

Abstract
HIV-1 infection causes AIDS, infecting millions worldwide. The virus can persist in a state of chronic infection due to its ability to become latent. We have previously shown a link between HIV-1 infection and exosome production. Specifically, we have reported that exosomes transport viral proteins and RNA from infected cells to neighboring uninfected cells. These viral products could then elicit an innate immune response, leading to activation of the Toll-like receptor and NF-κB pathways. In this study, we asked whether exosomes from uninfected cells could activate latent HIV-1 in infected cells. We observed that irrespective of combination antiretroviral therapy, both short- and long-length viral transcripts were increased in wild-type HIV-1–infected cells exposed to purified exosomes from uninfected cells. A search for a possible mechanism for this finding revealed that the exosomes increase RNA polymerase II loading onto the HIV-1 promoter in the infected cells. These viral transcripts, which include trans-activation response (TAR) RNA and a novel RNA that we termed TAR-gag, can then be packaged into exosomes and potentially be exported to neighboring uninfected cells, leading to increased cellular activation. To better decipher the exosome release pathways involved, we used siRNA to suppress expression of ESCRT (endosomal sorting complex required for transport) proteins and found that ESCRT II and IV significantly control exosome release. Collectively, these results imply that exosomes from uninfected cells activate latent HIV-1 in infected cells and that true transcriptional latency may not be possible in vivo, especially in the presence of combination antiretroviral therapy.
Keywords: endosomal sorting complexes required for transport (ESCRT), exosome (vesicle), human immunodeficiency virus (HIV), monocyte, small interfering RNA (siRNA), T-cell, transcription, latency
Introduction
Through the use of combination antiretroviral therapy (cART)4 against HIV-1, it is possible to inhibit active viral replication by > 99%. This renders infected cells into a perpetual state of clinical latency, which is defined as a state in which no infectious viral particles are observed in blood (1). Latency, which occurs once HIV-1 has fully integrated its genome into the host, is best understood in HIV-1-infected resting memory CD4+ T cells, one of several known reservoirs (which include macrophages) of latent HIV-1 infection (2). At least six mechanisms of HIV-1 latency have previously been observed: (i) the sequestration of host transcription factors, such as NF-κB, in the cytoplasm (3); (ii) epigenetic silencing, where nucleosomes 0 and 1 show histone modification that inhibits transcription through SWI/SNF (4–7); (iii) transcriptional interference, which occurs when one transcription factor inhibits another and can result in premature termination of RNA polymerase II (4); (iv) sequestration of p-TEFb and limiting amounts of cyclin T1 (2, 8); (v) antagonism between host protein BRD4 and viral Tat protein on p-TEFb (2); and (vi) use of viral non-coding RNA to bring about transcriptional silencing (3, 9, 10). Because of these mechanisms, HIV-1 can remain within the host immune system, thus causing a persistent reservoir and preventing clearance of the infection by the host (11).
Several compounds have been shown to reverse latency mechanisms and activate the latent viral genome, known as latency-reversing agents (LRAs). One group of these compounds are histone deacetylase inhibitors, such as valproic acid and vorinostat (9), which function by controlling histone deacetylases 1 and 3 and epigenetic silencing (2, 12, 13). Another group is histone methyltransferase inhibitors, such as cyproheptadine (14), which prevent methylation of histones (12). Other LRAs target transcription factors and include PKC modulators (i.e. bryostatin (15)), which activate PKC, allowing it to activate NF-κB and induce transcription (12, 13). LRAs also target p-TEFb, either through direct activation (i.e. hexamethylene bisacetamide (16)) or by inhibiting BRD4 (i.e. JQ1) (12, 17). These methods of activating latent HIV-1 all fall under the umbrella strategy of “shock and kill,” a general concept that HIV-1 can be activated from latent cells (“shocked”) and killed through various mechanisms, including cART, gene therapy, use of immunotoxins, and radiation therapy (3, 7, 18, 19).
