

Abstract
Mutations of the recombinase-activating genes 1 and 2 (RAG1 and RAG2) in humans are associated with a broad range of phenotypes. For patients with severe clinical presentation, hematopoietic stem cell transplantation (HSCT) represents the only curative treatment; however, high rates of graft failure and incomplete immune reconstitution have been observed, especially after unconditioned haploidentical transplantation. Studies in mice have shown that Rag−/− natural killer (NK) cells have a mature phenotype, reduced fitness, and increased cytotoxicity. We aimed to analyze NK cell phenotype and function in patients with mutations in RAG and in non-homologous end joining (NHEJ) genes. Here, we provide evidence that NK cells from these patients have an immature phenotype, with significant expansion of CD56bright CD16−/int CD57− cells, yet increased degranulation and high perforin content. Correlation was observed between in vitro recombinase activity of the mutant proteins, NK cell abnormalities, and in vivo clinical phenotype. Addition of serotherapy in the conditioning regimen, with the aim of depleting the autologous NK cell compartment, may be important to facilitate engraftment and immune reconstitution in patients with RAG and NHEJ defects treated by HSCT.
Keywords: natural killer cells, recombinase-activating genes, non-homologous end joining, immunodeficiency, CD56, interferon-γ, degranulation
Introduction
Natural killer (NK) cells are effector and immunomodulatory cells of the innate immune system. Through recognition of HLA class I molecules via activating and inhibitory killer cell immunoglobulin-like receptors (KIRs), and upon release of cytolytic granules, NK cells mediate killing of tumor and virus-infected cells and editing of dendritic cells, and play an important role in graft-versus-leukemia and graft rejection after hematopoietic stem cell transplantation (HSCT) (1–4). Furthermore, they can modulate immune responses by secreting chemokines and cytokines (5, 6).
Human peripheral blood contains two major NK cell subsets that can be distinguished based on the density of CD56 and CD16 expression on the cell surface: CD56bright CD16−/low and CD56dim CD16bright cells. These two NK cell subsets differ for the expression pattern of various other cell surface and intracellular molecules (7). In particular, CD56bright cells express NKp46, CD94/NKG2A, and CCR7 at higher levels than CD56dim NK cells, whereas CXCR1 and KIRs are more abundantly expressed by CD56dim cells. Furthermore, CD56bright and CD56dim NK cells have distinct functional properties, with CD56bright cells being potent producers of cytokines, and CD56dim cells being active mediators of natural and antibody-dependent cellular cytotoxicity, as also reflected by higher intracellular levels of perforin and granzymes (8, 9). In healthy adults, CD56bright cells comprise a minority (5–10%) of all circulating NK cells, but because they express CCR7, they are attracted to secondary lymphoid organs where they represent the predominant NK subset (10, 11). A subset of CD56low KIR+ NK cells, expressing CD57 represent terminally differentiated NK cells, whereas a further subset expressing the CD56− CD16+ CD57+ KIR+ phenotype are thought to represent exhausted NK cells (12).
RAG1 and RAG2 mutations in humans are associated with a broad spectrum of clinical and immunological phenotypes, including T− B− severe combined immune deficiency (SCID) (13), Omenn syndrome (OS) (14), atypical SCID (AS) (15–17), and combined immune deficiency with granuloma and/or autoimmunity (CID-G/A) (18–21). We have previously shown that the severity of the clinical and immunological phenotype in patients with RAG mutations correlates with the residual recombination activity of the mutant recombinase-activating gene (RAG) protein (22), which may differently affect diversity and composition of T and B cell receptor repertoires (23), whereas NK cell differentiation proceeds unaffected. For patients with severe RAG mutations presenting with SCID, OS, or AS, HSCT represents the only option of definitive cure; however, an increased rate of allograft rejection has been observed as compared to patients with other forms of SCID (24, 25). An important role of NK lymphocytes in mediating rejection of bone marrow allografts has been known for decades (26), but why patients with RAG deficiency would have a higher risk of graft rejection than other forms of NK+ SCID (such as IL7R or CD3 deficiency) remains unknown.
Although RAG genes are not required for NK cell development, data in mice indicate that Rag deficiency affects NK cell phenotype and function. It has been shown that expression of the RAG genes begins in common lymphoid progenitor cells that give rise to T, B, and NK cells (27–29). Studies in mice harboring transgenic reporters for Rag expression or recombinase activity have demonstrated the existence of two populations of mature NK cells: those that have been exposed to transient Rag expression during lymphoid differentiation (here termed as Ragpast) and NK cells that were not previously exposed to Rag (Ragnaive NK cells) (30). These two populations differ for their proliferative capacity and interleukin-2 (IL-2)-mediated Stat5 phosphorylation, and a progressive decrease in the proportion of Ragpast cells has been observed during NK cell differentiation (29). Furthermore, Ragnaive NK cells display an activated phenotype, increased cytotoxicity, and enhanced apoptosis, thereby resulting in poor survival and impaired DNA damage response as compared to their Ragpast counterpart (30). It has been postulated that Rag expression in lymphoid progenitors would favor selection of cells with optimal levels of expression of proteins involved in DNA break repair, including ARTEMIS and DNA ligase 4 (LIG4), thereby marking functionally distinct subsets of NK cells, and providing Ragpast NK cells with improved survival and “fitness.” If confirmed also in humans, the hypothesis that RAG deficiency results in the presence of NK cells with a distinctive phenotype and enhanced cytotoxic potential, would provide novel mechanistic insights to account for the high rate of primary graft failure, incomplete T cell reconstitution, and lack of B cell engraftment that are frequently observed following haploidentical HSCT for RAG deficiency, unless chemotherapy is used (25). To test this hypothesis, we have analyzed NK cell phenotype and function in a large cohort of patients with mutations in RAG and in other genes involved in non-homologous end joining (NHEJ), presenting with a variable severity of clinical and immunological manifestations, as compared to healthy donors of comparable age, and to patients with other forms of T or B cell lymphopenia.
Materials and Methods
Patients and Controls
The patient population consisted of 66 subjects with molecularly confirmed biallelic mutations in gene involved in V(D)J recombination, in particular 35 patients with RAG1, 11 with RAG2, 15 with DCLRE1C (ARTEMIS), 3 with LIG4, and 2 with NHEJ1 (Cernunnos/XLF) mutations. Based on the clinical and immunological phenotype (Table 1), these patients were assigned to the following subgroups: T− B− NK+ SCID (n = 19), OS (n = 11), AS (n = 13), and delayed onset combined immune deficiency (CID, n = 23). For the purposes of analysis, patients with OS and with AS, were combined into a single group (OS/AS, n = 24).
Table 1.
Clinical, molecular, and immunological features of patients with mutations in RAG or non-homologous end-joining (NHEJ) genes.
| Patient ID | Phenotype | Age | Gene defect and mutation | Lymphocyte subsets (cells/μL) | IgG (mg/dL) | IgA (mg/dL) | IgM (mg/dL) | IgE (kU/L) | Proliferation to phytohemagglutinin | Cytomegalovirus (CMV) status | EpsteinBarr virus (EBV) status | Infections | Autoimmunity | Granulomas | Skin rash | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3 | CD4 | CD8 | CD19 | CD16/56 | |||||||||||||||
