1371/journal.pone.0127671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, James Shen CK, Marsh JL (2015) Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington's disease. Hum Mol Genet 24:1602–1616. 10.1093/hmg/ddu574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TC, Langley B, Ko B, Wei N, Gazaryan IG, Zareen N, Yamashiro DJ, Willis DE, Ratan RR (2013) A screen for inducers of p21(waf1/cip1) identifies HIF prolyl hydroxylase inhibitors as neuroprotective agents with antitumor properties. Neurobiol Dis 49:13–21. 10.1016/j.nbd.2012.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D'Mello SR (2008) HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol 68:1076–1092. 10.1002/dneu.20637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, D'Mello SR (2014) JAZ (Znf346), a SIRT1-interacting protein, protects neurons by stimulating p21 (WAF/CIP1) protein expression. J Biol Chem 289:35409–35420. 10.1074/jbc.M114.597575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariani MJ, Spencer A, Graham JM Jr, Rimoin DL (2009) A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur J Med Genet 52:123–127. 10.1016/j.ejmg.2009.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poluha W, Poluha DK, Chang B, Crosbie NE, Schonhoff CM, Kilpatrick DL, Ross AH (1996) The cyclin-dependent kinase inhibitor p21 (WAF1) is required for survival of differentiating neuroblastoma cells. Mol Cell Biol 16:1335–1341. 10.1128/MCB.16.4.1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangone H, Pardo R, Colin E, Girault JA, Saudou F, Humbert S (2005) Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J Biol Chem 280:22021–22028. 10.1074/jbc.M407528200 [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM (1994) Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 62:376–379. [DOI] [PubMed] [Google Scholar]
- Ribases M, Gratacos M, Fernandez-Aranda F, Bellodi L, Boni C, Anderluh M, Cavallini MC, Cellini E, Di Bella D, Erzegovesi S, Foulon C, Gabrovsek M, Gorwood P, Hebebrand J, Hinney A, Holliday J, Hu X, Karwautz A, Kipman A, Komel R, et al. (2004) Association of BDNF with anorexia, bulimia and age of onset of weight loss in six European populations. Hum Mol Genet 13:1205–1212. [DOI] [PubMed] [Google Scholar]
- Ribases M, Gratacos M, Fernandez-Aranda F, Bellodi L, Boni C, Anderluh M, Cristina Cavallini M, Cellini E, Di Bella D, Erzegovesi S, Foulon C, Gabrovsek M, Gorwood P, Hebebrand J, Hinney A, Holliday J, Hu X, Karwautz A, Kipman A, Komel R, et al. (2005) Association of BDNF with restricting anorexia nervosa and minimum body mass index: a family-based association study of eight European populations. Eur J Hum Genet 13:428–434. [DOI] [PubMed] [Google Scholar]
- Santos ME, Athanasiadis A, Leitao AB, DuPasquier L, Sucena E (2011) Alternative splicing and gene duplication in the evolution of the FoxP gene subfamily. Mol Biol Evol 28:237–247. 10.1093/molbev/msq182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E (2014) Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet 384:545–555. 10.1016/S0140-6736(14)61010-2 [DOI] [PubMed] [Google Scholar]
- Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, Gilissen C, Pfundt R, Rappold GA, Brunner HG, Deriziotis P, Fisher SE (2016) Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet 25:546–557. 10.1093/hmg/ddv495 [DOI] [PubMed] [Google Scholar]
- Strauss R, Heisenberg M (1993) A higher control center of locomotor behavior in the Drosophila brain. J Neurosci 13:1852–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WY, Leung CK, Tong KK, Kwan KM (2011) Foxp4 is essential in maintenance of Purkinje cell dendritic arborization in the mouse cerebellum. Neuroscience 172:562–571. 10.1016/j.neuroscience.2010.10.023 [DOI] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E (2004) Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience 124:261–267. 10.1016/j.neuroscience.2003.11.036 [DOI] [PubMed] [Google Scholar]
- Tang B, Becanovic K, Desplats PA, Spencer B, Hill AM, Connolly C, Masliah E, Leavitt BR, Thomas EA (2012) Forkhead box protein p1 is a transcriptional repressor of immune signaling in the CNS: implications for transcriptional dysregulation in Huntington disease. Hum Mol Genet 21:3097–3111. 10.1093/hmg/dds132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Seredenina T, Coppola G, Kuhn A, Geschwind DH, Luthi-Carter R, Thomas EA (2011) Gene expression profiling of R6/2 transgenic mice with different CAG repeat lengths reveals genes associated with disease onset and progression in Huntington's disease. Neurobiol Dis 42:459–467. 10.1016/j.nbd.2011.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM (2008) The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc Natl Acad Sci U S A 105:15564–15569. 10.1073/pnas.0804249105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Kubo T, Matsuda K, Madura T, Yano K, Fujiwara T, Tanaka H, Tohyama M, Hosokawa K (2006) p21Cip1/WAF1 regulates radial axon growth and enhances motor functional recovery in the injured peripheral nervous system. Brain Res 1081:44–52. 10.1016/j.brainres.2006.01.120 [DOI] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW (2004) Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development 131:4477–4487. 10.1242/dev.01287 [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Bolk S, Silvestri JM, Chakravarti A (2002) Idiopathic congenital central hypoventilation syndrome: evaluation of brain-derived neurotrophic factor genomic DNA sequence variation. Am J Med Genet 107:306–310. [DOI] [PubMed] [Google Scholar]
- Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, Chuang DM (2001) Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington's disease. Neuroscience 106:603–612. 10.1016/S0306-4522(01)00311-6 [DOI] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev 90:905–981. 10.1152/physrev.00041.2009 [DOI] [PubMed] [Google Scholar]