However, there have been reports where patients infected with HIV-1 and under cART have expressed low levels of HIV-1 RNA in the blood as well as low, but sustained, levels of viremia in the absence of any LRAs (20, 21). This suggests that there is a biological mechanism(s) in vivo activating the virus from latency. One potential agent may be cytokines, because it has been reported that IL-2 and IL-15 both act to prime latent HIV-1–infected cells for recognition by CD8+ T cells (22). Activated T cells could also induce HIV-1 activation in both latently infected monocytes and macrophages (23, 24). Yet another possibility exists in the form of extracellular vesicles called exosomes, which previous studies have associated with transcriptional regulation and gene expression. For example, Muller et al. (25) have shown that exosomes regulate immune function-related gene transcription in T cells. Another study by Ung et al. (26) showed that exosomes can carry > 80 different proteins involved in gene expression, including transcription factors such as CREB1, HTATIP2, and STAT-1.
Exosomes are small vesicles, 30–120 nm in length, released from all cell types in the body, can be found in various bodily fluids, such as semen and urine (26–32), and are transported through the bloodstream and the lymphatic system (33). They are formed by inward folding of the endosomal membrane to form multivesicular bodies (MVBs), a process carried out by the endosomal sorting complex required for transport (ESCRT) (21, 28, 29, 31, 33, 34). Exosomes can also be formed and secreted through a non-ESCRT pathway utilizing Rab27 (35–37), Rab35 (38), ceramide (39), and Alix (40).
The contents of exosomes depend on the originating cell. It was first thought that exosomes simply carry waste material out of cells, but in recent years, exosomes have been discovered to carry functional proteins, mRNA, miRNA, and lipids between cells (26, 28, 41). Some membrane proteins that are typically found in exosomes, such as Alix, TSG101, CD63, CD81, and CD9, can be considered exosome markers (28, 29). Some miRNAs identified within exosomes can cause phenotypic changes in the recipient cells (30, 32, 42).
Previous studies have established a relationship between viral infection, both RNA and DNA viruses, of host cells and exosome production and packaging (43, 44). Following infection with HIV-1, two viral proteins, p24 (capsid) and Nef, and TAR RNA (a double-stranded non-coding RNA) have been reported to be present in exosomes secreted from the infected cell (28). EBV viral proteins, LMP1 and LMP2A, have also been found in exosomes secreted from EBV-infected cells (28). Furthermore, human T-lymphotropic virus-1 (HTLV-1) Tax protein, implicated in oncogenic activity, has been found in exosomes from HTLV-1-infected cells (28). Even viruses that do not enter the host's nucleus can utilize exosomes for packaging and transport of viral materials. For instance, hepatitis C virus has been demonstrated to transport entire virions from infected liver cells to uninfected liver cells, leading to productive infection in the recipient cells (45).
The transport of the aforementioned viral material in exosomes can cause a wide array of responses, including enhanced infectivity and increased pathogenesis in target cells (28). For instance, HIV-1 can elicit resting T cells to better replicate HIV-1 through the use of Nef and ADAM17 proteins (27). Exosomes containing the EBV protein LMP1 cause immunosuppressive activity by restricting T cell proliferation (28). Tax of HTLV-1 can modulate NF-κB, which in turn enhances cytokine production, and interferes with DNA repair, leading to DNA mutation and the potential for cancer development (26). It is important to note that whereas exosomes are not virions per se, in some cases, they can transmit infectious material from infected to uninfected cells through a non-receptor-mediated mechanism (i.e. hepatitis C virus and human herpes virus 6B) (45–50).
The role of the ESCRT pathway has also been studied in both viral and exosome release. In HIV-1 budding, TSG101 and Alix bind to the Gag polyprotein at the p6 domain (51). CHMP4B is then recruited to the site, followed by Vps4A, which disassembles the entire complex when fission occurs (51). Langelier et al. (52) found that knockdown of EAP20 and CHMP6 did not significantly decrease viral budding. They concluded that ESCRT II was not involved in viral release and that only ESCRT I, in combination with ESCRT III, was important for viral release (52). A later study, however, revealed that ESCRT II, both EAP20 and EAP45, knockdown partially decreased viral budding by 60 and 30%, respectively (53). Whereas these studies focused on viral release, they did not consider how the knockdown of these proteins affected exosomal release.