| P1 | Severe combined immune deficiency (SCID) | 9 months | RAG1: p.R332X; p.R561H | 185 | 74 | 24 | 0 | 166 | 883^ | <7 | <5 | <2 | Absent | neg | neg | Rotavirus, Pseudomonas, Enterococcus enteritis | No | No | No |
| P2 | SCID | 2 months | RAG2: p.I218N; p.I218N | 25 | 23 | 0 | 1 | 155 | <7 | <7 | <5 | nd | nd | neg | neg | PJP | No | No | No |
| P3 | SCID | 10 months | RAG2: p.T125Rfs*10; p.T125Rfs*10 | 230 | 230 | 4 | 0 | 352 | 929^ | <7 | <5 | <1 | Absent | neg | neg | PJP, metapneumovirus, oral candidiasis | No | No | No |
| P4 | SCID | 2 months | RAG1: p.R841Q; p.F974L | 34 | 30 | 4 | 265 | 395 | 371 | 22 | 67 | 38 | Markedly reduced | neg | neg | No | No | No | No |
| P5 | SCID | 2 weeks | RAG1: p.R507W; p.S966T | 286 | 111 | 190 | 16 | 1,027 | 235 | <15 | 18 | nd | Reduced | pos | nd | CMV viremia | No | No | No |
| P6 | SCID | 6 months | RAG1: p.E965X; p.E965X | 2 | 2 | 0 | 0 | 252 | <7 | <7 | <5 | nd | Absent | neg | neg | Klebsiella pneumonia | No | No | No |
| P7 | SCID | 2 months | RAG1: p.M605I/R561C; p.F972S | 2 | 1 | 0 | 0 | 275 | 325 | <7 | <5 | nd | nd | pos | neg | RSV bronchiolitis, adenovirus pneumonia, CMV | No | No | No |
| P8 | SCID | 6 months | RAG2: p.C41W; p.C41W | 5 | 1 | 0 | 1 | 804 | 71 | <7 | <5 | <1 | absent | neg | neg | Disseminated BCG | No | No | No |
| P9 | SCID | 5 months | RAG1: p.G720D; p.G720D | 9 | 9 | 0 | 0 | 449 | 766 | <7 | <5 | <1 | absent | neg | neg | Cutaneous S. aureus abscess | No | No | No |
| P10 | SCID | 5 months | RAG1: p.C470Lfs*55; p.C470Lfs*55 | 236 | 90 | 146 | 0 | 80 | 161 | <7 | <5 | nd | nd | neg | neg | No | No | No | No |
| P11 | SCID | 1 week | RAG1: p.C176F; p.C176F | 23 | 13 | 1 | 573 | 1,449 | 754 | <7 | <5 | 7.8 | Absent | neg | neg | No | No | No | No |
| P12 | SCID | 14 months | DCLRE1C: exon1_3del; exon1_3del | 11 | 0 | 4 | 10 | 1,248 | 722^ | 29 | 16 | <1 | Absent | neg | neg | Disseminated BCG | No | AFB + necrotizing granulomatous lymphadenitis | No |
| P13 | SCID | 3 months | DCLRE1C: exon1_3del; exon1_3del | 1,076 | 1,070 | 10 | 0 | 904 | 50 | <7 | <5 | nd | nd | neg | neg | RSV pneumonia | No | No | Yes |
| P14 | SCID | 5 months | DCLRE1C: p.K157Kfs*13; p.K157Kfs*13 | 215 | 152 | 61 | 40 | 287 | 540 | 38 | 106 | nd | Markedly reduced | neg | neg | Pseudomonas bacteremia, recurrent otitis, skin abscesses, oral candidiasis | No | No | Papular rash |
| P15 | SCID | 1 week | DCLRE1C: exon1_3del; exon1_3del | 0 | 0 | 0 | 10 | 840 | nd | nd | nd | nd | nd | nd | nd | Sepsis | No | No | No |
| P16 | SCID | 23 months | DCLRE1C: p.Y199X; p.Y199X | 107 | 50 | 63 | 0 | 498 | 1,350^ | 15 | <5 | <1 | Absent | neg | neg | PJP, RSV pneumonia, cutaneous Pseudomonas infection, candidiasis | No | No | No |
| P17 | SCID | 28 months | DCLRE1C: p.Y199X; p.Y199X | 46 | 39 | 7 | 0 | 291 | 1,060 | 620 | nd | nd | Absent | neg | neg | No | No | No | np |
| P18 | SCID | 4 months | DCLRE1C: p.S32C; IVS11, +5g>a | 7 | 1 | 0 | 1 | 598 | 115 | <7 | nd | nd | Absent | neg | neg | Adenovirus hepatitis, oral ulcers | No | No | No |
| P19 | SCID | 10 months | DCLRE1C: p.F393fs*8; p.F393fs*8 | 13,997 (maternal) | 9,851 | 3,221 | 29 | 274 | 830^ | <7 | nd | nd | nd | pos | nd | Oral candidiasis | No | No | Generalized erythroderma |
| P20 | Omenn syndrome (OS) | 2 months | RAG1: p.R396H; p.R396H | 15,760 | 6,390 | 9,465 | 0 | 1,958 | 300 | <7 | <5 | 1,744 | Reduced | pos | neg | Candidiasis, E. coli UTI | No | No | Generalized erythroderma |
| P21 | OS | 1 month | RAG1: p.W959X; p.W959X | 3,424 | 3,368 | 0 | 0 | 5,003 | 390 | 22 | 16 | <2 | Absent | neg | neg | Rhinovirus | No | No | Generalized erythroderma |
| P22 | OS | 2 months | RAG1: p.R410Q; p.M435V | 19,230 | 10,286 | 8,215 | 0 | 816 | 1,120 | <7 | 101 | >5,000 | Markedly reduced | neg | neg | Rhinovirus | No | No | Generalized erythroderma |
| P23 | OS | 2 months | RAG1: p.R561C; p.R716W | 4,667 | 3,976 | 696 | 0 | 7,231 | <7 | <7 | <5 | 233 | Absent | neg | neg | No | No | No | Generalized erythroderma |
| P24 | OS | 4 months | RAG1: p.R396C; p.R404Q | 15,751 | 8,850 | 6,957 | 0 | 1,389 | 258 | 216 | <5 | 2,181 | Absent | neg | neg | PJP, rhinovirus | No | No | Generalized erythroderma |
| P25 | OS | 2 months | RAG1: p.R561H; p.R624H | 11,140 | 6,204 | 4,931 | 0 | 4,564 | 100 | 6 | 302 | >5,000 | Markedly reduced | pos | neg | No | Anti-smooth muscle ab | No | Generalized erythroderma |
| P26 | OS | 6 months | RAG2: p.G95V; p.E480X | 2,671 | 2,351 | 855 | 0 | 36 | 102 | 116 | 235 | <5 | Markedly reduced | neg | neg | Impetigo, otitis | No | No | Generalized erythroderma |
| P27 | OS | 1 month | RAG1: p.R142X; p.M458fs*33 | 5,105 | 5,029 | 228 | 15 | 4,458 | 634* | 17 | 8 | <2 | Markedly reduced | pos | neg | Recurrent URTI/LRTI | No | No | Generalized erythroderma |
| P28 | OS | 1 month | RAG1: p.S259Afs*5; p.S259Afs*5 | 170 | 155 | 15 | 0 | 320 | 815 | <7 | 5 | 14.4 | Markedly reduced | neg | neg | Enterococcus faecalis bacteremia | Intermittent neutropenia | No | Generalized erythroderma |
| P29 | OS | 4 months | RAG1: p.R332X; p.L1025fs*39 | 34,115 | 22,416 | 8,364 | 0 | 1,338 | <7 | <7 | <5 | 600 | Reduced | neg | neg | Enterovirus pneumonia, Moraxella bacteremia | No | No | Generalized erythroderma |
| P30 | OS | 4 months | DCLRE1C: exon1_3del; spling defect intron6 | 26,295 | 5,085 | 19,382 | 0 | 1,498 | 120 | 602 | 26 | 192 | nd | neg | neg | PJP, rhinovirus | No | No | Generalized erythroderma |
| P31 | Atypical SCID (AS) | Not known | RAG2: p.R123C; p.R123C | 1,056 | 96 | 780 | 23 | 67 | 206 | 11 | 14 | 1 | nd | neg | pos | Skin abscess, recurrent pneumonia, candidiasis | Lymphadenopathy | No | No |
| P32 | AS | 4 months | RAG1: p.H375D; p.H375D | 870 | 610 | 210 | 350 | 390 | 370 | nd | 89 | nd | Absent | nd | nd | Candidiasis | No | No | Candida rash |
| P33 | AS | 17 months | RAG1: p.R396C; p.M435V | 360 | 180 | 150 | 31 | 370 | 1,036 | <7 | 145 | 77 | Markedly reduced | neg | neg | Vaccine strain varicella zoster virus (VZV), chronic diarrhea | AIHA, vasculitis | No | Vasculitis |
| P34 | AS | 3 years | RAG1: p.H612R; p.A857V | 2,063 | 225 | 878 | 612 | 1,068 | 2,080 | <7 | 172 | 1.9 | Reduced | pos | neg | Adenovirus pneumonia, Klebsiella pneumonia, candidiasis | ANA, TPO Ab | No | No |
| P35 | AS | 2 years | RAG1: p.F939C; p.F939C | 729 | 110 | 266 | 41 | 733 | 1,260 | 55 | 215 | 78 | normal | nd | nd | BCG infection | No | No | No |
| P36 | AS | 2 months | RAG1: p.T173fs*27; p.T173fs*27 | 399 | 93 | 209 | 3 | 1,205 | 392 | 10 | <5 | 113 | absent | pos | neg | Pneumonia | AIHA | No | No |
| P37 | AS | 16 months | RAG2: p.G35A; p.G35A | 716 | 277 | 102 | 105 | 209 | 229 | 75 | 327 | nd | Reduced | neg | pos | Recurrent pneumonia (Pseudomonas, Klebsiella, H. influenzae), nail candidiasis | AIHA, psoriasis | NO | Psoriasis |
| P38 | AS | 25 months | RAG1: p.R841W; p.R841W | 950 | 220 | 20 | 121 | 665 | 538^ | 146 | 54 | 2 | Normal | neg | neg | Adenovirus, rhinovirus | Tubulointerstitial nephritis with lymphocytic infiltrate | No | No |
| P39 | AS | 13 months | RAG1: p.K86Vfs*33; p.K86Vfs*33 | 1,010 | 80 | 840 | 620 | 460 | 2,420 | 194 | 328 | 5.6 | Markedly reduced | pos | pos | Pseudomonas sepsis, CMV, BCGitis | Miller-Fisher | No | No |
| P40 | AS | 4 years | DCLRE1C: p.T65I; p.T65I | 558 | 207 | 243 | 36 | 108 | 560 | 19 | 54 | 5 | nd | nd | nd | HPV | No | No | Warts |
| P41 | AS | 10 years | DCLRE1C: p.T65I; p.T65I | 683 | 268 | 390 | 24 | 403 | 1,190 | <7 | 87 | 5 | nd | nd | nd | HPV, recurrent URTI/LRTI | No | No | Warts |
| P42 | AS | 2 years | DCLRE1C: p.T65I; p.T65I | 547 | 217 | 268 | 22 | 226 | 240 | <7 | 35 | 5 | Absent | nd | nd | HPV, BCG | No | No | Warts |
| P43 | AS | 9 months | NHEJ1: p.R57X; p.R57X | 426 | 299 | 98 | 259 | 265 | 600 | <7 | 949 | nd | Reduced | neg | neg | Adenovirus | No | No | No |
| P44 | CID | 6 years | RAG2: p.T215I; p.T215I | 840 | 560 | 180 | 70 | 190 | 2,480^ | <7 | 170 | nd | Reduced | nd | pos | Recurrent URTI, intermittent diarrhea | Neutropenia | No | No |
| P45 | CID | 19 years | RAG2: p.G35V; p.M322T | 457 | 236 | 184 | 165 | 5,781 | <100 | <7 | <5 | nd | Reduced | neg | pos | Single episode of RTI requiring admission at age 13 years | No | Skin granulomatosis | Skin granulomas |
