

Abstract
Metalloenzymes catalyze complex and essential processes, such as photosynthesis, respiration, and nitrogen fixation. For example, bacteria and archaea use [NiFe]-hydrogenases to catalyze the uptake and release of molecular hydrogen (H2). [NiFe]-hydrogenases are redox enzymes composed of a large subunit that harbors a NiFe(CN)2CO metallo-center and a small subunit with three iron–sulfur clusters. The large subunit is synthesized with a C-terminal extension, cleaved off by a specific endopeptidase during maturation. The exact role of the C-terminal extension has remained elusive; however, cleavage takes place exclusively after assembly of the [NiFe]-cofactor and before large and small subunits form the catalytically active heterodimer. To unravel the functional role of the C-terminal extension, we used an enzymatic in vitro maturation assay that allows synthesizing functional [NiFe]-hydrogenase-2 of Escherichia coli from purified components. The maturation process included formation and insertion of the NiFe(CN)2CO cofactor into the large subunit, endoproteolytic cleavage of the C-terminal extension, and dimerization with the small subunit. Biochemical and spectroscopic analysis indicated that the C-terminal extension of the large subunit is essential for recognition by the maturation machinery. Only upon completion of cofactor insertion was removal of the C-terminal extension observed. Our results indicate that endoproteolytic cleavage is a central checkpoint in the maturation process. Here, cleavage temporally orchestrates cofactor insertion and protein assembly and ensures that only cofactor-containing protein can continue along the assembly line toward functional [NiFe]-hydrogenase.
Keywords: circular dichroism (CD), infrared spectroscopy (IR spectroscopy), metalloprotein, protein assembly, proteolytic enzyme
Introduction
More than one-third of all proteins in nature have been shown to bind metal cofactors (1). Metalloenzymes catalyze complex and essential processes, such as photosynthesis, respiration, and nitrogen fixation (2). Industrial interest stems from the excellent catalytic performance and specificity of certain metalloenzymes (e.g. in biotechnological and pharmaceutical applications) (3). Furthermore, metalloproteins receive growing attention due to the discovery of numerous diseases related to cofactor biosynthesis (4). Coordination of synthesis and assembly of multisubunit metalloproteins must be tightly controlled to ensure that only correctly folded and catalytically active enzymes are delivered to their subcellular destination. The physiological and molecular orchestration of events (e.g. coordination of protein and cofactor synthesis or assembly of cofactors into apoprotein) is not fully understood. Our comprehension of metal cofactor biosynthesis has been facilitated by the study of [NiFe]-hydrogenases, which served as excellent model systems (5, 6). Due to their capability to catalyze the reversible reduction of protons to molecular hydrogen (H2), hydrogenases have been discussed for potential bioenergy applications and industrial production of H2 (7).
Typically, [NiFe]-hydrogenases are heterodimers of a large and a small subunit. The “large subunit” binds the catalytic NiFe(CN)2CO cofactor (8, 9), and the “small subunit” contains three iron–sulfur (FeS) clusters involved in electron transfer. The nickel ion of the bimetallic active site is coordinated via four conserved cysteinyl thiolates, two of which additionally ligate an iron ion. The iron site carries one carbonyl (CO)2 and two cyanide (CN) ligands. These unique ligands are proposed to maintain the low-spin Fe(II) state important for H2 activation (10, 11). Fig. 1 presents our current working model for hydrogenase maturation and suggests the following sequence of events: (i) synthesis of CO and CN, ligation to iron, and incorporation of the fully coordinated iron into the large subunit pro-protein by the coordinated activity of HypCDEF (12); (ii) nickel insertion by concerted activity of HypA, HypB, and SlyD (6, 13) (these accessory proteins are not required at high Ni2+ concentrations) (5, 14); (iii) nickel acting as recognition site for a specific endopeptidase (15); cleavage of the C-terminal extension of the pro-protein that harbors the [NiFe]-cofactor, inducing conformational changes; and (iv) complex formation with the small subunit to yield active [NiFe]-hydrogenase (16, 17).
Figure 1.

Working model for maturation of the large subunit into functional [NiFe]-hydrogenase. i, incorporation of the Fe(CN)2CO moiety into the hydrogenase large subunit (pro-protein) by the coordinated activity of HypCDEF. ii, nickel insertion by concerted activity of HypA, HypB, and SlyD. iii, endoproteolytic cleavage of the C-terminal peptide, associated with conformational changes of the protein. iv, dimerization of large and small subunit forms functional [NiFe]-hydrogenase.
Escherichia coli synthesizes three distinct membrane-bound [NiFe]-hydrogenases: hydrogenase-1 (Hya), hydrogenase-2 (Hyb), and hydrogenase-3 (Hyc). The large subunit of each hydrogenases is synthesized as a pro-protein with a C-terminal extension, cleaved off by a specific endopeptidase. Besides the proteins mentioned above (HypABCDEF), orthologous accessory proteins have been described. For example, HybG is involved in delivery of the completed Fe(CN)2CO moiety to the large subunit of hydrogenase-2, whereas its homologue HypC delivers the cofactor to hydrogenase-3. Table 1 describes the proposed function of [NiFe]-hydrogenase maturation proteins from E. coli used in this study. Most enzymes and substrates involved in biosynthesis and assembly of the bimetallic cofactor have been identified. However, details about the coordination of protein synthesis and cofactor assembly are largely unknown.
Table 1.
Proteins involved in the maturation process of hydrogenase-2 from E. coli
| Protein | Molecular mass | Function/Properties |
|---|---|---|
| kDa | ||
| HybC | 63.5 | Large subunit of hydrogenase-2; contains the [NiFe] active site (50). |
| HybO | 39.6 | Small subunit of hydrogenase-2; contains one [3Fe-4S] and two [4Fe-4S] clusters that are involved in electron transfer from or to the active site (52). |
| HybF (HypA) | 12.5 | Involved in acquisition and insertion of the nickel into large subunit HybC (53). |
| HypB | 31.5 | GTPase activity; binds nickel and forms with SlyD the HypB–SlyD complex that activates nickel release and insertion into the large subunit (54,55). |
| HybG (HypC) | 8.7 | Small iron- and CO2-binding protein; proposed to deliver CO2 for reduction to CO by HypD (27). Forms with HypD the core complex of hydrogenase-2 maturation machinery (HybG–HypD), that is involved in delivering the completed Fe(CN)2CO moiety to the large subunit HybC (14). |
| HypD | 41.4 | The only FeS cluster-containing Hyp protein; acts as a scaffold for assembly of the Fe(CN)2CO moiety of the cofactor prior to its transfer to the large subunit (28). |
| HypE | 35 | HypE is a homodimer; forms a heterooligomer with HypCD (38) and a heterotetramer with HypF (56). HypEF heterotetramer generates CN ligands from carbamoyl phosphate in an ATP-dependent condensation reaction (25). |
| HypF | 82 | Carbamoyl phosphate phosphatase; carbamoyl-transferase; ATPase; involved in CN synthesis (26,57). |
| HybD | 17.6 | Endopeptidase; involved in C-terminal cleavage of the hydrogenase-2 large subunit (33). |
To identify the sequence of events in active site biosynthesis and investigate the individual role of each accessory protein, an approach is required that allows reconstructing the maturation pathway. Here, we developed an enzymatic in vitro assay to study the synthesis of active [NiFe]-hydrogenase 2 from E. coli. The starting point is the unprocessed, large subunit pro-protein (HybC) and five individual [NiFe]-hydrogenase maturation proteins in purified form. The experiments focus on the question of when and why the C-terminal extension of HybC is cleaved off. Making use of biochemical analysis, circular dichroism, and infrared spectroscopy, we found that proteolytic cleavage coordinates cofactor insertion and protein assembly during the maturation of [NiFe]-hydrogenases.
Results
The in vitro system to study [NiFe]-hydrogenase maturation
The anoxic reaction mixture was composed of purified HybC, the core maturation complex HybG–HypDE (GDE), purified accessory proteins HisHypE and HisHypF in stoichiometric amounts, and an activation mix including ATP, carbamoyl phosphate, NiCl2, FeSO4, and sodium dithionite. The reaction was started by the addition of endopeptidase HybD in catalytic amounts. After 30 min of incubation time at room temperature, aliquots of the reaction mixture were analyzed by SDS-PAGE for cleavage of HybC (∼63.5 kDa) into the processed form, HybC−15AA (∼62 kDa) (Fig. 2A). Maximal cleavage was observed for the complete reaction mix (lane 1). Although HypE is supplied to the reaction mix in the form of the GDE complex, without the addition of extra HisHypE (lane 2) to the reaction assay, only ∼60% cleavage was detected. The cleavage of ∼20% of HybC in the absence of HisHypF (lane 3) can be explained by the presence of Fe(CN)2CO on the GDE complex (14). Notably, the maximal cleavage shown in lane 1 was achieved by the addition of equimolar ratio of purified HisHypE and HisHypF to the reaction mix, and if either component of HypE or HypF is missing from reaction assay, the counterpart component cannot stimulate the maturation reaction. No cleavage of HybC could be observed when the reaction was performed in the absence of the GDE complex (lane 4), HybD endopeptidase (lane 5), or activation mix (lane 6) or under aerobic conditions (lane 7). No significant increase in cleavage after 60 min of incubation was observed. For longer incubation times, a decrease of enzyme activity was observed, potentially due to protein degradations that appear as a smear on SDS-PAGE rather than separate bands (data not shown). Infrared spectroscopy showed the CO/CN vibrational fingerprint of the cofactor for in vitro (complete) and in vivo matured hydrogenase-2 (Fig. 2B). The presence of two CO bands suggested co-population of two redox species (18, 19). We tentatively assign these peaks to the “activated” Ni-C and fully reduced Ni-R state (1965 and 1947 cm−1, respectively). Both Ni-C and Ni-R are reported to be involved in the catalytic cycle of [NiFe]-hydrogenase (7). In the absence of maturation proteins (GDE) or activation mix (AM), no formation of cofactor was observed (Fig. 2B). However, upon closer inspection, traces of putative CO/CN bands at lower wave numbers were detected.