Our current study is aimed at addressing three fundamental points regarding normal exosomes and their potential significance in HIV-1 transcriptional activation. First, we investigated the effect of exosomes derived from uninfected cells on latent HIV-1–infected cells. Second, we characterized the viral RNAs found within exosomes generated from infected cells. Third, we disrupted exosome formation in the infected cells using siRNA knockdown of ESCRT proteins to assess the role of exosomes in HIV-1 pathogenesis (44). We found that exosomes from uninfected cells can indeed activate HIV-1 virus from latency through phosphorylation of RNA polymerase II. This activation was seen not only in cell lines but also in infected primary cells. We also found multiple forms of incomplete viral transcripts containing truncated gag sequences incorporated into the exosomes that are released via ESCRT II and IV pathways and may potentially be involved in HIV-1 pathogenesis. The general implication of our findings is that a true “transcriptional latency” may not exist in vivo because cells are constantly in contact with exosomes from uninfected cells. Our results, in relation to transcription and viral RNA synthesis, will be discussed.
Results
Effect of exosomes from uninfected cells on HIV-1–infected cells
In previous studies, the effect of exosomes derived from HIV-1–infected cells on uninfected recipient cells showed an inhibition of the PKR/eIF2α pathway as well as activation of the Toll-like receptors by TAR RNA leading to activation of the NF-κB pathway (30, 44). Here, we asked whether exosomes from uninfected cells could potentially activate HIV-1 basal transcription in infected cells. Our rationale was that HIV-1–latently infected cells may still allow either short or long transcripts to be made in the presence of stimulatory molecules present in exosomes, which could partly explain a leaky HIV-1 transcriptional machinery or stochastic fluctuations in gene expression in infected cells. Exosomes are abundantly present in various bodily fluids and may contain various cytokines and transcription factors as well as kinases that could potentially activate transcription of cellular or viral genes (43, 54). Here, we isolated and purified exosomes from uninfected T cell lines (CEM and Jurkat) and monocytes (U937) prior to addition onto HIV-1–infected Jurkat E4 cells, a T cell line lacking the gag gene and having GFP inserted into the nef gene. Our read-out assays were for the presence of TAR RNA (short non-coding, double-stranded transcript; ∼23 bp) as well as GFP (coding spliced RNA) in the cells. All cells were grown in exosome-free media. Results in Fig. 1A indicate that all three types of exosomes increased TAR transcription at varying levels, dependent on the exosome concentration. CEM exosomes led to a 5.5-fold increase in TAR transcription at low dosage, whereas Jurkat exosomes elicited a 4-fold increase in TAR transcription with medium dosage, and U937 exosomes showed the greatest levels of TAR increase (5.5-fold) with the highest dosage used. However, there was no significant increase in GFP transcription regardless of exosome type or concentration used in these experiments (Fig. 1B).
Figure 1.

Exosomes from uninfected cells on latent HIV-1–infected cells increase short RNA levels. A, exosomes from CEM, Jurkat, and U937 cells were isolated using ultracentrifugation prior to addition to Jurkat E4 cells in 0.24-, 0.78-, and 2.4-unit/ml increments (determined using an AchE assay) once per day over the course of 3 days. The E4s were incubated for an additional 48 h prior to harvest, and total RNA was isolated and subjected to RT with a TAR-specific primer. qPCR was performed to quantify the total amount of TAR RNA copies. B, exosomes from CEM, Jurkat, and U937 cells were isolated using ultracentrifugation prior to addition to Jurkat E4 cells in 0.6-, 1.2-, and 1.8-unit/ml (determined using AchE assay) increments. The cells were then incubated for an additional 48 h prior to a GFP assay. Student's t tests compared untreated cells with cells treated with exosomes. In Figs. 1–7 and 10, all cells were grown in exosome-free medium. **, p < 0.01; ***, p < 0.001. Error bars, S.D.