| P46 | CID | 11 years | RAG1:p.R404Q; p.R404Q | 4,520 | 3,300 | 1,220 | 61 | 1,004 | <200 | <7 | 30 | nd | Absent | pos | pos | CMV retinitis, esophageal candidiasis, EBV | Colitis | Colon granulomatosis | No |
| P47 | CID | 8 years | RAG1: p.H612R; p.H612R | 526 | 361 | 144 | 373 | 106 | 859^ | <7 | <5 | 1 | Markedly reduced | na | na | Recurrent pneumonia, MAC, cellulitis | AIHA | No | No |
| P48 | CID | 17 years | RAG1: p.K86Vfs*33; p.H612R | 581 | 530 | 80 | 460 | 450 | 390 | <7 | 38 | <1 | Reduced | pos | pos | Herpes zoster, H. influenzae, Corynebacterium propinquum, Pseudomonas | ITP, vitiligo | Lung granulomatosis | No |
| P49 | CID | 11 years | RAG1: p.L514R; p.L514R | 2,831 | 1,658 | 837 | 0* | 309 | <140 | <7 | 130 | <18 | normal | neg | pos | Recurrent URTI/LRTI, warts, molluscum, EBV-LPD | Autoimmune cytopenias | Granulomas in liver and lungs | Warts, molluscum |
| P50 | CID | 31 years | RAG2: p.F62L; p.F62L | 380 | 225 | 162 | 78 | 215 | 1,000^ | <7 | 25 | nd | nd | na | na | Disseminated VZV, cryptococcus meningitis, URTI, Pseudomonas pneumonia | No | Lung granulomatosis | |
| P51 | CID | 12 years | RAG1: p.R624H; p.Y728H | 665 | 406 | 219 | 23 | 177 | 8 | <7 | 10 | <2 | Normal | neg | pos | Moderately severe VZV, zoster, recurrent URTI/LRTI | No | No | No |
| P52 | CID | 9 years | RAG2: p.V8I; p.D400H | 2,253 | 1,275 | 774 | 293 | 205 | 910 | 257 | 33 | 29 | Normal | neg | neg | S. pneumoniae sepsis, severe VZV infection, recurrent URTI/LRTI, perianal abscess | No | No | Severe atopic dermatitis |
| P53 | CID | 16 years | RAG1: p.H612R; p.H612R | 629 | 390 | 141 | 69 | 60 | 637 | 37 | 45 | nd | Normal | neg | neg | Recurrent otitris, recurrent pneumonia, S. aureus skin infection | Alopecia areata, AIHA, neutropenia | No | No |
| P54 | CID | 9 years | RAG1: p.R474C; p.L506F | 890 | 500 | 400 | 300 | 130 | 560 | 20 | 60 | 5 | Normal | neg | pos | VZV pneumonia, bronchiectasis | No | No | No |
| P55 | CID | 39 years | RAG1: p.R108X; p.W522C | 532 | 336 | 179 | 14 | 166 | 673 | 106 | 69 | <1 | nd | nd | nd | Pneumonia, warts, orolabial HSV, oral candidiasis, MRSA skin infection | Alopecia, autoimmune hypothyroidism, autoimmune hypogonadism | Vocal cord granuloma | No |
| P56 | CID | 16 years | RAG1: p.H375D; p.Y562C | 522 | 468 | 18 | 72 | 6 | 1,346^ | 420 | 320 | nd | Reduced | neg | neg | Pneumonia | ITP, neutropenia | Granulomatosis of skin, liver, spleen, lungs | No |
| P57 | CID | 10 years | RAG1: p.R410W; p.H375D | 273 | 169 | 42 | 56 | 150 | 597^ | 71 | 87 | nd | Normal | neg | neg | Recurrent LRTI | Alopecia totalis, vitiligo, myasthenia gravis | No | No |
| P58 | CID | 40 years | RAG1: p.R108X; p.W522C | 362 | 235 | 117 | 0* | 106 | 976 | 100 | 33 | 0 | nd | neg | neg | Recurrent URTI/LRTI, warts, mouth sores, oral and nail candidiasis | Alopecia, vitiligo | No | No |
| P59 | CID | 36 years | RAG2: p.N173S; p.E437K | 260 | 204 | 54 | 0* | 134 | 1,050^ | 17 | 17 | 0 | Reduced | neg | pos | Septis arthritis, recurrent pneumonia, warts | Alopecia, myositis | No | Psoriatic rash |
| P60 | CID | 30 months | DCLRE1C: p.G126D; p.L70del | 2,800 | 1,040 | 2,340 | 520 | 510 | 630^ | <7 | 210 | <2 | Markedly reduced | neg | pos | Echovirus, adenovirus | AIHA | no | Perianal rash |
| P61 | CID | 2 years | DCLRE1C: p.T65I; p.T65I | 540 | 160 | 240 | 16 | 1,320 | 135 | <7 | 15 | nd | nd | nd | nd | Recurrent URTI/LRTI, aphtous stomatitis | Lupus vulgaris | No | Lupus vulgaris |
| P62 | CID | 12 years | DCLRE1C: p.S147fs*6; p.S147fs*6 | 260 | 170 | 90 | 2 | 1,635 | 173 | <7 | 11 | 2 | nd | neg | neg | JC virus-associated PML | Vasculitis | Cutaneous granulomatous vasculitis | Vasculitis |
| P63 | CID | 7 years | NHEJ1: p.R57G; p.R57G | 693 | 319 | 330 | 44 | 195 | 60 | <7 | 104 | nd | Reduced | neg | neg | Recurrent URTI/LRTI | No | No | No |
| P64 | CID | 3 years | Ligase 4 (LIG4): p.R278H; p.R278H | 3,088 | 1,191 | 1,832 | 46 | 2,081 | 1,400^ | <7 | 167 | nd | Normal | neg | pos | Chronic calicivirus | No | No | No |
| P65 | CID | 8 years | LIG4: p.K424Rfs*20; p.R278H | 513 | 214 | 289 | 96 | 438 | 622 | <7 | <10 | 2 | Reduced | pos | pos | Pneumonia, otitis | No | No | No |
| P66 | CID | 17 years | LIG4: p.K449Q; p.R814X | 771 | 308 | 410 | 12 | 140 | 149 | <7 | <5 | <1 | Reduced | neg | neg | Recurrent URTI/LRTI, bronchiectasis, skin ringworm infection | No | No | No |
^, on immunoglobulin replacement therapy; Ab, antibody; AIHA, autoimmune hemolytic anemia; ANA, anti-nuclear antibodies; BCG, bacillus calmette-guerin; CMV, cytomegalovirus; HPV, human papillomavirus; ITP, immune thrombocytopenia; LPD, lymphoproliferative disease; LRTI, lower respiratory tract infection; PJP, Pneumocystis jiroveci pneumonia; RSV, respiratory syncitial virus; TPO, thyroid peroxidase; URTI, upper respiratory tract infection; UTI, urinary tract infection; VZV, Varicella zoster virus.
In addition, 22 patients with various other conditions characterized by numerical and/or functional defects of T lymphocytes served as a control for T cell deficiency. This group of other T cell defects (TCDs) included four patients with CD3G mutations, two patients with Di George syndrome, two patients with IL7R deficiency, and one patient each with mutations in MSN, JAK3, IL2RG, CD3D, CD3E, RMRP genes, or with MHC class II deficiency. In seven cases with T cell deficiency, the underlying diagnosis was unknown, including two infants with severe T cell lymphopenia diagnosed at birth with low T cell receptor excision circle (TRECs). Based on established criteria (31), 7 of these patients met definition of SCID (with <300 T cells/μL), and the remaining 15 had either less severe numerical TCDs or had functional T cell abnormalities. None of these patients with TCD carried mutations in RAG1/2, DCLRE1C, PRKDC, LIG4, or NHEJ1 genes.
As a control for B cell lymphopenia, nine patients with molecularly confirmed mutation in the BTK gene, causing X-linked agammaglobulinemia (XLA), were also studied.
Finally, 19 healthy infants (age ≤2 years) and 29 healthy subjects of >2 years of age served as normal controls for patients with SCID and OS/AS, or for patients with CID, respectively.
The study was approved by the Institutional Review Boards of Boston Children’s Hospital, the National Institute of Allergy and Infectious Diseases (protocols 93-I-0119 and 16-I-N139), and of all other referring centers. Blood samples were obtained upon written informed consent of the subject or, in the case of minors, of their parents or legal guardians. Because of limitations in blood volume and/or in the number of cells, not all analyses could be performed in each individual included in the study.
Analysis of NK Cell Phenotype
Peripheral blood was collected in EDTA- or heparin-containing vacutainers. Red blood cells were lysed from 200 µl of EDTA-blood using RBC lysis buffer (eBioscience). Cells were washed with FACS buffer (PBS containing 2% FBS), then equally distributed in five tubes and incubated for 1 h at 4°C with primary antibodies (Ab), as described below. After washing with FACS buffer, cells were incubated for 30 min at 4°C with secondary antibodies, washed again with FACS buffer, and incubated for 30 min at 4°C with conjugated antibodies. After washing, cells were re-suspended in FACS buffer and immediately analyzed on LSR Fortessa Flow Cytometer (BD) using FACS Diva v.6.1.3 software (BD). NK cells were defined as CD56+ CD3− CD20− CD14− cells. Final analysis on the expression of NK cell markers was performed using FloJo v.10.2 (TreeStar). The antibodies used to define NK cell phenotype were either commercially available or were generated in the laboratories of Alessandro Moretta and Silvia Parolini.
To analyze the expression of NK cell surface markers, five different tubes were prepared (Table 2), each of which contained FITC-conjugated antibodies directed against CD3, CD20, and CD14, to gate out T cells, B cells, and monocytes. Furthermore, tube #1 contained isotype controls, whereas tubes #2–5 contained antibodies to NK cell markers.
Table 2.