Figure 2.
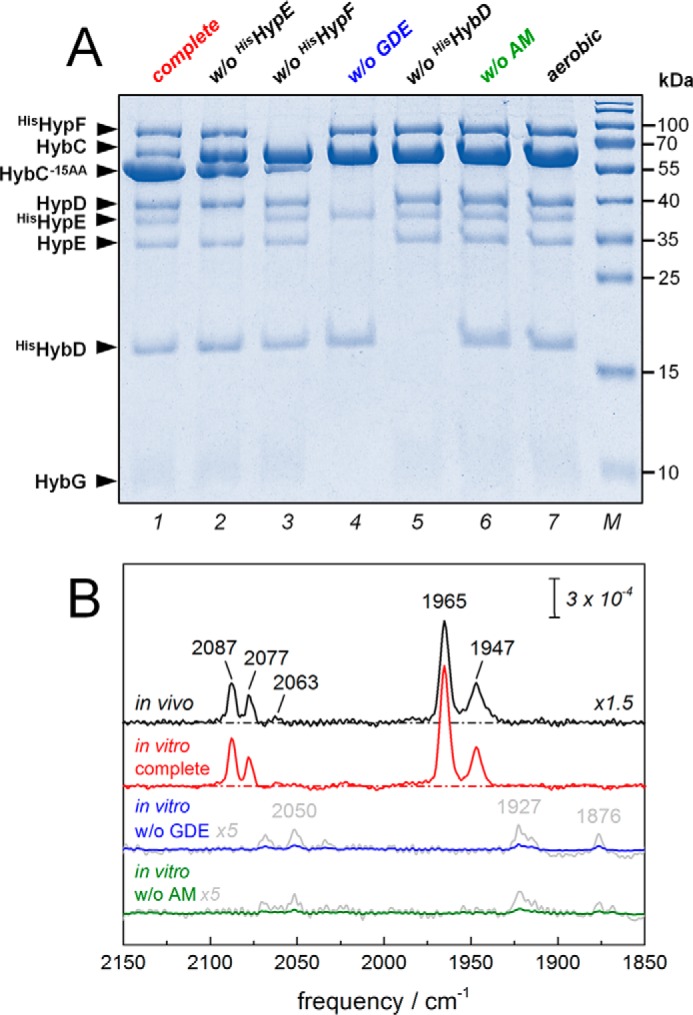
In vitro system to study the maturation of [NiFe]-hydrogenase 2 from E. coli. A, SDS-PAGE analysis of the in vitro assay. The complete assay was composed of purified HybC; accessory proteins HybG–HypDE complex (GDE), HisHypE, and HisHypF; the endopeptidase HisHybD; and the AM. After 30 min of incubation, aliquots of reaction mixture were analyzed for cleavage of HybC by SDS-PAGE. The complete assay in lane 1 predominantly showed cleavage of HybC (∼63.5 kDa) into HybC−15AA (∼62 kDa). Approximately 60 and 20% cleavage activity was observed in the absence of HisHypE and HisHypF, respectively (lanes 2 and 3). Note the visible presence of native HypE in lane 2, which originates from the GDE. In the absence of the HybG–HypDE complex (without (w/o) GDE), endopeptidase HybD (w/o HybD), or the activation mixture (w/o AM), no cleavage was observed (lanes 4–6). Lane 7 shows that aerobiosis disrupted cleavage as well. B, FTIR analysis of the CO/CN vibrational fingerprint of the cofactor. Both in vivo (black) and in vitro (complete, red) matured hydrogenase-2 signatures were identical and comprised two redox species at least (CN frequencies 2087, 2077, and 2063 cm−1; CO frequencies 1965 and 1947 cm−1). Efficiently, no formation of cofactor was observed in the absence of the HybG–HypDE complex (in vitro w/o GDE, blue) or activation mixture (in vitro w/o AM, green). Only residual traces of CO/CN bands can be observed (gray, magnified ×5).
Analysis of hydrogenase-2 activity matured in vitro
Although HybC potentially harbors a [NiFe]-center, the large subunit alone does not exhibit hydrogenase activity. The presence of hydrogenase-2 small subunit HybO is required for enzyme activity (16). After maturation of HybC as described above, we therefore added a fraction of enriched HybO to HybC and analyzed for hydrogen conversion activity by ATR FTIR spectroscopy. Here, in vitro (complete) matured hydrogenase-2 was reduced by deuterium gas (D2), and the release of deuterium ions into the H2O bulk was followed in real time (Fig. 3A). Recombination of D+ and OH− ions gave rise to intense bands at 2505 and 1445 cm−1 that can be assigned to semi-heavy water (O–D stretch and HDO bending, respectively). These bands are a highly specific marker for hydrogen uptake activity. See the legend to Fig. 3 for further details on the observed difference spectrum, which also comprises spectral features of film swelling. Fig. 3B shows comparable activity of hydrogenase-2 matured in vivo. In the absence of the GDE complex or AM, hydrogenase-2 was limited to residual D2 oxidation activity. The D2-minus-N2 difference spectrum of in vitro maturated hydrogenase-2 shows cofactor reduction upon D2 uptake (Fig. 3B, inset). At least three reduced species in the fingerprint region of the cofactor were populated (CO bands 1951, 1936, and 1927 cm−1), and a similar red shift was observed in the CN region (2075 and 2063 cm−1). Future work will identify the involved redox species in greater detail. Additionally, the samples were analyzed for H2 uptake activity by following hydrogenase-dependent reduction of benzyl viologen photometrically (Fig. 3C). The specific activity of purified in vivo maturated hydrogenase-2 was 1.88 ± 0.11 μmol of H2 oxidized/min/mg. This value represents 100% activation and serves as an estimate of the maximum hydrogenase-2 activity that can be restored in vitro. Under the same reaction conditions, the highest activity obtained was in the presence of all components of the in vitro assay (Fig. 3C, complete). Without the addition of an equimolar ratio of HisHypE and HisHypF to the reaction mix, only ∼70 and ∼30% of enzyme activity could be restored, respectively. Only background activity that varied between an undetectable level and 6.8% of maximal activity was observed when GDE, HybD, HybO, or AM was omitted. The background activity is most probably due to nonspecific assembly of cofactor in trace amounts (compare Fig. 2B). HybO alone shows no hydrogenase activity. Furthermore, incubation performed under aerobic conditions completely abolished H2 uptake activity.
Figure 3.