Because the Jurkat E4 cell line is infected with a virus lacking the gag and nef genes, we decided to utilize different cell lines chronically infected with wild-type viruses. ACH2 (infected T cell line), U1 (infected monocyte line), and OM10.1 (infected premyeloid line) cells were treated with purified exosomes from uninfected T cells and monocytes. Specifically, we added Jurkat- and U937-derived exosomes to the infected cells at varying concentrations and performed RT-qPCR for the presence of short and long transcripts. Data in Fig. 2 (A and B) indicate that upon the addition of exosomes to U1 cells, there is a significant increase in TAR RNA transcription at the highest dosages used. Specifically, there was an ∼16-fold increase with the use of exosomes from T cells and monocytes. We observed a similar trend upon the addition of U937-derived exosomes to ACH2 cells at high dosage with an ∼11-fold increase of TAR (Fig. 2C). However, when Jurkat exosomes were added to the ACH2 cells at high dosage, there was a significant decrease in the TAR levels (Fig. 2D). This could possibly be explained by the idea that a lower amount of Jurkat exosomes was needed to increase TAR levels due to the similar background between the donor exosome and infected cell. Interestingly, within the OM10.1 cells, no significant changes in TAR RNA transcription were observed regardless of exosome type or concentration used (Fig. 2, E and F). It is important to note that OM10.1 cells are a latently HIV-1–infected clone of human leukemic myeloblast HL-60 as compared with U1-infected cells, which are derived from a monoblast U937 cell line.
Figure 2.

Presence of short RNA transcripts in wild-type HIV-1–infected T cells and monocytes. Exosomes from Jurkat and U937 cells were isolated using ultracentrifugation prior to being added to ACH2, U1, or OM10.1 cells once per day over the course of 3 days. The cells were then allowed to incubate for an additional 48 h prior to harvest. Total RNA was isolated and then subjected to RT with a primer specific for TAR. qPCR was performed to quantify the total amount of TAR RNA copies. A, U1 cells were treated with 2.79, 13.95, and 41.85 milliunits/ml U937 exosomes. B, U1 cells were treated with 2.90, 14.5, and 43.5 milliunits/ml Jurkat exosomes. C, ACH2 cells were treated with 2.42, 12.10, and 36.30 milliunits/ml U937 exosomes. D, ACH2 cells were treated with 1.75, 8.75, and 26.25 milliunits/ml Jurkat exosomes. E, OM10.1 cells were treated with 2.42, 12.10, and 36.30 milliunits/ml U937 exosomes. F, OM10.1 cells were treated with 1.75, 8.75, and 26.25 milliunits/ml Jurkat exosomes. All exosome concentrations were determined by an AchE assay. Student's t tests compare exosome-treated cells with untreated cells. Error bars, S.D.
Next, we examined whether there were any changes in the full-length genomic RNA levels following treatment with uninfected exosomes. Data in Fig. 3A indicate that upon the addition of Jurkat exosomes to ACH2 cells, there was a ∼5-fold increase in the HIV genomic RNA. U1 cells treated with Jurkat or U937 exosomes showed an increase of ∼20- and ∼31-fold of genomic RNA, respectively. We finally asked whether an increase in genomic RNA could also result in increased pr55 or p24 protein levels. Data in Fig. 3B indicate that p24 levels were slightly increased when the infected cells lines were treated with exosomes derived from uninfected cells. However, the increased levels of RNA did not correlate to a similar increase in protein levels (in ACH2, an increase of ∼10.5–11-fold was observed, and in U1, an increase of ∼6–10.5-fold was observed), suggesting that most of the exosomal effects may be at the transcriptional activation stage as opposed to an increase in translation. Collectively, these data indicate that exosomes from uninfected immune cells modulate transcription of HIV-1 in infected cells and, in most cases, lead to an increase of accumulation of short and long RNA transcripts. However, there is no direct correlation with the level of translation of Gag proteins, suggesting that many of these transcripts are defective in translation or incompletely processed to mRNA.
Figure 3.