Combination of antibodies used to characterize natural killer cell phenotype.
| Tube # | Antibody | Source |
|---|---|---|
| 1 | IgG1 isotype control | Biolegend |
| IgG2a isotype control | Biolegend | |
| IgG2b isotype control | Biolegend | |
| PE-conjugated goat anti-mouse IgG1 | Southern Biotech | |
| PE-conjugated goat anti-mouse IgG2a | Southern Biotech | |
| PE/Cy7-conjugated goat anti-mouse IgG2b | Southern Biotech | |
| FITC-conjugated anti-CD3 | Beckman Coulter | |
| PC5-conjugated anti-CD56 | Beckman Coulter | |
| FITC-conjugated anti-CD14 | Beckman Coulter | |
| FITC-conjugated anti-CD20 | Biolegend | |
| APC/Cy7-conjugated IgG1 | Biolegend | |
| BV510-conjugated IgG1 | Biolegend | |
| BV421-conjugated mouse IgM | Becton-Dickinson | |
| 2 | Anti-NKG2A (Z199) [IgG2b] | A. Moretta, S. Parolini |
| Anti-KIR2 DL1/S1 (AZ115) [IgG1] | A. Moretta, S. Parolini | |
| Anti-KIR2 DL2/S2 (GL183) [IgG1] | A. Moretta, S. Parolini | |
| Anti-KIR3D (AZ158) [IgG2a] | A. Moretta, S. Parolini | |
| PE-conjugated goat-anti-mouse IgG1 | Southern Biotech | |
| PE-conjugated goat anti-mouse IgG2a | Southern Biotech | |
| PE/Cy7-conjugated goat anti-mouse IgG2b | Southern Biotech | |
| FITC-conjugated anti-CD3 | Beckman Coulter | |
| PC5-conjugated anti-CD56 | Beckman Coulter | |
| FITC-conjugated anti-CD14 | Beckman Coulter | |
| FITC-conjugated anti-CD20 | Biolegend | |
| APC/Cy7-conjugated anti-CD16 | Biolegend | |
| BV510-conjugated anti-CD69 | Biolegend | |
| BV421-conjugated anti-CD57 | Becton-Dickinson | |
| 3 | Anti-SIGLEC7 (Z176) [IgG2b] | A. Moretta, S. Parolini |
| Anti-LIR1 (F278) [IgG1] | A. Moretta, S. Parolini | |
| PE-conjugated goat-anti-mouse IgG1 | Southern Biotech | |
| PE/Cy7-conjugated goat anti-mouse IgG2b | Southern Biotech | |
| FITC-conjugated anti-CD3 | Beckman Coulter | |
| PC5-conjugated anti-CD56 | Beckman Coulter | |
| FITC-conjugated anti-CD14 | Beckman Coulter | |
| FITC-conjugated anti-CD20 | Biolegend | |
| APC/Cy7-conjugated anti-CD16 | Biolegend | |
| BV510-conjugated anti-CD69 | Biolegend | |
| BV421-conjugated anti-CD57 | Becton-Dickinson | |
| 4 | Anti-NKG2C [IgG2b] | R&D Systems |
| Anti-KIR2 DL1/S1 (11PB6) [IgG1] | A. Moretta, S. Parolini | |
| Anti-KIR2 DL2/S2 (GL183) [IgG1] | A. Moretta, S. Parolini | |
| Anti-KIR3D (AZ158) [IgG2a] | A. Moretta, S. Parolini | |
| PE-conjugated goat-anti-mouse IgG1 | Southern Biotech | |
| PE-conjugated goat anti-mouse IgG2a | Southern Biotech | |
| PE/Cy7-conjugated goat anti-mouse IgG2b | Southern Biotech | |
| FITC-conjugated anti-CD3 | Beckman Coulter | |
| PC5-conjugated anti-CD56 | Beckman Coulter | |
| FITC-conjugated anti-CD14 | Beckman Coulter | |
| FITC-conjugated anti-CD20 | Biolegend | |
| APC/Cy7-conjugated anti-CD16 | Biolegend | |
| BV510-conjugated anti-CD69 | Biolegend | |
| BV421-conjugated anti-CD57 | Becton-Dickinson | |
| 5 | Anti-CXCR1 [IgG1] | Santa Cruz |
| PE-conjugated goat-anti-mouse IgG1 | Southern Biotech | |
| FITC-conjugated anti-CD3 | Beckman Coulter | |
| PC5-conjugated anti-CD56 | Beckman Coulter | |
| FITC-conjugated anti-CD14 | Beckman Coulter | |
| FITC-conjugated anti-CD20 | Biolegend | |
| APC/Cy7-conjugated anti-CD16 | Biolegend | |
| Pacific Blue-conjugated anti-CCR7 | Biolegend |
Analysis of Perforin Expression
Intracellular content of perforin was analyzed in a limited number of patients with SCID/OS/AS due to RAG/NHEJ defects and in healthy infants. Briefly, PBMCs were first stained with a mixture of FITC-conjugated mAbs directed against CD3, CD20, and CD14, as well as with PC5-labeled anti-CD56 mAb, and incubated for 30 min at 4°C. After treatment with 200 µl of Cytofix/Cytoperm (BD-Bioscience, Pharmigen CA, USA) for 20 min at 4°C, cells were washed with 1 ml of saponin (0.1% solution in PBS), and then stained with 5 µl of purified R-PE-labeled anti-perforin mAb (Ancell). After washing, the proportion of CD3− CD14− CD20− CD56+ cells expressing perforin and the mean fluorescent intensity (MFI) of perforin were immediately analyzed on LSR Fortessa Flow Cytometer (BD) using FACSDiva software (BD). Final analysis was done using FloJo v.10.2 (TreeStar).
Analysis of NK Cell Degranulation
Natural killer cell degranulation activity was tested against the K562 erythroleukemia human cell line. In particular, PBMCs derived from patients and from healthy donors were obtained upon Ficoll separation of heparinized blood samples, and incubated with or without 100 U/mL recombinant human IL-2 (NIH) at 37°C overnight. Cells were then incubated with target cells at an effector:target ratio of 1:3 in a final volume of 200 µl in round-bottomed 96-well plates at 37°C and 5% CO2 for 4 h in culture medium supplemented with anti-CD107a-PE (BD Biosciences Pharmingen, San Diego, CA, USA) monoclonal antibody. Cells were then surface-stained with FITC anti-CD3, PC5 anti-CD56, FITC anti-CD14 (Beckman Coulter), FITC anti-CD20, and APC/Cy7 anti-CD16 (BD Biosciences Pharmingen, San Diego, CA, USA) Ab for 30 min at 4°C. The cells were washed, and the proportion of CD3− CD14− CD20− CD56+ cells expressing CD107a was analyzed immediately on LSR Fortessa Flow Cytometer (BD) using FACSDiva v6.1.3 software (BD Biosciences, Mountain View, CA, USA). Final analysis was performed using FloJo v.10.2 (TreeStar). The threshold to define CD107a expression in cells co-cultured with K562 target cells (in the presence or absence of IL-2) was set up on cells cultured with IL-2 alone, without K562 cells.
Analysis of Interferon-γ (IFN-γ) Production
To detect intracellular production of IFN-γ, PBMCs from patients and healthy donors were incubated overnight at 37°C with IL-12 (0.5 ng/ml), or IL-12 (0.5 ng/ml) and IL-18 (0.1 ng/ml) combined. Surface staining was done by incubating the cells with FITC anti-CD3, PC5 anti-CD56, FITC anti-CD14 (Beckman Coulter), FITC anti-CD20, and APC/Cy7 anti-CD16 (BD) mAbs for 30 min at 4°C. Cells were then washed, fixed, and permeabilized with BD Cytofix/Cytoperm kit (BD Biosciences Pharmingen). IFN-γ production was detected by subsequent intracellular staining with PE-conjugated anti-IFN-γ (BD Biosciences Pharmingen). After washing, the proportion of CD3− CD14− CD20− CD56+ cells expressing IFN-γ was immediately analyzed on LSR Fortessa Flow Cytometer (BD) using FACSDiva software (BD). Final analysis was done using FloJo v.10.2 (TreeStar).
Statistical Analysis
Statistical analysis of the in vitro recombination activity of mutant RAG and ARTEMIS proteins, and of the expression of NK cell markers in patients and controls was performed using Mann–Whitney test for non-parametric variables.
Results
Demographic and Clinical Features
At the time of evaluation, mean age (±SEM) was significantly lower in patients with SCID (7.1 ± 1.72 months; range: 0.25–28 months) and in those with OS/AS (15.96 ± 5.41 months; range: 24–480 months) than in patients with CID (177.2 ± 28.22 months; range: 24–480 months; p < 0.0001).
A clinical history of significant infections was present in 60 patients with RAG/NHEJ gene defects (Table 1). Candida (n = 11), Pseudomonas (n = 7), human papillomavirus (HPV, n = 7), adenovirus (n = 6), varicella zoster virus (VZV, n = 6), Pneumocystis jiroveci (n = 5), Bacillus Calmette-Guerin (BCG, n = 5), and rhinovirus (n = 5) were the most common pathogens. In particular, P. jiroveci pneumonia was observed only in patients with SCID or OS/AS, whereas severe VZV infection was mostly restricted to patients with CID. Cytomegalovirus (CMV) and Epstein–Barr virus (EBV) infections were documented in 13 patients each, but were clinically significant only in 4 and 3 patients, respectively. Two SCID patients (P4 and P11) were diagnosed at birth following newborn screening, and infections were effectively prevented with isolation and prophylactic antimicrobials.
Autoimmunity and/or autoantibody production were documented in 23 patients, and were more common in patients presenting with CID (14 out of 21 patients) than in those with SCID (0/19) or with OS/AS (9/24). Cytopenias were the most frequent manifestation of autoimmunity and were documented in 11 patients (4 with OS/AS and 7 with CID). Alopecia and/or vitiligo were observed in six CID patients. Granulomatous disease was present in eight patients with CID. Finally, 12 patients (P20–P31) had typical features of OS (generalized erythroderma, lymphadenopathy, hepatosplenomegaly).