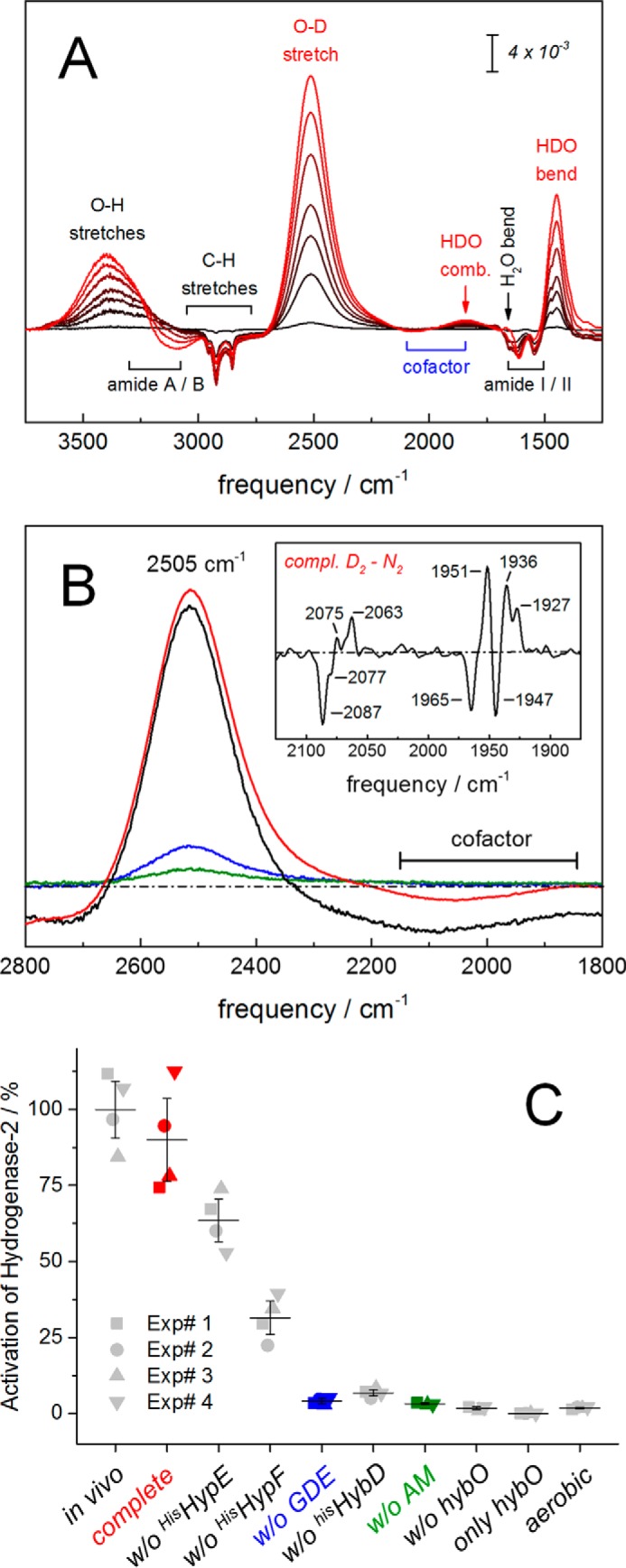
Analysis of hydrogenase activity matured in vitro. A, FTIR analysis of oxidation activity of in vitro maturated hydrogenase-2 in the presence of H2O and D2. Catalytic activity is followed via D2 oxidation and release of deuterium ions into the H2O bulk. This induces a hydrogenase-specific increase of HDO (bands at 2505, 1850, and 1445 cm−1; red labels). Further signals show a slight increase of liquid water (O–H stretches at 3400 cm−1 and H2O bending around 1660 cm−1) and a concomitant decrease of protein bands (i.e. CH stretches from 3000 to 2750 cm−1, amide I/II vibrations from 1650 to 1500 cm−1, and a decrease of N–H stretches from 3300 to 3100 cm−1 (amide A/B) (60). B, matured hydrogenase-2 as isolated (in vivo, black) or as synthesized in vitro (in vitro complete, red) did not differ significantly in D2 uptake activity. Residual activity was observed in the absence of the HybG–HypDE complex (in vitro w/o GDE, blue) or activation mixture (in vitro w/o AM, green). Note the D2-N2 difference spectrum of in vitro synthesized hydrogenase-2 (inset). Here, the population of at least three reduced species is shown (positive CO frequencies at 1951, 1936, and 1927 cm−1). C, kinetic analysis of H2 uptake activity. The complete assay was composed of purified HybC, GDE, HypE, HypF, the endopeptidase HybD, HybO, and AM. Reactions were conducted in the absence of one or more substrates at a time, as indicated. The samples were analyzed for activity by following H2-dependent reduction of benzyl viologen photometrically under the same reaction conditions. The specific activity of purified hydrogenase-2 (in vivo) was 1.88 μmol of H2 oxidized/min/mg. This activity represents 100% activation and provides an estimate of the maximum activity expected for the in vitro matured hydrogenase. The maximal attainable activity obtained under this condition was in the presence of all components of the in vitro assay (complete). Without the addition of an equimolar ratio of HisHypE and HisHypF to the reaction mix, ∼70 or 30% of enzyme activity could be restored, respectively. Only background activity was observed when GDE, HybD, AM, or HybO was omitted or when the incubation was performed under aerobic conditions. HybO alone shows no hydrogenase activity. Four independent experiments and the S.D. value is shown.
The C-terminal peptide on HybC is essential for interaction with the hydrogenase maturation machinery
HybC is known to form a stable complex with the accessory protein HybG (20). HybG and HypD are the core complex of the hydrogenase-2 maturation machinery. With our in vitro system, we addressed the question of whether the 15-amino acid C-terminal extension on HybC is required for interaction with the HybG protein. Complex formation of Strep-tagged HybG with HybC, HybC−15AA, or HybC+5AA was investigated. HybC−15AA is a genetic construct equal to truncated HybC (i.e. without the 15 amino acids at the C terminus). HybC+5AA equals HybC but includes an artificial extension of five amino acids (Gly-Leu-Cys-Gly-Arg) at the C terminus (Fig. 4A).
Figure 4.

Interaction of HybC variants with maturation proteins HybGStrep and HypDEF. A, schematic representation of HybC+5AA (black), HybC (red), and HybC−15AA (blue). HybC+5AA is a genetic construct with an additional five amino acids at the C terminus. The HybC−15AA variant lacks the C-terminal extension. B, the respective proteins were overproduced in E. coli, and subsequently HybGStrep was pulled out from the cell extracts by Strep-Tactin affinity chromatography. Shown is SDS-PAGE analysis of complexes HybGStrep and HybC−15AA (lane 1) or HybGStrep and HybC (lane 2). Note that HybC but not HybC−15AA forms a complex with HybGStrep. C, SDS-PAGE analysis of 20 μg of HybGStrep complexes pulled out of extracts containing overproduced HybGStrep–HypDEF(GDEF) and either overproduced HybC+5AA or HybC or HybC−15AA (lanes 1–3). The experiment was repeated with extracts, in which either HypF (GDE ΔhypF; lanes 4–6) or HypE (GDF ΔhypE; lanes 7–9) was lacking. Again, only HybC formed a complex with HypGStrep. HypF and HypE were found to be not essential for the interaction of HybGStrep and HybC (lanes 5 and 8).
The plasmid p-hybG was expressed together with either p-hybC or p-hybC−15AA plasmid in the strain FTD-G (Table 2). The respective cell extracts were subjected to chromatography on a Strep-Tactin affinity column. The result shows that HybG interacted exclusively with HybC but not with the truncated variant HybC−15AA (Fig. 4B). Furthermore, we addressed the question of whether the C-terminal extension on HybC is important for recruitment of other proteins involved in maturation. To probe this, the plasmid pT-hybGStrep-hypDEF was expressed together with either HybC−15AA, HybC, or HybC+5AA. Analysis of the isolated protein complexes after Strep-Tactin affinity chromatography revealed that only HybC interacted with the core complex HybG–HypD, whereas both HybC−15AA and HybC+5AA did not interact (Fig. 4C, lanes 1–3). To determine whether HypE and/or HypF are required for HybG and HypD to form a complex with HybC, the experiment was repeated in a hypF or hypE deletion background. The plasmid pT-hybG-hypDE was expressed into hypF deletion strain FTD-F, whereas plasmid pT-hybG-hypDF was expressed in the hypE deletion strain FTD-E. The lack of HypF or HypE (Fig. 4C, lanes 4–6 (ΔhypF) and lanes 7–9 (ΔhypE)) appeared to have no negative impact on the ability of HybC to interact with the HybG–HypD core complex (Fig. 4C, lanes 5 and 8). Our study also demonstrates that extension of the C terminus by five amino acids (HybC+5AA) prevents interaction with HybG–HybD (Fig. 4C, lanes 1, 4, and 7). Altogether, these results indicated that the C-terminal extension on HybC is important for interaction with HybG–HypD of the maturation machinery. HybG and HypD form the central maturation complex, with neither HypE nor HypF being essential for complex formation with HybC.
Table 2.
Strains and plasmids used in this study
| Strains/plasmids | Genotype | Source/Reference |
|---|---|---|
| Strain | ||
| MC4100 | F− araD139 α (argF-lac)U169 ptsF25 deoC1 relA1 flbB5301 rspL150− | Ref. 58 |
| BL21(DE3) | F-ompT hsdSB (rB-,mB-)gal dcm (DE3) | Novagen |
| FTD147 | MC4100 ΔhyaB ΔhybC ΔhycE | Ref. 21 |
| FTD-F | MC4100 ΔhyaB ΔhybC ΔhypF::KanR | This study |
| FTD-E | MC4100 ΔhyaB ΔhybC ΔhypE::KanR | This study |
| FTD-G | MC4100 ΔhyaB ΔhybC ΔhycE ΔhybG::KanR | This study |
| FTD-O | MC4100 ΔhyaB ΔhybC ΔhycE ΔhybO::KanR | Ref. 16 |
| Plasmid | ||
| pT-hybG-hypDEF | pT7-7, hypD, hypE, his-TEV-hybG, hypF, kmR | This study |
| pT-hybG-hypDEF | pT7-7, hypD, hypE, hybG-Strep, hypF, AmpR | This study |
| pT-hybG-hypDE | pT7-7, hypD, hypE, hybG-Strep, AmpR | This study |
| pT-hybG-hypDF | pT7-7, hypD, hybG-Strep, hypF, AmpR | This study |
| p-hypF | pET28A, hypF, encodes His-HypF, kmR | Ref. 39 |
| p-hypE | pET28A, hypE, encodes His-HypE, kmR | Ref. 39 |
| pC-hybO | pET28A, hybO, encodes His-HybO, kmR | This study |
| pC-hybD | pCA24N, hybD, encodes His-HybD | This study |
| p-hybC+5AA | pCA24N, hybC, encodes His-HybC+5AA (GLCGR), CmR | Ref. 59 |
| p-hybC | Like p-hybC+5AA but with a stop codon, which removes the codons encoding GLCGR | Ref. 32 |
| p-hybC−15AA | Like p-HybC but a stop codon at position encoding codon V553, which removes the 15-amino acid C-terminal peptide | This study |
| pASK-HybC | pASK-IBA5, hybC, encodes Strep-HybC, AmpR | Ref. 39 |
| p-hybG | pASK-IBA3, encodes hybG-Strep, AmpR | Ref. 27 |
Cleavage of the C-terminal peptide on HybC after cofactor assembly is essential for the formation of active hydrogenase
We investigated at which stage during maturation the C-terminal extension of HybC is cleaved off. The individual compositions of protein complexes of the in vitro assays were analyzed at different maturation steps by SDS-PAGE (Fig. 5A). Lane 1 shows the complete reaction mix but lacking HybD endopeptidase. The reaction was started by the addition of HybD, and immediately afterward, an aliquot was analyzed (lane 2, t0). No HybC−15AA was detected. After t = 10, 20, and 30 min of incubation at room temperature, HybC−15AA was observed in increasing amounts (lanes 3–5). No additional processing of HybC after 20 min was observed. Both unprocessed StrepHybC and processed StrepHybC−15AA were isolated from the reaction mix by Strep-Tactin affinity chromatography (lane 6). Fig. 5B shows SDS-PAGE analysis of an enriched HisHybO fraction (lane 1). The HisHybO fraction was added to a fraction containing StrepHybC and StrepHybC−15AA (lane 2). Subsequently, active holoenzyme StrepHybC–HisHybO was isolated by histidine affinity chromatography (lane 3). These results demonstrate that the processed StrepHybC−15AA, but not the unprocessed HybC protein, forms a heterodimer with the small subunit HybO. Removal of the C-terminal extension appears to be a prerequisite for formation of catalytically active [NiFe]-hydrogenase.