The addition of uninfected exosomes to wild-type HIV-1–infected cells increases long transcript levels. A, exosomes from Jurkat and U937 cells were isolated using ultracentrifugation prior to addition to ACH2 and U1 cells at concentrations of 26.25 milliunits/ml (lane 2), 43.50 milliunits/ml (lane 4), and 41.85 milliunits/ml (lane 5) (all were determined by an AchE assay). The exosomes were added once per day for 3 days prior to incubation for another 48 h. The cells were then harvested; total RNA was isolated and subjected to RT with a primer specific to the 3′-end of the HIV-1 genome. qPCR was performed with gag-specific primers to quantify the levels of genomic mRNA. B, exosomes from Jurkat and U937 cells were isolated using ultracentrifugation before addition to ACH2 and U1 cells at concentrations of 26.25 milliunits/ml (lanes 4 and 6) and 41.85 milliunits/ml (lanes 5 and 7), respectively. The exosomes were added once per day for 3 days prior to incubation for another 48 h. The cells were then harvested and lysed, and the resulting lysates were run on a gel, transferred, and subjected to Western blotting for the presence of pr55 and p24. β-Actin was used as a control. Student's t tests compare exosome-treated ACH2 cells with untreated ACH2 cells, whereas a second t test compares exosome-treated U1 cells with untreated U1 cells. Whole-cell extract (WCE) was used as positive control for Western blots. Molecular weight (MW) is shown in lane 3. Error bars, S.D.
Exosomes from uninfected cells affect HIV-1 transcription in latently infected cells under cART treatment
Long-term cART significantly reduces HIV-1 titers in blood and enhances latency (44). We asked whether RNA transcription levels could be increased in recipient cells, cultured under cART after treatment with exosomes from uninfected cells. The infected cells, including U1, ACH2, and OM10.1, were treated with purified exosomes from uninfected T cells and monocytes. Specifically, we added Jurkat- and U937-derived exosomes to the infected cells, which were pretreated with a cART mixture for several days (as described under “Materials and methods”), at varying concentrations and performed RT-qPCR for the presence of short and long transcripts. Results from Fig. 4A indicate that exosomes from Jurkat and U937 increased TAR RNA transcription in U1 cells by 4- and 9-fold, respectively, whereas the addition of Jurkat exosomes to ACH2 cells increased TAR RNA transcription by 11-fold. However, the addition of exosomes from U937 cells to ACH2 cells did not result in increased short RNA transcript levels. This could be due to the effect of cART on exosomal entry. Along these lines, it has previously been reported that the protease inhibitor indinavir inhibits matrix metalloprotease 2, which may affect exosomal entry into cells (55). Data in Fig. 4 (B and C) indicate that the addition of Jurkat or U937 exosomes to OM10.1 cells did not result in a dramatic change of transcription in short RNA products.
Figure 4.

Presence of short RNA transcripts in wild-type HIV-1–infected T cells and monocytes treated with cART. Exosomes from Jurkat and U937 cells were isolated using ultracentrifugation. The aforementioned exosomes were added to either ACH2 cells or U1 cells under cART treatment. cART consisted of an equal-parts (10 μm) mixture of indinavir (IDV), lamivudine (3TC), tenofovir disoproxil fumarate (TDF), and emtricitabine (FTC). A, the exosomes were added at concentrations of 1.60 milliunits/ml (lanes 2 and 4) and 1.88 milliunits/ml (lanes 3 and 5) once per day over the course of 3 days. The cells were allowed to incubate an additional 48 h prior to harvest; their total RNA was isolated and subjected to RT with a TAR-specific primer. qPCR was performed to quantify the total amount of TAR RNA copies. B, OM10.1 cells were treated with exosomes derived from Jurkat cells. The concentration of exosomes added to each sample was 0.107 milliunits/ml (lane 2), 0.535 milliunits/ml (lane 3), or 1.60 milliunits/ml (lane 4). C, OM10.1 cells were treated with exosomes derived from U937 cells. The concentration of exosomes added to each sample was as follows: 0.125 milliunits/ml (lane 2), 0.626 milliunits/ml (lane 3), or 1.88 milliunits/ml (lane 4). All exosome concentrations were determined by an AchE assay. Student's t test compares exosome-treated ACH2 cells with untreated ACH2 cells, whereas a second t test compares exosome-treated U1 cells with untreated U1 cells. A third t test compares exosome-treated OM10.1 cells with untreated OM10.1 cells. Error bars, S.D.