Immunological Phenotype
The count of circulating CD3+, CD19+, and CD3− CD56+ cells in patients belonging to the various groups (SCID, OS/AS, and CID) is shown in Figure 1. With the exception of one SCID patient (P19) who had a high count of maternal T cells (13997 cells/μl), all others had severe T cell lymphopenia (Figure 1A). By contrast, a higher count of autologous T cells was recorded in patients with CID and especially in patients with OS/AS. B cell lymphopenia was observed in the vast majority of patients, irrespective of the clinical phenotype (Figure 1B). By contrast, NK cell count was either normal or increased in most patients (Figure 1C).
Figure 1.
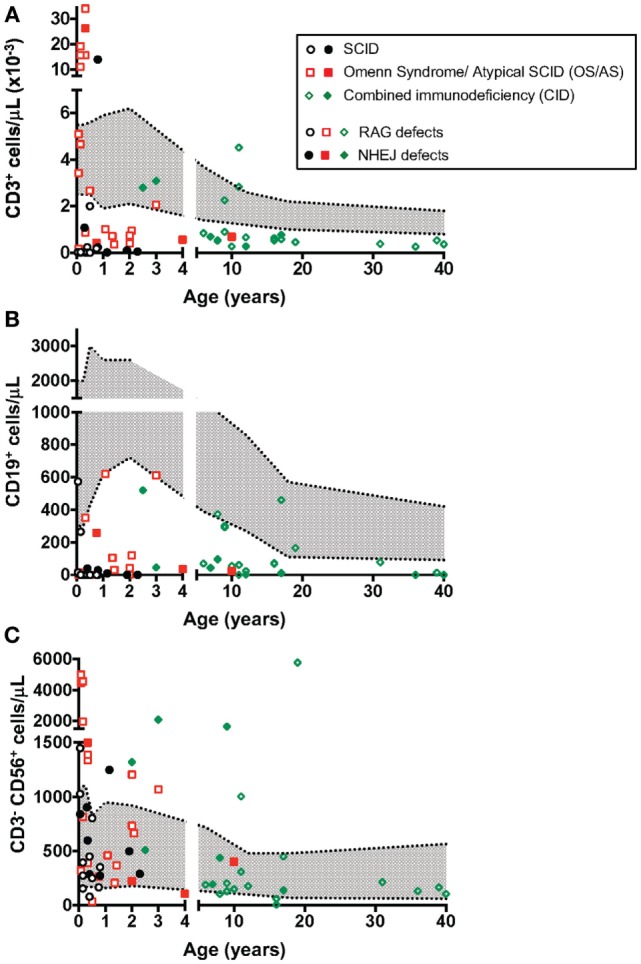
Absolute count of T (A), B (B), and natural killer (NK) (C) lymphocytes at the time of evaluation of NK cell phenotype and function. The area shown in shaded gray represents normal values for age. Different symbols (circle, square, diamond) are used to indicate patients with severe combined immune deficiency (SCID), Omenn syndrome/atypical SCID (OS/AS), and delayed onset combined immune deficiency (CID), respectively. Open circles identify patients with mutations of the recombinase-activating gene (RAG), and close symbols identify patients with mutations in genes involved in non-homologous end joining (NHEJ).
Data on in vitro proliferation to phytohemagglutinin (PHA) were available in 51 patients. Response was absent (<10% of lower limit of normal, LLN) in 17, markedly reduced (10–30% of LLN) in 11, reduced (>30% of LLN, but lower than LLN) in 14, and normal in 9 patients. T cell proliferation to PHA was better preserved in patients with CID; among 18 such patients for whom information was available, 15 had either normal (n = 7) or reduced (n = 8) response.
Low serum levels of IgA and IgM were detected in the majority of SCID patients (Table 1); serum IgG in this group was less informative because of the presence of maternally derived immunoglobulins. Immunoglobulin levels were more variable in patients with OS/AS and in those with CID. In particular, among 23 CID patients, 17 had either low serum IgG (n = 9) or were under immunoglobulin replacement therapy (n = 8) and 15 had undetectable serum IgA.
Correlation between Clinical Phenotype and Recombination Activity Sustained by the Mutant Alleles
Molecular analysis of the patients included in this study identified a total of 41 RAG1, 16 RAG2, 11 DCLRE1C, 4 LIG4, and 2 NHEJ distinct gene mutations (Table 1). Using a flow cytometry-based assay, we have previously reported correlation between recombination activity supported by RAG and DCLRE1C mutant alleles and the clinical and immunological disease phenotype (22, 32). For the patient cohort included in this study, data on in vitro recombination activity were available for 36 RAG1, 9 RAG2, and 6 DCLRE1C mutant alleles. Upon plotting for each patient the recombination activity associated with each of the two mutant alleles, a significant genotype–phenotype correlation was observed, with higher residual recombination activity for mutant alleles associated with a CID phenotype than for those associated with OS/AS or SCID (Figure 2).
Figure 2.
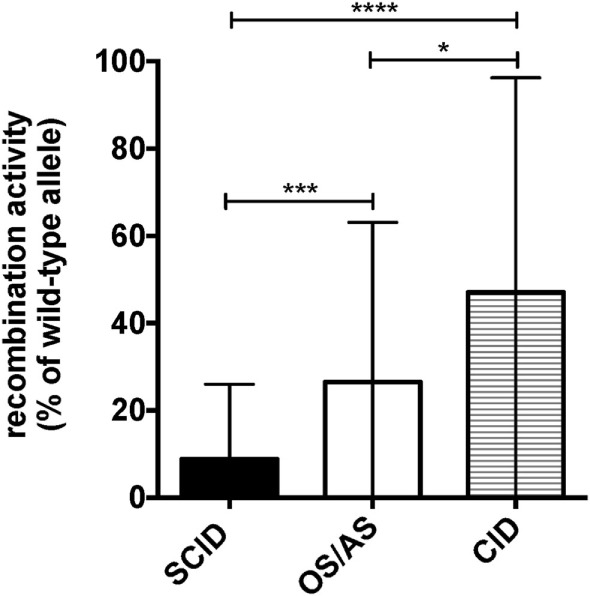
Graphical representation of the recombination activity (mean ± SD) of RAG and DCLRE1C mutant alleles in patients with severe combined immune deficiency (SCID), Omenn syndrome/atypical SCID (OS/AS), and delayed onset combined immune deficiency (CID). *p < 0.05; ***p < 0.005; ****p < 0.001.
Circulating NK Cells from Healthy Infants Comprise a High Proportion of CD56bright Cells
According to a widely accepted scheme, human NK cell differentiation is marked by changes in the expression of cell surface and intracytoplamsic markers, so that five distinct stages of NK cell development are recognized (33). In peripheral blood from healthy controls, both stage 4 (CD56bright CD16−) and stage 5 (CD56+ CD16hi) NK cells are detectable. In particular, in normal adults, CD56bright CD16− cells account for only 5–10% of all circulating NK cells. By contrast, a higher percentage of CD56bright cells was detected in healthy infants (Figures 3A,B). Moreover, the proportion of CD16+ cells was lower in infants than in healthy controls greater than 2 years of age (Figures 3A,C). Finally, NK cells from healthy infants included a lower proportion of cells expressing CD57 (Figures 3A,D), KIRs (Figures 3A,E), and CXCR1 (Figures 3A,F), whereas the percentage of NK cells expressing NKG2A was higher in infants than in older healthy controls (Figures 3A,G).
Figure 3.

Flow cytometry analysis of natural killer cell phenotype in healthy infants (<2 years of age) and healthy donors >2 years of age. (A) Representative analysis of the expression of CD56, CD16, CD57, killer cell immunoglobulin-like receptors (KIRs), CXCR1, and NKG2A in a healthy infant and a healthy donor >2 years of age; (B–G) percentage of CD56bright (B), CD16+ (C), CD57+ (D), KIR+ (E), CXCR1+ (F), and NKG2A+ (G) cells upon gating on CD3− CD19− CD14− CD56+ cells. For NKG2A, different gates are shown to indicate NKG2A+, NKG2A++, and NKG2A+++ cells. For each marker, positivity was defined as fluorescence intensity above that of isotype control. Bars represent mean ± SD values. **p < 0.01; ****p < 0.001.
Markedly Increased Proportion of CD56bright NK Cells in the Peripheral Blood of Patients with RAG and NHEJ Defects Correlates with the Clinical Phenotype
A recent study in Rag−/− mice has unexpectedly disclosed abnormalities of NK phenotype and function, with increased proportion of mature, activated cells with increased cytotoxic activity and reduced survival (30). In order to assess whether similar abnormalities exist in patients with RAG or NHEJ defects, and to establish whether such abnormalities, if present, correlate with the severity of the clinical phenotype and with residual function of the mutant protein, we have analyzed the expression of CD56 and CD16 on the surface of CD3− CD14− CD20− peripheral blood NK cells from patients and controls. As shown in Figures 4A,B, patients with SCID due to RAG/NHEJ defects had a high proportion of CD56bright CD16− NK cells (mean ± SEM: 34.7 ± 4.4), that exceeded what was observed in healthy infants (13.9 ± 3.3; p = 0.0007). Similarly, the proportion of CD56bright CD16− NK cells was higher in patients with CID than in healthy controls greater than 2 years of age (10.4 ± 1.8 vs. 5.1 ± 0.7; p = 0.0431; Figures 4A,B). Among infants with RAG/NHEJ defects, the proportion of CD56bright CD16− NK cells was higher in those presenting with SCID than in those with OS/AS (p = 0.0008; Figure 4B). In order to investigate whether the increase in the proportion of CD56bright NK cells was secondary to numerical and/or functional abnormalities of T and/or B cells, we also analyzed a group of patients with other forms of TCD or with XLA. The proportion of CD56bright CD16− NK cells in these groups was similar to that of healthy controls, and significantly lower than in patients with SCID due to RAGH/NHEJ defects (Figure 4B). Among patients with TCD, no correlation was observed among the proportion of CD56bright NK cells and the severity of T cell lymphopenia, indicating that the increase of CD56bright NK cells observed in SCID patients with RAG/NHEJ defects is not simply secondary to T cell lymphopenia. Furthermore, the proportion of CD56bright CD16int NK cells, that are considered to represent an intermediate step of differentiation between CD56bright CD16− and CD56dim CD16hi cells, was significantly higher in infants with SCID due to RAG/NHEJ defects than in healthy infants, patients with TCD or with XLA (p < 0.0001; Figure 4C). A higher percentage of CD56bright CD16int NK cells was also observed in patients with CID than in healthy controls greater than 2 years of age (p < 0.05; Figure 4C). Finally, patients with SCID due to RAG/NHEJ defects had a lower proportion of CD56dim CD16hi NK cells when compared to healthy infants (p = 0.0007; Figure 4D). Similar results were obtained when comparing patients with CID versus healthy controls older than 2 years of age (p = 0.0011).