Figure 5.

SDS-PAGE analysis of protein compositions of in vitro assays at different maturation steps. A, in vitro processing of hydrogenase-2 catalytic subunit HybC. Lane 1, reaction mix contained HybC, GDE complex, HypE, and HypF in stoichiometric amounts (2 μm each) and AM. Lane 2, the reaction was started by the addition of endopeptidase HybD in catalytic amounts, and immediately an aliquot of the reaction mix (∼25 μg) was analyzed (t0). No HybC−15AA was detected. Lanes 3–5, aliquots of the reaction mix were analyzed after 10, 20, and 30 min of incubation at room temperature, respectively (t10, t20, and t30). HybC−15AA formation was observed over time. After 20 min, no additional processing of HybC was observed. Lane 6, isolation of both unprocessed StrepHybC and processed StrepHybC−15AA from the reaction mix by Strep-Tactin affinity chromatography. B, formation of HybC–HybO holoenzyme. Lane 1, HisHybO-enriched fraction. Lane 2, the StrepHybC and StrepHybC−15AA fraction isolated from reaction mix was supplemented with the HisHybO-enriched fraction. Lane 3, the StrepHybC–HisHybO heterocomplex was isolated by a histidine affinity column. Only the processed large subunit (HybC−15AA) can form a heterodimer with the small subunit HybO.
To determine whether the C-terminal extension of HybC is required for synthesis of active hydrogenase also in vivo, plasmids encoding either N-terminal His-tagged HybC or equally tagged HybC−15AA were introduced into a hybC deletion strain of E. coli (21). After anaerobic growth, recombinant HybC and HybC−15AA proteins were isolated by histidine affinity chromatography (Co2+-charged resin). The HybC preparation exhibited hydrogenase activity (Fig. 3, B and C; in vivo) and showed the (CN)2CO signature of a [NiFe]-center (Fig. 2B; in vivo), but the HybC−15AA preparation was inactive and devoid of cofactor (data not shown). These results support the aforementioned findings that truncated HybC (HybC−15AA) is not able to recruit the hydrogenase maturation machinery and thus hinders maturation into active enzyme also in vivo.
Conformational changes of HybC upon removal of the C-terminal peptide
To detect possible conformational changes associated with cleavage of pro-protein, we purified and analyzed HybC, HybC−15AA, and HybC+5AA by CD and native gradient gel electrophoresis. Far-UV CD spectroscopy (150–250 nm) is sensitive to changes in secondary structure of proteins, whereas near-UV CD (250–350 nm) monitors alterations in the environment of aromatic amino acid side chains (i.e. changes in the tertiary structure of proteins) (22–24). Notably, the C-terminal extension does not contain any aromatic amino acids. Spectra of HybC (red) and HybC+5AA (black) were found to be almost identical (Fig. 6). Thus, the five-amino acid extension of the C terminus of HybC does not lead to structural changes in the protein. In contrast, the truncated HybC−15AA protein showed significant spectral differences in far- and near-UV CD spectroscopy (Fig. 6, A and B). Changes of far CD spectra are probably due to differences in the content of regular secondary structural features, such as the α-helix and β-sheet. Near-CD spectra indicate changes in the environment of aromatic amino acid side chains (e.g. changes in tertiary structure of the protein after removal of the C-terminal extension). Native PAGE (Fig. 6C, left) showed clear changes in migration behavior of HybC after removal of the C-terminal extension. The five-amino acid extension of HybC did not lead to changes in migration behavior, because both HybC+5AA and HybC migrated faster and showed broad, diffuse protein bands. In contrast, upon removal of C-terminal extension, the HybC−15AA migrated slower and showed a sharper protein band. SDS-PAGE was used to resolve and confirm the small differences in mass between HybC variants (Fig. 6C). In sum, these results indicate that conformational changes in secondary and tertiary structure of HybC take place upon removal of C-terminal extension.
Figure 6.
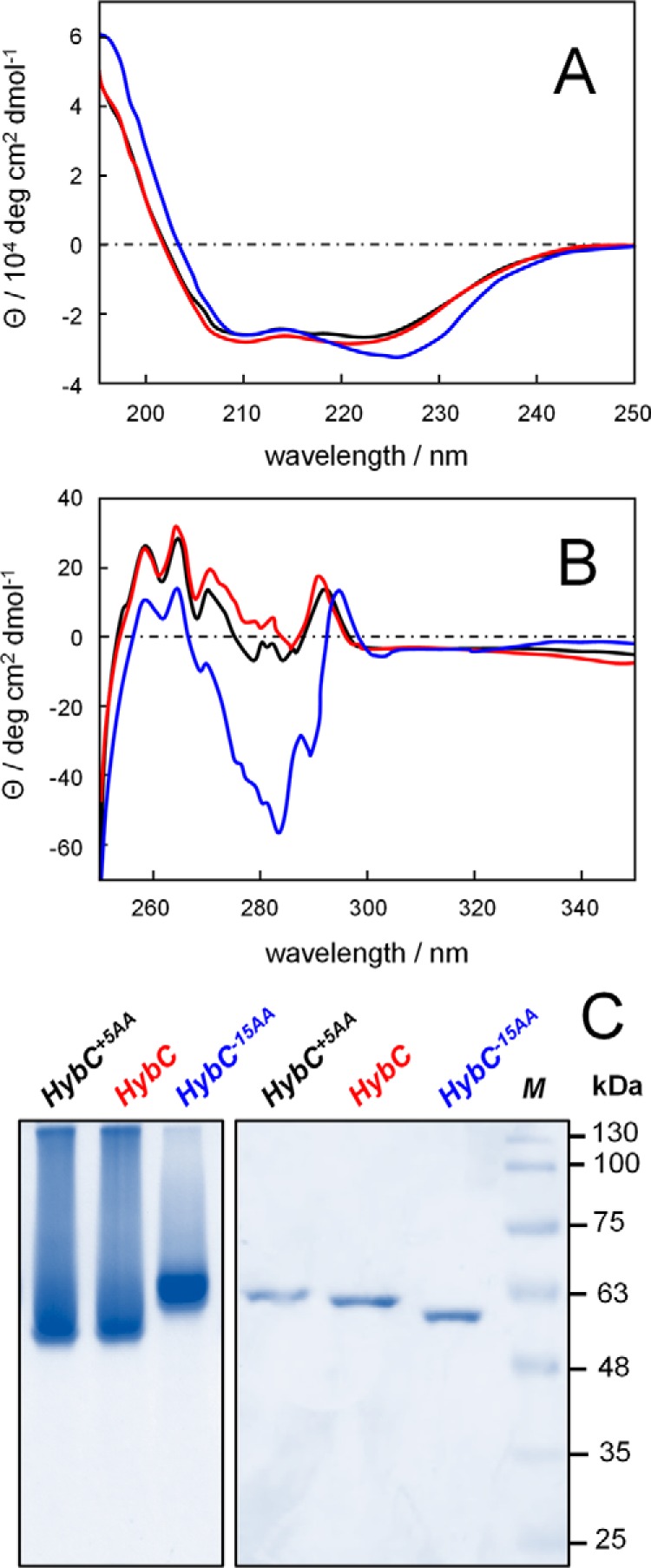
Conformational changes of HybC upon removal of the C-terminal peptide. A, far-UV CD spectra; B, near-UV CD spectra of HybC+5AA (black), HybC (red), and HybC−15AA (blue). The five-amino acid extension on HybC does not lead to significant conformational changes. In contrast, removal of the C-terminal extension induces clear conformational changes in secondary and tertiary structure (compare red and black versus blue). C, native PAGE analysis (left) shows clear changes in migration behavior after removal of the C-terminal extension. The HybC−15AA protein band is sharper and migrates slower than HybC and HybC+5AA variants, indicative of a different conformation. SDS-PAGE (right) reveals the only small differences in mass between HybC variants.