We next assayed for the presence of polyadenylated coding RNA in cART-treated infected cells. Data in Fig. 5A demonstrate results of the treatment of cART-pretreated chronically HIV-1–infected T cells (ACH2) and monocytes (U1) with exosomes from uninfected cells. Upon the addition of exosomes from Jurkat and U937 cells, the ACH2 cells displayed a significant increase in coding HIV-1 RNA transcription (shown with gag-specific primers in Fig. 5A). Similarly, data in Fig. 5B also show an increase of coding RNA levels within U1 cells. Taken together, these data further imply that uninfected T cells and monocyte-derived exosomes can activate transcription of both short non-coding and coding RNA transcripts from the provirus, demonstrating that there is a low level of transcriptional activity in cART-treated cells.
Figure 5.

Effect of uninfected exosomes on long RNA transcripts in wild-type HIV-1–infected cells under cART conditions. Exosomes from Jurkat and U937 cells were isolated using ultracentrifugation prior to addition to ACH2 (A) and U1 cells (B); cells were under cART treatment, which consisted of an equal-parts (10 μm) mixture of IDV, 3TC, TDF, and FTC. Concentrations of exosomes were 1.60 milliunits/ml (lane 2) and 1.88 milliunits/ml (lane 3). Exosomes were added once per day for 3 days. Cells were allowed to incubate for an additional 48 h. The cells were then harvested; total RNA was isolated and subjected to RT with a primer specific to the 3′-end of the HIV-1 genome. RT-qPCR was performed to quantify the levels of genomic RNA. Student's t test was performed to compare exosome-treated lanes with untreated lanes. Experimental results in B (lane 3) were run on a plate different from that used for lanes 1 and 2 and thus cannot be statistically compared with lane 1. Error bars, S.D.
To test whether this trend is observed in primary cells, PBMCs from three healthy donors were prepared and treated with exosomes as indicated in Fig. 6A. Briefly, exosomes were isolated from the PBMCs prior to infecting the same cells with dual-tropic HIV-1 (strain 89.6). The cells were then treated with IL-7, to induce latency, as well as with cART 3 days postinfection. After 1 week, the exosomes isolated from each uninfected donor sample were added to the infected cells at varying concentrations. The cells were allowed to incubate for 3 days prior to harvest and assayed for TAR RNA and genomic RNA levels by RT-qPCR. Results from Fig. 6B indicate that in all three independent donor samples, the addition of exosomes from PBMCs led to increased TAR RNA levels reaching 106 RNA copies/ml. Data from Fig. 6C indicate that exosome addition to the PBMCs from two of the three donors led to an increase in genomic RNA levels by at least 1 log. The samples from Donors 2 and 3 contained high background levels of genomic RNA, indicating that combined cART and IL-7 treatment for 1 week may not have been sufficient to reduce transcription of the HIV-1 genome and induce cells into latency. Interestingly, the data from donor 1 imply that exosomes from PBMCs led to increased levels of both short and long viral transcripts in cART-treated HIV-1–infected primary cells. Collectively, these data imply that primary PBMC exosomes are capable of inducing HIV-1 basal transcription in infected cells.
Figure 6.

The addition of uninfected exosome causes increase of short and long RNA transcripts in wild-type HIV-1–infected PBMCs treated with cART. A, diagram of the experimental design used for primary cell infection and exosome treatment. Briefly, PBMCs from three different donors were treated with phytohemagglutinin (PHA) and IL-2 and allowed to grow for 1 week. Exosomes were then isolated from 25 ml of supernatant from each of the PBMCs prior to infection with HIV-1 (89.6); 3 days later, cells were treated with IL-7, to induce latency, as well as with cART, which consisted of an equal-parts (10 μm) mixture of IDV, 3TC, TDF, and FTC. Twelve days postinfection, the exosomes were added to each of their respective PBMCs, and the cells were allowed to incubate for an additional 72 h prior to harvest. Total RNA was isolated and then subjected to RT with a TAR-specific primer (B) and a primer specific to the 3′-end of the HIV-1 genome (C). qPCR was performed to quantify the total amount of TAR RNA and genomic RNA copies. Student's t test was used to compare untreated cells with exosome-treated cells for each donor. Error bars, S.D.