Figure 4.

Flow cytometry analysis of CD56 and CD16 expression in natural killer cells from patients and controls. (A) Representative analysis of the expression of CD56 and CD16 in CD3− CD19− CD14− CD56+ cells from one patient each belonging to the severe combined immune deficiency (SCID), Omenn syndrome (OS), delayed-onset combined immune deficiency (CID), other forms of T cell defect (TCD), and X-linked agammaglobulinemia (XLA), respectively. (B–D) Percentage of CD56bright CD16− (B), CD56bright CD16int (C), and CD56dim CD16hi (D) cells among CD3− CD19− CD14− CD56+ cells. For each marker, positivity was defined as fluorescence intensity above that of isotype control. Bars represent mean ± SD values. *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
CD56bright NK Cells from Infants with RAG/NHEJ Defects Have an Immature Phenotype
The data reported above on CD56 and CD16 surface expression indicate that patients with RAG/NHEJ defects have an abnormal distribution of NK cell subsets, with an increased proportion of more immature cells as compared to what is observed in healthy controls. In order to further confirm this, we analyzed the expression of additional surface markers that are differentially expressed in CD56bright versus more mature CD56dim NK cells. As compared to healthy controls >2 years of age, patients with CID had a lower proportion of CD57+ NK cells (p = 0.0022; Figures 5A,B). Furthermore, patients with SCID had fewer CD57+ NK cells than patients with other forms of TCD (p = 0.0041), and a similar trend was also observed when compared to infant controls, although the difference did not reach statistical significance (Figure 5B).
Figure 5.

Flow cytometry analysis of the expression of CD57 (A,B), NKG2A (C,D), and CXCR1 (E,F), and killer cell immunoglobulin-like receptors (KIRs) (G,H) among CD3− CD19− CD14− CD56+ cells. In panels (A,C,E,G), representative examples are shown for one patient each belonging to the severe combined immune deficiency (SCID), Omenn syndrome (OS), and delayed-onset combined immune deficiency (CID) subgroups. For each marker, positivity was defined as fluorescence intensity above that of isotype control. Bars represent mean ± SD values. *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
In normal subjects, circulating stage 4 CD56bright NK cells are characterized by high levels of expression of NKG2A and of the chemokine receptor CCR7, which facilitates homing to lymphoid tissues. By contrast, expression of KIRs and of the chemokine receptor CXCR1 is virtually absent in CD56bright NK cells. As compared to healthy controls, the proportion of NK cells expressing NKG2A at high density was increased in patients with SCID and CID due to RAG/NHEJ defects (Figures 5C,D), and by contrast, the percentage of CXCR1+ NK cells was reduced (Figures 5E,F). Furthermore, patients with SCID due to RAG/NHEJ defects had a lower proportion of NK cells expressing KIR than healthy infants, patients with OS and those with other forms of TCD (Figures 5G,H). While the proportion of CCR7+ cells was higher in healthy infants than in healthy controls >2 years of age (Figure 6), no significant differences were observed in the percentage of CCR7+ NK cells between patients with RAG/NHEJ defects and healthy controls. Notably, CCR7 expression was downregulated during the transition from CD56bright CD16− to CD56bright CD16int cells (data not shown). Furthermore, the proportion of NK cells expressing Siglec-7, leukocyte Ig-like receptor (LIR)-1, or the activation marker CD69 was similar in patients and healthy controls, although interestingly a high proportion of Siglec-7+ NK cells were detected in the control group of patients with XLA (Figure 6). Overall, these data support the notion that the CD56bright NK cells that are more abundant in patients with RAG/NHEJ defects represent immature NK cells and provide evidence for a genotype–phenotype correlation in this group of diseases.
Figure 6.

Percentage of CD3− CD19− CD14− CD56+ cells expressing CCR7 (left panel), Siglec-7 (middle panel), or CD69 (right panel) in patients and controls. For each marker, positivity was defined as fluorescence intensity above that of isotype control. Bars represent mean ± SD values. *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
Impact of Viral Infections on NK Cell Phenotype
Recombinase-activating gene and NHEJ defects lead to increased susceptibility to viral infections, including CMV, EBV, and VZV. It has been reported that viral infections (and CMV in particular) may lead to significant changes of NK cell phenotype and function, with accumulation of cytotoxic CD56dim NKG2C+ NKG2A− KIR+ CD57+ cells (34). Expansion of these cells does not require T cells, as shown in an IL7R-deficient SCID infant after CMV infection (35). A significant proportion (26/66, 39.4%) of the patients with RAG/NHEJ defects included in this study had a documented history of CMV, EBV, VZV or severe JC virus (JCV) infection (Table 1). The proportion of NK cells expressing NKG2C was not statistically different in patients with SCID, OS/AS, or CID versus healthy controls (Figure 7A). Furthermore, when patients were divided into two groups according to their history of CMV, EBV, VZV, or severe JCV infection, no differences were observed in the proportion of NKG2C+ NK cells (Figure 7B). Even when patients with SCID/OS/AS or with CID were divided into two subgroups based on their CMV infection history, no difference was observed in the proportion of NKG2C+ (Figure 7C) and of CD56dim (Figure 7D) NK cells. Finally, although a higher percentage of “exhausted” NK cells with a CD56− CD16+ CD57+ phenotype was detected in patients with SCID, OS/AS, and TCD than in healthy infants (Figure 7E), no correlation was observed with a history of viral infections.
Figure 7.

(A) Percentage of NKG2C+ cells among CD3− CD19− CD14− CD56+ cells from patients and controls. (B) Percentage of NKG2C+ cells among natural killer (NK) cells from patients with recombinase-activating gene (RAG)/non-homologous end-joining (NHEJ) defects with positive (+) or negative (−) history of infections due to cytomegalovirus (CMV), Epstein–Barr virus (EBV), varicella zoster virus (VZV), or JC virus (JCV). (C) Percentage of NKG2C+ cells among NK cells from patients with RAG/NHEJ defects presenting with severe combined immune deficiency (SCID), Omenn syndrome/atypical SCID (SCID/OS/AS), or with delayed onset combined immune deficiency (CID), and divided into two groups according to the presence (+) or absence (−) of a history of CMV infection. (D) Percentage of CD56dim CD16+ cells among NK cells from patients with RAG/NHEJ defects presenting with SCID/OS/AS or with CID, and with either positive (+) or negative (−) history of CMV infection. (E) Percentage of CD56− CD16+ CD57+ cells among CD3− CD14− CD20− peripheral blood mononuclear cells. Bars represent mean ± SD values. *p < 0.05; **p < 0.01; ***p < 0.005.
CD56bright Cells from Patients with RAG/NHEJ Defects Are Potent Producers of IFN-γ
Circulating CD56bright NK cells serve an immunoregulatory function through secretion of various cytokines (IFN-γ, TNF-α, IL-10) (36). In order to assess this aspect of NK cell function, we performed in vitro stimulation with IL-12 and IL-18, and analyzed intracellular expression of IFN-γ upon gating separately on CD56bright vs. CD56dim NK cells. As expected, in both patients and controls CD56bright cells were more potent producers of IFN-γ than CD56dim cells, but both subsets were capable of producing this cytokine (Figure 8). However, CD56dim cells from patients with SCID and with OS/AS due to RAG/NHEJ defects showed impaired IFN-γ production when compared to equivalent cells from healthy infants or patients with TCD. A similar trend was observed in patients with CID versus healthy controls greater than 2 years of age (Figure 8).
Figure 8.
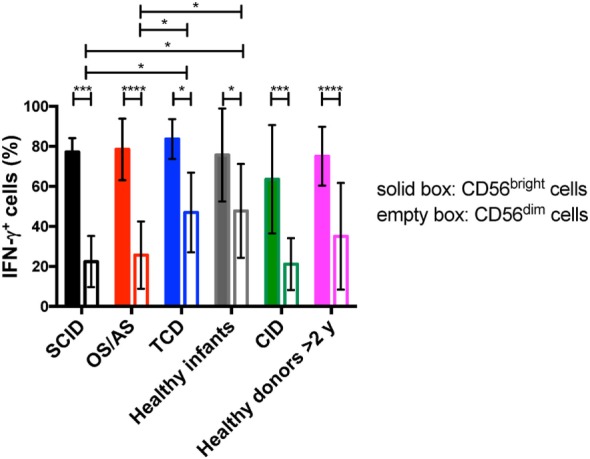
Percentage of interferon-γ (IFN-γ) expressing cells among CD56bright and CD56dim natural killer cells from patients and controls, upon in vitro stimulation with IL-12 and IL-18. Bars represent mean ± SD values. *p < 0.05; ***p < 0.005; ****p < 0.001.