Discussion
Maturation of [NiFe]-hydrogenases in vitro provides the possibility to prove and refine our understanding of [NiFe]-cofactor biosynthesis. The presented approach exclusively relies on purified components and allows studying the order of events individually. Our maturation assay included HypE and HypF, both of which have been shown to synthesize the CN ligands from carbamoyl phosphate, as supplied with the activation mixture (25, 26). Although HypE and HypF are absolutely required for synthesis of the cyano-ligands, partial cleavage of HybC took place in their absence. This is due to the presence of HypE that was supplied to the reaction mix in the form of the GDE complex. The reason for cleavage in the absence of HypF is the presence of Fe(CN)2CO on the GDE complex that allows for partial assembly of [NiFe]-cofactor (14). Only the complete assay, including HypE and HypF, allows for multiple maturation turnover, as supported by formation of [NiFe]-cofactor and maturation of hydrogenase-2 in vitro at activity levels near those of the in vivo matured enzyme. Lenz and co-workers (61) recently identified the accessory protein HypX that synthesizes the CO ligand of O2-tolerant [NiFe]-hydrogenases from formyl-tetrahydrofolate. However, the anaerobic route of biosynthesis remains ambiguous because E. coli lacks HypX or orthologue enzymes (27).
An interesting aspect of performing in vitro reconstitution with purified proteins is whether a particular protein has a direct participation in the maturation process or not and which minimal combination of components is sufficient to achieve complete maturation. This capability provides a rapid means of “quality control” with regard to the reproducibility of the purification procedures for the individual enzymes by confirming the activity of the purified proteins individually. The functionality of the purified GDE complex, HypE, HypF, HybD, HybC, and HybO was confirmed by activity measurements of maturation assays complemented with the corresponding missing protein, as shown in Fig. 3C. FTIR analysis performed in previous and current studies indicates that anaerobically purified GDE complex carries two cyano-ligands, one CO ligand and bound CO2 (14). Whereas HypD alone shows infrared bands assigned to one CO and two CN ligands (28), purified HybG exhibits a band characteristic of bound CO2 (27). Metal analysis determined by inductively coupled plasma mass spectrometry (ICP-MS) showed that the GDE complex contained ∼4.4 iron ions/mol of ternary complex, whereas HybG contained substoichiometric amounts of iron (∼0.3 mol of iron/mol of HybG). The amount of nickel, copper, cobalt, and zinc was below the detection limit (14). Employing the GDE complex that does not harbor the Fe(CN)2CO moiety could not restore hydrogenase activity. Therefore, the presence of the Fe(CN)2CO moiety seems to be necessary for hydrogenase-2 maturation in vitro. No hydrogenase activity could be measured when the GDE complex was replaced by HybG, HypD, or HypE individually or in combination after separate purification (14). This finding indicates that the physical ternary complex carrying the Fe(CN)2CO moiety is essential for successful activation of hydrogenase-2. Anerobically purified HypD protein preparations typically contain five equivalents of iron and four sulfides per protein molecule (28). Four of these iron ions are associated with a low-potential [4Fe-4S] cluster (29). Electron paramagnetic resonance (EPR) and Mossbauer spectroscopy on HypD revealed a diamagnetic [4Fe-4S]2+ cluster (30). The fifth iron ion was proposed to coordinate the (CN)2CO ligands (28). HypD is the only Hyp [4Fe-4S] cluster containing protein and is therefore likely to be capable of performing redox chemistry and delivering electrons for CO and CN ligand generation (29). During the course of this study, HypD was found to catalyze the CO-dependent reduction of the electron acceptor methyl viologen with a maximal rate of 24 milliunits/mg of protein. However, the reverse reaction, CO2-dependent oxidation of reduced methyl viologen, performed in the presence of ATP could not be observed (data not shown). Analysis of anaerobically purified HypF revealed that the protein contained only substoichiometric amounts of iron and zinc (∼0.1 mol of iron and 0.4 mol of zinc per HypF monomer). The UV-visible spectrum did not indicate the presence of any cofactor. Similar results were obtained for the aerobically purified HypF (26). The carbamoyl phosphate phosphatase activity of HypF was routinely confirmed by the release of inorganic phosphate directly in non-denaturing polyacrylamide gels (31). The functionality of purified HypE and HypF was also routinely verified by observation of the cyanation on HypE from carbamoyl phosphate in an ATP-dependent condensation reaction using ATR FTIR (25). The addition of only HypF or HypE to the reconstitution reaction had no effect on the level of hydrogenase activity, but the addition of both (complete reaction) increased the hydrogenase activity about 2-fold. This suggests that an equimolar ratio of HypE and HypF is needed for full reconstitution of hydrogenase activity. Anerobically purified HybC precursor isolated from any hydrogenase deletion mutant of hypF, hypE, hypD, or hybD was unprocessed, was not associated with the small subunit HybO, and lacked enzyme activity. These HybC preparations were colorless and lacked cofactor or any associated metal, as indicated by UV-visible spectrum and ICP-MS analysis. In a Hyp-competent genetic background, the HybC precursor can be matured in vivo and in vitro and delivers active hydrogenase-2. In contrast, the isolated HybC+5AA could not be matured, was brown, and had substoichiometric amounts of an oxygen-sensitive iron–sulfur cluster (0.2 mol of iron/mol of HybC+5AA). This cluster is coordinated by the conserved cysteinyl residues that normally ligate the NiFe(CN)2CO cofactor, as was shown by UV-visible spectroscopy and EPR (32).
Isolation of the small subunit HybO in its active form was more challenging. The pC-hybO plasmid that encodes for N-terminal His-tagged protein (HisHybO) was transformed in E. coli strain FTD-O, which lacks the genes encoding HybO and the large subunit of hydrogenase-2 (Table 2). Overproduction and purification of HisHybO was not possible due to inclusion body formation. A low level of soluble protein without isopropyl 1-thio-β-d-galactopyranoside induction (∼0.70 mg of protein per 5 g wet weight of anaerobically grown cells) could be isolated by histidine affinity chromatography, but HisHybO was labile due to the imidazole-containing elution buffer and could not restore hydrogenase activity in vitro. We therefore separated the soluble fraction containing HisHybO by ultracentrifugation and subsequent ion-exchange chromatography. The HisHybO fraction was brown and most likely present as holoprotein with three iron–sulfur clusters. It should be noted that the HybO protein concentration in hydrogenase deletion mutants was usually at a low level, particularly if the large subunit is absent (17). This can be due to rapid turnover of HybO. These results suggest that direct physical contact between the large and small subunit is required to ensure stability of the small subunit against degradation.
The results provide evidence that the C-terminal extension of the large subunit HybC is removed only after cofactor assembly and before complex formation with the small subunit HybO has occurred. Proteolytic cleavage is thus a checkpoint that guarantees formation of catalytically active [NiFe]-hydrogenase exclusively. The underlying mechanism was shown to be a larger conformational change associated with removal of the 15 C-terminal amino acids. We suggest that proteolytic cleavage of HybC triggers structural changes and induces formation of the [NiFe]-center from the Fe(CN)2CO moiety and nickel (33). It has been hypothesized that changes in conformation precede the internalization of the metal center within the large subunit (5). Crystal structures show that the [NiFe]-cofactor lies close to the interface with the small subunit (34).
The data presented in this study suggest that the role of the C-terminal extension is maintaining a “correct” conformation of the HybC protein that facilitates interaction with accessory proteins HybG–HypDEF. We could demonstrate that maturation proteins, particularly HybG, interact specifically with HybC but are incapable of interacting with truncated HybC−15AA. Most likely, the different conformation adopted by HybC−15AA prevents the interaction. During synthesis of hydrogenase-3, HypC (a homologue of HybG) forms a stable complex with the pro-protein of the large subunit of hydrogenase-3. This complex was found to accumulate in mutants unable to synthesize the NiFe(CN)2CO cofactor (35). Therefore, it is likely that complex formation between the unprocessed large subunit and the hydrogenase maturation machinery makes HybC accessible for the incoming cofactor (5). Because we observe formation of the HybC–HybO heterodimer exclusively with processed HybC, it seems that the C-terminal extension on HybC prevents complex formation with HybO. Upon removal of the C-terminal peptide, far-CD spectroscopy indicates rather insignificant conformational changes in secondary structure. In contrast, global changes in tertiary structure of the protein are suggested by the observed difference in the near-UV spectra between HybC and HybC−15AA. This difference is most likely due to the tertiary folding of the polypeptide chain, which can place the aromatic amino acid side chains in chiral environments, thus giving rise to circular dichroism (22, 24). This is in good agreement with the proposed refolding of HybC upon cleavage of the C-terminal peptide (5). The fully folded and cofactor-containing HybC large subunit can then engage with the small subunit to form catalytically active heterodimer. This would ensure that in wild-type cells, only a large subunit with a complete [NiFe]-cofactor proceeds along the maturation path. A similar function was suggested for the C-terminal extension of hydrogenase-3 of E. coli and the membrane-bound [NiFe]-hydrogenase of Ralstonia eutropha (36, 37).
Biosynthesis and insertion of [NiFe]-cofactor involves a multistep pathway catalyzed by different compositions of accessory proteins (20, 38). Precursors of [NiFe]-hydrogenase are unstable in aqueous solutions and highly sensitive to oxygen (39, 40); thus, it seems likely that accessory and pro-protein form a supramolecular complex during maturation. A stable “supercomplex” consisting of HybG, HypD, HypE, and HybC was isolated in this work. Furthermore, multiprotein complexes containing HypA, HypB, SlyD, and pro-protein of hydrogenase-3 were observed earlier (49).