Potential mechanism of increased HIV-1 transcription in cells treated with exosomes
We next investigated a possible mechanism for how transcription could be increased on the HIV-1 promoter using a ChIP assay. For our initial set of experiments, we isolated and purified exosomes from U937 cell culture prior to addition to U1 cells under cART. A ChIP assay was performed on cross-linked DNA utilizing antibodies specific for RNA polymerase II large subunit (pol II), p65 (NF-κB component), and IgG (as control). When U937 exosomes were added to the U1 cells, RNA polymerase II loading onto the DNA increased by 4-fold over the control (data not shown). Interestingly, the amount of DNA bound by NF-κB p65 did not change between treated and untreated cells, indicating that the effect of the exosomes on transcription may occur at late initiation or early elongation.
We then performed a second series of ChIP assays using antibodies against phosphorylated RNA polymerase II (Ser-2/5) and Cdk9. Following cross-linking, the immunoprecipitated DNA was isolated and quantified by qPCR. Data from Fig. 7A indicate that in ACH2 cells treated with exosomes from Jurkat cells, there was an increased amount of pol II (Ser-2/5) bound to the HIV-1 LTR promoter as compared with the untreated cells. Specifically, pol II (Ser-2/5) loading onto the DNA increased by a factor of 89 over the IgG control. This was accompanied by an increase of Cdk9 loading onto the DNA as compared with the untreated cells. A similar increase of pol II (Ser-2/5) loading was observed when U937-derived exosomes were added to ACH2 cells. Specifically, pol II (Ser-2/5) loading onto the HIV-1 LTR promoter increased 1 log over the IgG control. This was further accompanied by an increase of Cdk9 loading onto the DNA as compared with untreated cells.
Figure 7.

Presence of increased RNA pol II and Cdk9 on HIV-1 genome when treated with exosomes from uninfected cells. Exosomes from Jurkat and U937 cells were isolated by ultracentrifugation and added to ACH2 (A) and U1 (B) cells under cART treatment. The exosomes were added at a concentration of 0.403 millunits/ml once per day for 3 days. Cells were allowed to incubate for an additional 48 h. The cells were cross-linked prior to ChIP assay utilizing antibodies for phosphorylated RNA polymerase II (Ser-2/5), Cdk9, and IgG. DNA was then quantified using qPCR. The primers for the PCR were NF-κB1–2F and TAR +59-R (supplemental Fig. 2). Student's t test was used to compare samples with the IgG control. Error bars, S.D.
Results in Fig. 7B indicate that in U1 cells treated with Jurkat exosomes, loading of pol II (Ser-2/5) onto the DNA increased by a factor of 200 compared with the control, whereas loading of Cdk9 onto the DNA also increased. In U1 cells treated with U937-derived exosomes, 1200 times more pol II (Ser-2/5) was loaded onto the DNA than in untreated cells, whereas Cdk9 loading onto the DNA also increased. Collectively, these data indicate that exosomes derived from uninfected cells lead to an increase in transcription by potentially increasing the pol II and/or Cdk9 occupancy on the HIV-1 promoter.
Various RNA transcripts are present in latently HIV-1–infected cells and extracellular vesicles
We next tested the variability of viral RNA transcripts in cells latently infected with HIV-1 as well as in the extracellular environments. Patients receiving antiretroviral therapy for a long time may have low or undetectable genomic viral RNA; however, few data exist to date describing the presence of short RNA transcripts in these patients (56, 57). To test whether various HIV-1 transcripts are present in cells and extracellular vesicles circulating in the plasma of aviremic patients, we utilized a number of well-characterized samples from the Women's Interagency HIV Study (WIHS), specifically from four HIV-1–infected patients under various cART treatments, as indicated in Fig. 8A. Patient demographics can be found in supplemental Fig. 3. Both intracellular and extracellular RNA from PBMCs and serum samples were analyzed from each of the four patients. The serum samples were incubated with NT080/082 beads overnight at 4 °C to enrich for extracellular vesicles (58). Total RNA from both PBMCs and exosomes was isolated and subjected to RT with primers specific for TAR and full genomic RNA. Data in Fig. 8B indicate that proviral DNA was present in all samples tested, and more intracellular TAR RNA was found in comparison with genomic RNA. Higher levels of TAR and genomic RNA were found in two of the four patients.
Figure 8.