NK Cells from Patients with SCID due to RAG/NHEJ Defects Have Increased Degranulation Capacity in the Absence of IL-2-Mediated Stimulation, and Express a Higher Amount of Intracellular Perforin than NK Cells from Healthy Infant Controls
Previous data had shown that NK cells from rag−/− mice have increased cytotoxic activity (30). To investigate the cytolytic machinery of NK cells, we have analyzed NK cell degranulation in response to K562 target cells, in the absence or presence of IL-2. It is well known that human CD56bright NK cells have reduced cytotoxic activity when compared to CD56dim cells (8). In spite of the markedly increased proportion of CD56bright NK cells, we observed that in the absence of stimulation with IL-2, NK cells from patients with SCID due to RAG/NHEJ defects have increased degranulation capacity when compared to NK cells from healthy infants (Figure 9A, left panel). In particular, CD56dim NK cells from patients with SCID showed increased degranulation when compared to the equivalent subset of NK cells from healthy infants (Figure 9A, middle panel). A similar trend was observed also for CD56bright NK cells, but the difference between SCID patients and controls did not reach statistical significance (Figure 9A, middle panel). Stimulation with IL-2 significantly increased NK cell degranulation capacity to similar levels in patients and controls (Figure 9A, right panel). Although the amount of intracellular perforin could be measured only in a small number of patients with SCID, a significantly higher mean fluorescent intensity (MFI) was observed in unstimulated NK cells from patients than from healthy infants (Figure 9B).
Figure 9.

(A) Natural killer (NK) cells degranulation, expressed as percentage of CD107a+ cells, upon co-culture of peripheral blood mononuclear cells from patients with severe combined immune deficiency (SCID) and healthy infants with K562 target cells, in the absence (left and middle panels) or presence (right panel) of stimulation with exogenous interleukin-2 (IL-2). The middle panel shows degranulation of CD56bright (left) and CD56dim (right) NK cells from SCID patients and healthy infants in the absence of IL-2 stimulation. (B) Representative example (left) of perforin expression in resting NK cells from a patient with SCID due to RAG defect (red) and a healthy infant (blue), and mean fluorescent intensity (MFI) of perforin expression (right) in patients with SCID/OS/AS due to recombinase-activating gene (RAG)/non-homologous end-joining (NHEJ) defects and healthy infants. Bars represent mean ± SD values. *p < 0.05; **p < 0.01.
Discussion
In this manuscript, we have demonstrated that NK cells from healthy infants comprise a higher proportion of CD56bright CD16−/int CD57− cells than what is observed in older healthy subjects, confirming previous observations (37–39). Several lines of evidence indicate that CD56bright cells represent precursors to CD56dim cells. In particular, it has been demonstrated that appearance of CD56bright cells precedes that of CD56dim cells after HSCT (40) and that it is possible to induce differentiation of CD56bright into CD56dim NK cells in vitro in response to signaling via fibroblast growth factor receptor 1 (41). On the other hand, the model according to which CD56bright cells precede CD56dim cells during NK cell development has been challenged by NK cell lineage tracking experiments with genetic bar coding in macaques, which have suggested that these subsets may have distinct ontogeny (42).
In any case, the demonstration that composition of the peripheral NK cell compartment varies with age indicates that appropriate age-matched controls should be used when analyzing NK cell phenotype in pathological conditions.
In particular, by performing an extensive phenotypic analysis, we have shown that NK cells from patients with RAG/NHEJ defects comprise a higher proportion of CD56bright CD16−/int NKG2A+++ cells, and a reduced percentage of CD56dim CD16hi cells expressing CD57, KIRs, and CXCR1, than observed in age-matched healthy controls. Altogether, these observations suggest that NK cells from patients with RAG/NHEJ defects have a more immature phenotype when compared to age-matched healthy controls, in spite of the fact that NK cells from CID patients showed signs of progressive differentiation as compared to NK cells from patients with SCID/OS/AS. These data contrast with recent findings in Rag−/− mice, whose peripheral NK cell compartment is characterized by a predominance of cells expressing KLRG1, a marker of mature NK cells (30). Furthermore, peripheral NK cells from Rag−/− mice are largely CD62L−, indicating an in vivo activated phenotype (30). By contrast, we did not observe increased expression of CD69, an activation marker (43), in NK cells from patients with RAG/NHEJ defects, including those with OS, a condition characterized by accumulation of in vivo-activated autologous T cells. While the reasons for these discrepancies are not clear, murine and human NK cells differ substantially, also in regard to phenotypic markers that are differentially expressed during development.
By affecting the process of V(D)J recombination, mutations in RAG and NHEJ genes profoundly affect generation of T and B lymphocytes. Consistent with this, patients with RAG/NHEJ gene defects had a markedly reduced number of circulating B and T cells, with the exception of patients with OS, some of which had a normal or even increased number of oligoclonal and activated T cells, as typically observed in this disease. Previous studies in TCRβδ−/− and in μMT−/− mice had demonstrated that lack of T and B cells have opposite effects on the count of splenic NK cells. In particular, T cell deficiency is associated with a twofold to threefold increase, and B cell deficiency with a twofold to threefold decrease of NK cell count as compared to C57BL/6 wild-type mice (44). The patients with RAG and NHEJ defects included in this study had either normal or increased count of circulating NK cells. In order to rule out the possibility that their abnormalities of NK cell phenotype could be secondary to T and B cell lymphopenia, we have included in this study patients with XLA (who lack circulating B cells) and with other forms of TCD, including seven patients with SCID not due to RAG/NHEJ defects. Even when compared to these two control groups, patients with SCID due to RAG/NHEJ gene defects had a markedly increased proportion of CD56bright CD16− CD57− NKG2A+++ cells. Similar differences were also observed also when comparing NK cells from patients with OS/AS and those with TCD or XLA, with an increased percentage of CD56bright CD16− CD57− cells in the former. Altogether, these data indicate that the increased representation of immature cells within the peripheral NK cell compartment of patients with RAG/NHEJ defects is not secondary to T and B cell lymphopenia.
Because of their severe T cell immunodeficiency, patients with RAG/NHEJ defects are highly prone to viral infections, including CMV. LIR-1 is a member of the immunoglobulin superfamily which has been shown to bind the human CMV MHC class I homolog UL-18 protein (45). NKG2A and NKG2C represent inhibitory and activating forms of CD94, recognizing non-classical HLA-E molecules (46). CMV infection can drive expansion of KIR+ and/or LIR-1+, NKG2A− NKG2C+ NK cells (34). Furthermore, rapid differentiation of CD56dim NKG2C+ NKG2A− CD57+ cells has been observed after CMV reactivation in recipients of HSCT (47). It has been shown that upon transfer from CMV-seropositive donors into seropositive HSCT recipients, NKG2C+ cells undergo expansion and produce high amounts of IFN-γ as compared to NKG2C+ cells transfused into CMV-seronegative recipients, implying that NKG2C+ cells may represent “memory” NK cells capable of a prompt response upon re-exposure to CMV (48). Finally, rapid expansion of NKG2C+ NK cells has been reported in infants acquiring perinatal CMV infection (39). Although several of the patients included in this study had a history of CMV infection, no particular expansion of NKG2C+ and/or LIR-1+ cells was observed. Monocytes are apparently required to allow expansion of adaptive/memory-like NK cells (49). Lack of expansion of NKG2C+ cells in CMV-infected, RAG-/NHEJ mutated SCID patients may reflect a requirement also for T cell help in this process and may further contribute to poor control of CMV infection in these patients.
CD56bright and CD56dim NK cells differ in their immunomodulatory and effector function. In particular, CD56bright cells are potent producers of IFN-γ, but have lower content of perforin and display reduced cytotoxic activity as compared to CD56dim cells. Analysis of IFN-γ production by CD56bright and CD56dim NK cells from patients and controls confirmed that CD56bright cells were more potent producers of IFN-γ, irrespective of the underlying diagnosis. However, the proportion of CD56dim NK cells producing IFN-γ was lower in patients with SCID/OS/AS due to RAG/NHEJ defects than in healthy infants or in patients with other forms of TCD, and a similar trend was observed for CD56dim cells from CID patients. These results suggest that RAG/NHEJ defects are associated with abnormalities of NK cells that are not restricted to their phenotype, but may also involve NK cell function. In this regard, it is particularly significant that NK cells from the patients displayed enhanced degranulation in the absence of previous stimulation with IL-2, and that CD56bright cells from patients with SCID/OS/AS had a higher content of perforin than CD56bright cells from healthy infants. Although we did not perform a chromium release assay to investigate NK cytolytic function, these data suggest that RAG/NHEJ defects are associated with enhanced effector function, similarly to what was previously reported for Rag−/− mice (30).
Expression of RAG may initiate prior to T and B cell differentiation, as indicated by the presence of incomplete rearrangements at the immunoglobulin heavy chain or T cell receptor loci in a minority of murine and human NK cells (29, 50). It has been suggested that introduction of DNA double strand breaks in lymphoid progenitor cells would facilitate selection of cells with more efficient DNA repair machinery (30). In the absence of this mechanism, NK cells from Rag-deficient animals and humans would have reduced cellular fitness. Our data indicate that indeed patients with mutations in RAG or in NHEJ genes share similar abnormalities of NK cell phenotype and function. While it is not known whether the immunoglobulin and T cell receptor loci represent the only sites in the genome that are targeted by RAG during early stages of lymphoid development, it is interesting to note that selective loss of CD56dim cells, with relative preservation of the CD56bright NK cell compartment, has been reported in patients with mutations of MCM4 and POLE2 genes, all of which control DNA replication (51–53).
In conclusion, we have identified for the first time abnormalities of NK cell phenotype and function in patients with mutations in RAG and in genes involved in NHEJ. These abnormalities are more pronounced in patients with severe mutations associated with a more severe clinical and immunological phenotype, and are not secondary to T and B cell lymphopenia. Demonstration of enhanced degranulation activity and higher amount of perforin in NK cells from patients with SCID due to RAG/NHEJ defects suggests that inclusion of serotherapy targeting NK cells in the HSCT conditioning regimen may help reduce the risk of graft rejection that has been reported in these diseases.
Ethics Statement
The study was approved by the Institutional Review Boards of Boston Children’s Hospital, the National Institute of Allergy and Infectious Diseases (protocols 93-I-0119 and 16-I-N139), and of all other referring centers. Blood samples were obtained upon written informed consent of the subject or, in the case of minors, of their parents or legal guardians.