The C-terminal extension of the large subunit varies greatly both in sequence and length among different hydrogenases and organisms. Intriguingly, there are examples of hydrogenases whose large subunit lacks a C-terminal extension including H2-sensory and energy-converting [NiFe]-hydrogenases (41, 42). During the course of this study, the large subunit of [NiFe]-hydrogenase from Carboxydothermus hydrogenoformans (CooH) or Thermoanaerobacter tengcongensis (EchE), in which the C-terminal extension is missing (43, 44), could not be synthesized in an active form in E. coli. In reciprocal heterologous complementation studies, anaerobically purified heterologous HybG–HypDE complex, where HypD was replaced by HypD either from C. hydrogenoformans or from T. tengcongensis, was not capable of assembling and maturing active hydrogenase-2 in E. coli (data not shown). How insertion of the [NiFe]-cofactor and subunit assembly are coordinated in these enzymes remains to be investigated. However, it is possible that the hydrogenase-specific maturation machinery in these organisms encodes for a peptide that functionally replaces the C-terminal extension. In support of this is the finding that the extension must not be covalently linked to the large subunit but can be part of a separate polypeptide (45).
In conclusion, this study highlights the role of the C-terminal extension on the hydrogenase catalytic subunit. The 15-amino acid peptide promotes the complete sequence of maturation events by directing and binding maturation proteins and ensuring that only the cofactor-containing large subunit can continue on the assembly line toward active [NiFe]-hydrogenase.
Experimental procedures
Bacterial strains and growth conditions
All E. coli strains and plasmids used are listed in Table 2. Strains were generally grown anaerobically in LB or in modified TB medium. The antibiotics kanamycin, chloramphenicol, and ampicillin were added to the medium at final concentrations of 50, 15, and 100 μg/ml respectively. For overproduction of proteins, E. coli strains, BL21(DE) (46), FTD147 (21), or the indicated strains were transformed with the appropriate plasmid and grown anaerobically in modified TB medium at 37 °C until the optical density at 600 nm (A600) reached 0.3. Gene expression was induced by the addition of 0.2 μg/ml anhydrotetracycline or 150 μm isopropyl 1-thio-β-d-galactopyranoside followed by incubation at 30 °C for 3–5 h or after the culture reached an optical density at 600 nm between 0.7 and 0.9 (exponential phase cultures). Cells were harvested by centrifugation at 10,000 × g for 15 min at 4 °C. Large-scale protein purification was performed with 20 liters of anaerobic TB medium (3–4 g of cells/liter culture).
Strain and plasmid construction
Strains were constructed by the introduction of mutations from E. coli donor strains into recipient strains of MC4100 derivatives by P1kc-mediated phage transduction. Plasmid p-hybC served as DNA template for the introduction of mutation into the hybC gene using the QuikChange site-directed mutagenesis strategy of Stratagene. The oligonucleotide primers used to introduce a stop codon at position Val-553 of HybC were 5′-GCCTGTGCGGTACACTAAGTGGATGCTGACGGC-3′ and 5′-GCCGTCAGCATCCACTTAGTGTACCGCACAGGC-3′. To generate the pT-hybG-hypDF and pT-hybG-hypDE plasmids, the pT-hybG-hypDEF plasmid (14) was digested with BamHI or HindIII, which released either the complete hypE gene or the complete hypF gene, respectively. The p-hybG-hypDEF plasmid was used as a template for amplification of hypD, hypE, hypF, and hybG genes via PCR. The resulting PCR fragments were digested with NdeI and HindIII and ligated into NdeI-/HindIII-digested pET28A to generate the plasmid pT-hybG-hypDEF.
Preparation of crude extracts and affinity purification of protein
All steps were carried out under a nitrogen/hydrogen mixture (95:5) in an anaerobic chamber (Coy Laboratories) and at 4 ºC unless stated otherwise. Cells containing Strep-tagged protein were resuspended at a ratio of 1:3 (w/v) in buffer W (100 mm Tris/HCl and 150 mm NaCl, pH 8.0) including 2 mm sodium dithionite, 5 mg/liter DNase, and 0.2 mm PMSF. Cells were disrupted by sonication (40 watts for 16 min with 0.5-s pulses). Unbroken cells and debris were removed by centrifugation for 30 min at 10,000 × g at 4 °C to deliver the crude extract. Three-milliliter crude extracts were applied to a 1-ml Strep-Tactin-Sepharose column (IBA Technologies) using gravity flow. Unbound proteins were removed from the column by washing with 5 column volumes of buffer W. Specifically bound proteins were eluted with buffer W including 5 mm desthiobiotin. Desthiobiotin was subsequently removed by passage through a Hi-Prep 26/10 desalting column (GE Healthcare) equilibrated with buffer W without reducing agents and connected to an ÄKTA apparatus (GE Healthcare). Proteins were concentrated by centrifugation at 5000 × g using centrifugal filters (Amicon Ultra, 50 K, Millipore) and stored under liquid nitrogen. His-tagged protein variants were purified from cells transformed with the appropriate plasmid derivative (Table 2). Wet cell paste was resuspended at a ratio of 1:4 (w/v) in buffer A (50 mm Tris/HCl, pH 8.0, 300 mm NaCl) including 5 mg/liter DNase and 0.2 mm PMSF. Crude extracts derived from ∼10 g of wet cell paste were loaded onto a 1.5 × 10-cm column of Co2+-charged resin (Talon resin, Clontech). The column, which had been previously equilibrated with buffer A, was washed with 10 column volumes of the same buffer, followed by 5 column volumes of buffer A supplemented with 10 mm imidazole and then 3 column volumes of buffer A supplemented with 20 mm imidazole to remove nonspecifically bound proteins. His-tagged proteins were subsequently eluted with buffer A containing 300 mm imidazole. Imidazole was removed by a desalting column, and proteins were concentrated as described above.
Enrichment of an active small subunit HybO required for analysis of hydrogenase activity
The pC-hybO plasmid that encodes for N-terminal His-tagged protein (HisHybO) was transformed in E. coli strain FTD-O (Table 2) and grown anaerobically in LB medium at 25 °C without induction until the optical density at 600 nm reached 0.7. The cells were disrupted by three passages through a French pressure cell. HisHybO purified by histidine affinity chromatography could not restore hydrogenase activity in vitro. We therefore separated soluble fractions containing HisHybO by ultracentrifugation at 150,000 × g for 2 h. Subsequently, the supernatant was loaded onto a Q-Sepharose HiLoad column, equilibrated with 100 mm Tris buffer, pH 8. The protein was eluted in a stepwise NaCl gradient. Enriched active holoprotein HisHybO was recovered in the fractions eluted with 0.2 m NaCl.
The in vitro system for maturation of [NiFe]-hydrogenase-2 from E. coli
The 100-μl complete reaction mixture in 50 mm MOPS KOH, pH 7.0, contained 4 μm purified Strep-tagged HybC (39), 2 μm purified GDE complex (14), 2 μm HisHypE, and 2 μm HisHypF (14). An activation mix with ATP, carbamoyl phosphate, NiCl2, FeSO4, and sodium–dithionite was added at final concentrations of 2.5 mm, 50 μm, 100 μm, 50 μm, and 2 mm, respectively. The reaction mixtures were incubated at room temperature under nitrogen (95%) hydrogen (5%) in an anaerobic chamber. The reaction was started by the addition of a 1 μm concentration of the endopeptidase HisHybD. After a 30-min incubation, 20-μl aliquots of the reaction mixture were analyzed for cleavage of HybC by SDS-PAGE using the ImageQuant software (Molecular Dynamics). Aliquots of reaction mix (80 μl) were supplemented with 0.5 mg of the small subunit HybO-enriched fraction. After 15 min, the samples were analyzed for activity photometrically.
Isolation of in vitro matured hydrogenase-2
To isolate preparative amounts of active hydrogenase-2 that was matured in vitro, the reaction mix was scaled up to 5 ml. Matured StrepHybC was isolated from the reaction mix by Strep-Tactin affinity chromatography. StrepHybC was supplemented with the HisHybO-enriched fraction. Active StrepHybC–HisHybO heterocomplex was isolated by histidine affinity chromatography (Co2+-charged resin). Protein samples were concentrated and analyzed by ATR FTIR as reported earlier (47).
Isolation of in vivo matured hydrogenase-2
Hydrogenase-2 was isolated from FTD-147 strain, in which a fully active enzyme was restored by introducing genes coding for HybC together with maturation proteins HybG–HypDEF on an expression plasmids pASK-hybC and pT-hybG-hypDEF. The procedure for isolating an active enzyme was according to Ref. 48.
Determination of hydrogenase activity
H2 uptake activity was determined by following the reduction of benzyl viologen at 578 nm using an Uvicon 900 dual-wavelength spectrophotometer. The 0.8-ml assays contained 4 mm benzyl viologen in 50 mm MOPS buffer, pH 7.0. Cuvettes were allowed to equilibrate with a 100% H2 headspace. One unit of activity corresponds to oxidation of 1 μmol of H2/min.
Polyacrylamide gel electrophoresis
A defined amount of purified protein (3–20 μg) was separated by SDS-PAGE using 15% (w/v) polyacrylamide gels (49). Non-denaturating PAGE was performed using 7.5% (w/v) polyacrylamide gels including 0.1% (w/v) Triton X-100. Protein samples (5–100 μg) were incubated with 4% (w/v) Triton X-100 prior to application to the gels (50).
Non-heme iron and acid-labile sulfide determination
Iron and acid-labile sulfide of the GDE complex, HypD, and HybG were determined due to established protocols (51). Metal content of the GDE complex, HypD, HybG, HypE, HypF, and HybC variants was determined by ICP-MS, as described previously (32). For ICP-MS analysis, 0.1–0.2 mg of purified protein samples were analyzed for iron, nickel, zinc, cobalt, and copper.
Infrared spectroscopic characterization of HybC variants
All FTIR spectroscopy was conducted on a rapid-scan Tensor 27 spectrometer (Bruker Optik, Ettlingen, Germany) equipped with a three-reflection ZnSe/silicon crystal ATR cell (Smiths Detection, Wiesbaden Germany) as described earlier (27, 28, 47). The spectrometer was situated in an anaerobic gas chamber (Coy Laboratories, Grass Lake, MI) in a water-free atmosphere of typically 99% N2 and 1% H2.
Further spectroscopic characterization of HybC
Protein concentrations were determined spectroscopically using an extinction coefficient of EM (280 nm) = 97,310 m−1 cm−1. Spectra were measured at a protein concentration of 1 mg/ml in 25 mm Tris-HCl buffer, pH 7.5, containing 150 mm NaCl using a Jasco J815 spectropolarimeter in the far-UV region (light path 0.1 mm, data accumulation 16×) and near-UV region (light path 1 cm, data accumulation 32× or 64×), respectively. For all measurements, it was ensured that the proteins were stable under the given conditions.
Quality control and metal content
No hydrogen turnover activity is associated with any of the purified maturases: GDE complex, HypE, HypF, HybD, or HybO (14, 16). Thus, catalytic competence and cofactor integrity of these enzymes had to be probed alternatively. This included the use of the maturation assay established in this study, UV-visible spectroscopy, FTIR spectroscopy, ICP-MS, and different biochemical activity assays. For example, the phosphatase activity of HypF was routinely confirmed by measuring the release of inorganic phosphate from carbamoyl phosphate (31). The specific interplay of HypE and HypF was probed by observing cyanation of HypE from inorganic phosphate in an ATP-dependent condensation reaction via surface-enhanced IR absorption spectroscopy, as established earlier (25). Furthermore, the integrity of the Fe(CN)2CO cofactor as associated with HypD and the GDE complex was routinely probed by ATR FTIR (14, 28). The presence of an iron ion and CO2 absorption band on HybG was confirmed by ICP-MS, UV-visible spectroscopy, and ATR FTIR (27).
Author contributions
B. S. and S. T. S. designed the research; B. S., S. T. S., and M. S. performed the research; and B. S. and S. T. S. analyzed the data and wrote the paper.
Acknowledgments
We gratefully acknowledge Rudolf Thauer for helpful discussions, Joachim Heberle and Ramona Schlesinger for ongoing support, Ute Lindenstrauss for excellent technical assistance, and Hauke Lilie for assistance with the CD spectroscopy measurements.
This work was supported by the Deutsche Forschungsgemeinschaft through the priority program “Iron-Sulfur for Life,” Grant SO 1325/5-1 (to B. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- CO
- carbonyl
- CN
- cyanide
- ICP-MS
- inductively coupled plasma mass spectrometry
- GDE
- HybG–HypDE.
References
- 1. Waldron K. J., and Robinson N. J. (2009) How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 7, 25–35 [DOI] [PubMed] [Google Scholar]
- 2. Lu Y., Yeung N., Sieracki N., and Marshall N. M. (2009) Design of functional metalloproteins. Nature 460, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Currin A., Swainston N., Day P. J., and Kell D. B. (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem. Soc. Rev. 44, 1172–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lill R., and Mühlenhoff U. (2008) Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700 [DOI] [PubMed] [Google Scholar]
- 5. Böck A., King P. W., Blokesch M., and Posewitz M. C. (2006) Maturation of hydrogenases. Adv. Microb. Physiol. 51, 1–71 [DOI] [PubMed] [Google Scholar]
- 6. Lacasse M. J., Douglas C. D., and Zamble D. B. (2016) Mechanism of selective nickel transfer from HypB to HypA, Escherichia coli [NiFe]-hydrogenase accessory proteins. Biochemistry 55, 6821–6831 [DOI] [PubMed] [Google Scholar]
- 7. Lubitz W., Ogata H., Rüdiger O., and Reijerse E. (2014) Hydrogenases. Chem. Rev. 114, 4081–4148 [DOI] [PubMed] [Google Scholar]
- 8. Vignais P. M., and Billoud B. (2007) Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 [DOI] [PubMed] [Google Scholar]
- 9. Thauer R. K., Kaster A. K., Goenrich M., Schick M., Hiromoto T., and Shima S. (2010) Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H-2 storage. Annu. Rev. Biochem. 79, 507–536 [DOI] [PubMed] [Google Scholar]
- 10. Happe R. P., Roseboom W., Pierik A. J., Albracht S. P., and Bagley K. A. (1997) Biological activation of hydrogen. Nature 385, 126. [DOI] [PubMed] [Google Scholar]
- 11. Pierik A. J., Roseboom W., Happe R. P., Bagley K. A., and Albracht S. P. (1999) Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of [NiFe]-hydrogenases. NiFe(CN)2CO, biology's way to activate H2. J. Biol. Chem. 274, 3331–3337 [DOI] [PubMed] [Google Scholar]
- 12. Soboh B., Stripp S. T., Muhr E., Granich C., Braussemann M., Herzberg M., Heberle J., and Gary Sawers R. (2012) [NiFe]-hydrogenase maturation: isolation of a HypC-HypD complex carrying diatomic CO and CN− ligands. FEBS Lett. 586, 3882–3887 [DOI] [PubMed] [Google Scholar]
- 13. Watanabe S., Kawashima T., Nishitani Y., Kanai T., Wada T., Inaba K., Atomi H., Imanaka T., and Miki K. (2015) Structural basis of a Ni acquisition cycle for [NiFe] hydrogenase by Ni-metallochaperone HypA and its enhancer. Proc. Natl. Acad. Sci. U.S.A. 112, 7701–7706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soboh B., Lindenstrauss U., Granich C., Javed M., Herzberg M., Thomas C., and Stripp S. T. (2014) [NiFe]-hydrogenase maturation in vitro: analysis of the roles of the HybG and HypD accessory proteins. Biochem. J. 464, 169–177 [DOI] [PubMed] [Google Scholar]
- 15. Theodoratou E., Paschos A., Magalon A., Fritsche E., Huber R., and Böck A. (2000) Nickel serves as a substrate recognition motif for the endopeptidase involved in hydrogenase maturation. Eur. J. Biochem. 267, 1995–1999 [DOI] [PubMed] [Google Scholar]
- 16. Pinske C., Krüger S., Soboh B., Ihling C., Kuhns M., Braussemann M., Jaroschinsky M., Sauer C., Sargent F., Sinz A., and Sawers R. G. (2011) Efficient electron transfer from hydrogen to benzyl viologen by the [NiFe]-hydrogenases of Escherichia coli is dependent on the coexpression of the iron-sulfur cluster-containing small subunit. Arch. Microbiol. 193, 893–903 [DOI] [PubMed] [Google Scholar]
- 17. Thomas C., Muhr E., and Sawers R. G. (2015) Coordination of synthesis and assembly of a modular membrane-associated [NiFe]-hydrogenase is determined by cleavage of the C-terminal peptide. J. Bacteriol. 197, 2989–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pardo A., De Lacey A. L., Fernández V. M., Fan H. J., Fan Y., and Hall M. B. (2006) Density functional study of the catalytic cycle of nickel-iron [NiFe] hydrogenases and the involvement of high-spin nickel(II). J. Biol. Inorg. Chem. 11, 286–306 [DOI] [PubMed] [Google Scholar]
- 19. Ash P. A., Hidalgo R., and Vincent K. A. (2017) Proton transfer in the catalytic cycle of [NiFe] hydrogenases: insight from vibrational spectroscopy. ACS Catal. 7, 2471–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Butland G., Zhang J. W., Yang W., Sheung A., Wong P., Greenblatt J. F., Emili A., and Zamble D. B. (2006) Interactions of the Escherichia coli hydrogenase biosynthetic proteins: HybG complex formation. FEBS Lett. 580, 677–681 [DOI] [PubMed] [Google Scholar]
- 21. Redwood M. D., Mikheenko I. P., Sargent F., and Macaskie L. E. (2008) Dissecting the roles of Escherichia coli hydrogenases in biohydrogen production. FEMS Microbiol. Lett. 278, 48–55 [DOI] [PubMed] [Google Scholar]
- 22. Sreerama N., and Woody R. W. (2004) Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 383, 318–351 [DOI] [PubMed] [Google Scholar]
- 23. Kelly S. M., and Price N. C. (2000) The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1, 349–384 [DOI] [PubMed] [Google Scholar]