Author Contributions
JM, EM, AM, SP, and LN designed the study, interpreted the data, and wrote the manuscript; KD, GT, EC, OP, PM, SG, and DM performed experiments, acquired and analyzed the data; WA-H, CC, MC, JB, CB, DB, SB, TC, JC, VD-C, LOdB, MTdlM, GM, AF, RG, RKG, AH, SH, C-HH, MK, AlKi, BK, AnKl, TK, BL, VL, MiMa, IM, MeMo, BN, S-YP, NP, AP, SP, IR, JS, RS, TT, Y-JK, JW, AG, and SK contributed patient samples and clinical and immunological data; all authors have revised the work for its intellectual content, have approved its final version and have agreed to be accountable for all aspects related to the accuracy and integrity of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial of financial relationship that could be construed as a potential conflict of interest.
Footnotes
Funding. Funding for this study was provided in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. This work was also supported by grant 2R01AI100887 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (to JM). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ (2008) 15(2):226–33. 10.1038/sj.cdd.4402170 [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 3.Lam VC, Lanier LL. NK cells in host responses to viral infections. Curr Opin Immunol (2016) 44:43–51. 10.1016/j.coi.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaffer BC, Hsu KC. How important is NK alloreactivity and KIR in allogeneic transplantation? Best Pract Res Clin Haematol (2016) 29(4):351–8. 10.1016/j.beha.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caligiuri MA. Human natural killer cells. Blood (2008) 112(3):461–9. 10.1182/blood-2007-09-077438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cichocki F, Schlums H, Theorell J, Tesi B, Miller JS, Ljunggren HG, et al. Diversification and functional specialization of human NK cell subsets. Curr Top Microbiol Immunol (2016) 395:63–94. 10.1007/82_2015_487 [DOI] [PubMed] [Google Scholar]
- 7.Montaldo E, Del Zotto G, Della Chiesa M, Mingari MC, Moretta A, De Maria A, et al. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A (2013) 83(8):702–13. 10.1002/cyto.a.22302 [DOI] [PubMed] [Google Scholar]
- 8.Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol (2001) 31(10):3121–7. [DOI] [PubMed] [Google Scholar]
- 9.Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev (2006) 214:73–91. 10.1111/j.1600-065X.2006.00457.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol (2001) 166(11):6477–82. 10.4049/jimmunol.166.11.6477 [DOI] [PubMed] [Google Scholar]
- 11.Ferlazzo G, Thomas D, Lin SL, Goodman K, Morandi B, Muller WA, et al. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol (2004) 172(3):1455–62. 10.4049/jimmunol.172.3.1455 [DOI] [PubMed] [Google Scholar]
- 12.Bjorkstrom NK, Ljunggren HG, Sandberg JK. CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol (2010) 31(11):401–6. 10.1016/j.it.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 13.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science (1996) 274(5284):97–9. 10.1126/science.274.5284.97 [DOI] [PubMed] [Google Scholar]
- 14.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell (1998) 93(5):885–96. 10.1016/S0092-8674(00)81448-8 [DOI] [PubMed] [Google Scholar]
- 15.de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest (2005) 115(11):3291–9. 10.1172/JCI25178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kuhr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest (2005) 115(11):3140–8. 10.1172/JCI25221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol (2011) 141(1):73–82. 10.1016/j.clim.2011.05.007 [DOI] [PubMed] [Google Scholar]
- 18.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med (2008) 358(19):2030–8. 10.1056/NEJMoa073966 [DOI] [PubMed] [Google Scholar]
- 19.De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood (2010) 116(8):1263–71. 10.1182/blood-2010-02-267583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson LA, Frugoni F, Hopkins G, de Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol (2013) 132(4):969–71.e1–2. 10.1016/j.jaci.2013.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2016) 126(11):4389. 10.1172/JCI91162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol (2014) 133(4):1099–108. 10.1016/j.jaci.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency.Sci Immunol (2016) 1(6). 10.1126/sciimmunol.aah6109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pai SY, Cowan MJ. Stem cell transplantation for primary immunodeficiency diseases: the North American experience. Curr Opin Allergy Clin Immunol (2014) 14(6):521–6. 10.1097/ACI.0000000000000115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood (2014) 123(2):281–9. 10.1182/blood-2013-01-476432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy WJ, Kumar V, Bennett M. Rejection of bone marrow allografts by mice with severe combined immune deficiency (SCID). Evidence that natural killer cells can mediate the specificity of marrow graft rejection. J Exp Med (1987) 165(4):1212–7. 10.1084/jem.165.4.1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Igarashi H, Gregory SC, Yokota T, Sakaguchi N, Kincade PW. Transcription from the RAG1 locus marks the earliest lymphocyte progenitors in bone marrow. Immunity (2002) 17(2):117–30. 10.1016/S1074-7613(02)00366-7 [DOI] [PubMed] [Google Scholar]
- 28.Borghesi L, Hsu LY, Miller JP, Anderson M, Herzenberg L, Herzenberg L, et al. B lineage-specific regulation of V(D)J recombinase activity is established in common lymphoid progenitors. J Exp Med (2004) 199(4):491–502. 10.1084/jem.20031800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilbeam K, Basse P, Brossay L, Vujanovic N, Gerstein R, Vallejo AN, et al. The ontogeny and fate of NK cells marked by permanent DNA rearrangements. J Immunol (2008) 180(3):1432–41. 10.4049/jimmunol.180.3.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell (2014) 159(1):94–107. 10.1016/j.cell.2014.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol (2014) 133(4):1092–8. 10.1016/j.jaci.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felgentreff K, Lee YN, Frugoni F, Du L, van der Burg M, Giliani S, et al. Functional analysis of naturally occurring DCLRE1C mutations and correlation with the clinical phenotype of ARTEMIS deficiency. J Allergy Clin Immunol (2015) 136(1):140–50.e7. 10.1016/j.jaci.2015.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med (2006) 203(4):1033–43. 10.1084/jem.20052507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71. 10.1182/blood-2004-05-2058 [DOI] [PubMed] [Google Scholar]
- 35.Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. Human NK cells can control CMV infection in the absence of T cells. Blood (2008) 112(3):914–5. 10.1182/blood-2008-05-157354 [DOI] [PubMed] [Google Scholar]
- 36.Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, et al. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol (1999) 162(8):4511–20. [PubMed] [Google Scholar]
- 37.Sundstrom Y, Nilsson C, Lilja G, Karre K, Troye-Blomberg M, Berg L. The expression of human natural killer cell receptors in early life. Scand J Immunol (2007) 66(2–3):335–44. 10.1111/j.1365-3083.2007.01980.x [DOI] [PubMed] [Google Scholar]
- 38.Almeida-Oliveira A, Smith-Carvalho M, Porto LC, Cardoso-Oliveira J, Ribeiro Ados S, Falcao RR, et al. Age-related changes in natural killer cell receptors from childhood through old age. Hum Immunol (2011) 72(4):319–29. 10.1016/j.humimm.2011.01.009 [DOI] [PubMed] [Google Scholar]
- 39.Goodier MR, White MJ, Darboe A, Nielsen CM, Goncalves A, Bottomley C, et al. Rapid NK cell differentiation in a population with near-universal human cytomegalovirus infection is attenuated by NKG2C deletions. Blood (2014) 124(14):2213–22. 10.1182/blood-2014-05-576124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dulphy N, Haas P, Busson M, Belhadj S, Peffault, de Latour R, et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol (2008) 181(3):2227–37. 10.4049/jimmunol.181.3.2227 [DOI] [PubMed] [Google Scholar]
- 41.Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179(1):89–94. 10.4049/jimmunol.179.1.89 [DOI] [PubMed] [Google Scholar]
- 42.Wu C, Li B, Lu R, Koelle SJ, Yang Y, Jares A, et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell (2014) 14(4):486–99. 10.1016/j.stem.2014.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lanier LL, Buck DW, Rhodes L, Ding A, Evans E, Barney C, et al. Interleukin 2 activation of natural killer cells rapidly induces the expression and phosphorylation of the Leu-23 activation antigen. J Exp Med (1988) 167(5):1572–85. 10.1084/jem.167.5.1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeannet G, Coudert JD, Held W. T and B lymphocytes exert distinct effects on the homeostasis of NK cells. Eur J Immunol (2006) 36(10):2725–34. 10.1002/eji.200636011 [DOI] [PubMed] [Google Scholar]
- 45.Vitale M, Castriconi R, Parolini S, Pende D, Hsu ML, Moretta L, et al. The leukocyte Ig-like receptor (LIR)-1 for the cytomegalovirus UL18 protein displays a broad specificity for different HLA class I alleles: analysis of LIR-1+ NK cell clones. Int Immunol (1999) 11(1):29–35. 10.1093/intimm/11.1.29 [DOI] [PubMed] [Google Scholar]
- 46.Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9. 10.1038/35869 [DOI] [PubMed] [Google Scholar]
- 47.Della Chiesa M, Falco M, Podesta M, Locatelli F, Moretta L, Frassoni F, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood (2012) 119(2):399–410. 10.1182/blood-2011-08-372003 [DOI] [PubMed] [Google Scholar]
- 48.Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189(10):5082–8. 10.4049/jimmunol.1201964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia (2016) 30(2):456–63. 10.1038/leu.2015.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fronkova E, Krejci O, Kalina T, Horvath O, Trka J, Hrusak O. Lymphoid differentiation pathways can be traced by TCR delta rearrangements. J Immunol (2005) 175(4):2495–500. 10.4049/jimmunol.175.4.2495 [DOI] [PubMed] [Google Scholar]
- 51.Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest (2012) 122(3):821–32. 10.1172/JCI61014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest (2012) 122(3):814–20. 10.1172/JCI60224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frugoni F, Dobbs K, Felgentreff K, Aldhekri H, Al Saud BK, Arnaout R, et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J Allergy Clin Immunol (2016) 137(2):635–8.e1. 10.1016/j.jaci.2015.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]