- 24. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stripp S. T., Lindenstrauss U., Sawers R. G., and Soboh B. (2015) Identification of an isothiocyanate on the HypEF complex suggests a route for efficient cyanyl-group channeling during [NiFe]-hydrogenase cofactor generation. PLoS One 10, e0133118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paschos A., Bauer A., Zimmermann A., Zehelein E., and Böck A. (2002) HypF, a carbamoyl phosphate-converting enzyme involved in [NiFe] hydrogenase maturation. J. Biol. Chem. 277, 49945–49951 [DOI] [PubMed] [Google Scholar]
- 27. Soboh B., Stripp S. T., Bielak C., Lindenstrauss U., Braussemann M., Javaid M., Hallensleben M., Granich C., Herzberg M., Heberle J., and Sawers R. G. (2013) The [NiFe]-hydrogenase accessory chaperones HypC and HybG of Escherichia coli are iron- and carbon dioxide-binding proteins. FEBS Lett. 587, 2512–2516 [DOI] [PubMed] [Google Scholar]
- 28. Stripp S. T., Soboh B., Lindenstrauss U., Braussemann M., Herzberg M., Nies D. H., Sawers R. G., and Heberle J. (2013) HypD is the scaffold protein for Fe-(CN)2CO cofactor assembly in [NiFe]-hydrogenase maturation. Biochemistry 52, 3289–3296 [DOI] [PubMed] [Google Scholar]
- 29. Roseboom W., Blokesch M., Böck A., and Albracht S. P. (2005) The biosynthetic routes for carbon monoxide and cyanide in the Ni-Fe active site of hydrogenases are different. FEBS Lett. 579, 469–472 [DOI] [PubMed] [Google Scholar]
- 30. Blokesch M., Albracht S. P., Matzanke B. F., Drapal N. M., Jacobi A., and Böck A. (2004) The complex between hydrogenase-maturation proteins HypC and HypD is an intermediate in the supply of cyanide to the active site iron of [NiFe]-hydrogenases. J. Mol. Biol. 344, 155–167 [DOI] [PubMed] [Google Scholar]
- 31. Mizuno Y., Ohba Y., Fujita H., Kanesaka Y., Tamura T., and Shiokawa H. (1989) Activity staining of acylphosphatase after gel electrophoresis. Anal. Biochem. 183, 46–49 [DOI] [PubMed] [Google Scholar]
- 32. Soboh B., Kuhns M., Braussemann M., Waclawek M., Muhr E., Pierik A. J., and Sawers R. G. (2012) Evidence for an oxygen-sensitive iron-sulfur cluster in an immature large subunit species of Escherichia coli [NiFe]-hydrogenase 2. Biochem. Biophys. Res. Commun. 424, 158–163 [DOI] [PubMed] [Google Scholar]
- 33. Theodoratou E., Huber R., and Böck A. (2005) [NiFe]-hydrogenase maturation endopeptidase: structure and function. Biochem. Soc. Trans. 33, 108–111 [DOI] [PubMed] [Google Scholar]
- 34. Volbeda A., Charon M. H., Piras C., Hatchikian E. C., Frey M., and Fontecilla-Camps J. C. (1995) Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373, 580–587 [DOI] [PubMed] [Google Scholar]
- 35. Drapal N., and Bock A. (1998) Interaction of the hydrogenase accessory protein HypC with HycE, the large subunit of Escherichia coli hydrogenase 3 during enzyme maturation. Biochemistry 37, 2941–2948 [DOI] [PubMed] [Google Scholar]
- 36. Binder U., Maier T., and Böck A. (1996) Nickel incorporation into hydrogenase 3 from Escherichia coli requires the precursor form of the large subunit. Arch. Microbiol. 165, 69–72 [DOI] [PubMed] [Google Scholar]
- 37. Massanz C., Fernandez V. M., and Friedrich B. (1997) C-terminal extension of the H2-activating subunit, HoxH, directs maturation of the NAD-reducing hydrogenase in Alcaligenes eutrophus. Eur. J. Biochem. 245, 441–448 [DOI] [PubMed] [Google Scholar]
- 38. Watanabe S., Matsumi R., Atomi H., Imanaka T., and Miki K. (2012) Crystal structures of the HypCD complex and the HypCDE ternary complex: transient intermediate complexes during [NiFe] hydrogenase maturation. Structure 20, 2124–2137 [DOI] [PubMed] [Google Scholar]
- 39. Soboh B., Krüger S., Kuhns M., Pinske C., Lehmann A., and Sawers R. G. (2010) Development of a cell-free system reveals an oxygen-labile step in the maturation of [NiFe]-hydrogenase 2 of Escherichia coli. FEBS Lett. 584, 4109–4114 [DOI] [PubMed] [Google Scholar]
- 40. Stripp S. T., Lindenstrauss U., Granich C., Sawers R. G., and Soboh B. (2014) The influence of oxygen on [NiFe]-hydrogenase cofactor biosynthesis and how ligation of carbon monoxide precedes cyanation. PLoS One 9, e107488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Friedrich B., Buhrke T., Burgdorf T., and Lenz O. (2005) A hydrogen-sensing multiprotein complex controls aerobic hydrogen metabolism in Ralstonia eutropha. Biochem. Soc. Trans. 33, 97–101 [DOI] [PubMed] [Google Scholar]
- 42. Hedderich R., and Forzi L. (2005) Energy-converting [NiFe] hydrogenases: more than just H2 activation. J. Mol. Microbiol. Biotechnol. 10, 92–104 [DOI] [PubMed] [Google Scholar]
- 43. Soboh B., Linder D., and Hedderich R. (2002) Purification and catalytic properties of a CO-oxidizing:H2-evolving enzyme complex from Carboxydothermus hydrogenoformans. Eur. J. Biochem. 269, 5712–5721 [DOI] [PubMed] [Google Scholar]
- 44. Soboh B., Linder D., and Hedderich R. (2004) A multisubunit membrane-bound [NiFe] hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 150, 2451–2463 [DOI] [PubMed] [Google Scholar]
- 45. Sorgenfrei O., Linder D., Karas M., and Klein A. (1993) A novel very small subunit of a selenium containing [NiFe] hydrogenase of Methanococcus voltae is postranslationally processed by cleavage at a defined position. Eur. J. Biochem. 213, 1355–1358 [DOI] [PubMed] [Google Scholar]
- 46. Studier F. W., and Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 47. Senger M., Mebs S., Duan J., Wittkamp F., Apfel U. P., Heberle J., Haumann M., and Stripp S. T. (2016) Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics. Proc. Natl. Acad. Sci. U.S.A. 113, 8454–8459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lukey M. J., Parkin A., Roessler M. M., Murphy B. J., Harmer J., Palmer T., Sargent F., and Armstrong F. A. (2010) How Escherichia coli is equipped to oxidize hydrogen under different redox conditions. J. Biol. Chem. 285, 3928–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 50. Ballantine S. P., and Boxer D. H. (1985) Nickel-containing hydrogenase isoenzymes from anaerobically grown Escherichia coli K-12. J. Bacteriol. 163, 454–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fish W. W. (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364 [DOI] [PubMed] [Google Scholar]
- 52. Menon N. K., Chatelus C. Y., Dervartanian M., Wendt J. C., Shanmugam K. T., Peck H. D. Jr., and Przybyla A. E. (1994) Cloning, sequencing, and mutational analysis of the hyb operon encoding Escherichia coli hydrogenase 2. J. Bacteriol 176, 4416–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hube M., Blokesch M., and Böck A. (2002) Network of hydrogenase maturation in Escherichia coli: role of accessory proteins HypA and HybF. J. Bacteriol. 184, 3879–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maier T., Lottspeich F., and Böck A. (1995) Gtp hydrolysis by Hypb is essential for nickel insertion into hydrogenases of Escherichia coli. Eur. J. Biochem. 230, 133–138 [PubMed] [Google Scholar]
- 55. Khorasani-Motlagh M., Lacasse M. J., and Zamble D. B. (2017) High-affinity metal binding by the Escherichia coli [NiFe]-hydrogenase accessory protein HypB is selectively modulated by SlyD. Metallomics 9, 482–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shomura Y., and Higuchi Y. (2012) Structural basis for the reaction mechanism of S-carbamoylation of HypE by HypF in the maturation of [NiFe]-hydrogenases. J. Biol. Chem. 287, 28409–28419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petkun S., Shi R., Li Y., Asinas A., Munger C., Zhang L., Waclawek M., Soboh B., Sawers R. G., and Cygler M. (2011) Structure of hydrogenase maturation protein HypF with reaction intermediates shows two active sites. Structure 19, 1773–1783 [DOI] [PubMed] [Google Scholar]
- 58. Casadaban M. J. (1976) Transposition and fusion of lac genes to selected promoters in Escherichia coli using bacteriophage-λ and bacteriophage-μ. J. Mol. Biol. 104, 541–555 [DOI] [PubMed] [Google Scholar]
- 59. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., and Mori H. (2005) Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299 [DOI] [PubMed] [Google Scholar]
- 60. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 61. Bürstel I., Siebert E., Frielingsdorf S., Zebger I., Friedrich B., and Lenz O. (2016) CO synthesized from the central one-carbon pool as source for the iron carbonyl in O2-tolerant [NiFe]-hydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 14722–14726 [DOI] [PMC free article] [PubMed] [Google Scholar]