Presence of viral RNA in exosomes from patient PBMCs. A, PBMCs and serum were obtained from HIV-1-infected patients on cART from the Women's Interagency HIV Study (WIHS) (76, 77). Each patient was receiving a different cART treatment. The drugs used were zidovudine (AZT), efavirenz (EFV), nevirapine (NVP), 3TC, FTC, and TDF. B, serum was incubated with a 30% NT080/082 slurry overnight at 4 °C. Total RNA was isolated from the nanoparticles and from the PBMC cells and subjected to RT with primers specific for TAR and the 3′-end. Total DNA was also isolated from the PBMC cells. RT-qPCR was performed on the cDNA with TAR and gag primers and on the isolated DNA with GAPDH primers (for normalization). The results are in percentage of the negative control. Error bars, S.D. of three independent measurements. C, proportion of HIV-1 provirus to GAPDH DNA for each patient.
Analysis of viral RNAs in extracellular vesicles concentrated with nanotrap beads from the serum of the same patients revealed both short TAR RNA transcripts and coding RNAs; however, TAR RNA was in higher abundance (Fig. 8B). Finally, we normalized the proviral DNA to the cellular gene GAPDH to assess the relative number of cells containing HIV-1 provirus. We found that there was a rather high rate of infected cells (1.4–8.2% of all tested PBLs) (Fig. 8C). We assume that the sensitivity of our PCR assay may not be high enough to detect very low levels of RNA in one infected patient sample (participant 4). Collectively, these data suggest that the presence of both TAR and coding viral RNAs in cells is proportional to the number of integrated HIV-1 genomes.
Exosomes from uninfected cells cause release of various HIV-1 RNAs from infected cells
We have previously shown that exosomes from infected cells contain both short and long RNAs (44). To test whether the exosomes from uninfected (donor) cells could affect the profile of viral transcripts within infected (recipient) cells and whether the HIV-1 RNA profiles in extracellular vesicles are identical to cellular RNA profiles, we treated latently infected monocytes (U1) with exosomes isolated from parental uninfected monocytes (U937) and isolated RNA at 48 h post-treatment. We utilized nanoparticles (NT080/082) to trap and concentrate exosomes, followed by RNA isolation and analysis, which included quantifying the amounts of viral RNA transcripts of various lengths in exosomes. The RT reaction was performed with reverse primers specific for U5 LTR, gag, pol, env, and poly(A)–3′-LTR; the resulting cDNA was quantitated by real-time PCR with the primer set specific for the R-U5 LTR sequence. Comparison of the RNA profiles of exosomes derived from untreated and U937 exosome–treated U1 cells indicates that this treatment dramatically increased (> 100-fold) the release of viral RNA packaged into vesicles (Fig. 9, A and B). The ratio of short non-coding (TAR) and longer (incomplete coding) RNA transcripts is similar in the treated and untreated cells. Interestingly, treatment with the exosomes led to increased incorporation of longer transcripts. Particularly, truncated forms of coding RNA containing the full gag sequence is 80% higher in the exosomes from U1 cells treated with the U937 exosomes.
Figure 9.

Exosomes contain various viral RNA transcripts. A, U937-derived exosomes were added to U1 cells and incubated for 3 days. The cell supernatant was then collected, passed through a 0.22-μm filter, and incubated with NT080/082 overnight. Total RNA was isolated from the nanoparticles and subjected to RT with primers specific for TAR, U5, gag, pol, env, and the 3′-end of the HIV-1 genome. RT-qPCR was performed with primers specific for TAR (with the 3′-RT, gag primers were used for qPCR). B–D, U1, OM10.1, and J1.1 cells were grown for 4 days. The cell supernatant was then collected, filtered through a 0.22-μm filter, and incubated with NT080/082 overnight. Total RNA was isolated from the nanoparticles and subjected to RT with primers specific for TAR, U5, gag, pol, env, and the 3′-end of the HIV-1 genome. RT-qPCR was performed with primers specific for TAR (with the 3′-RT, gag primers were used for qPCR). Error bars, S.D.
Additionally, we observed a novel RNA transcript making up between 15.5 and 26.1% of the total RNA transcripts found within these exosomes, which we termed “TAR-gag”. This RNA contains the complete R (including TAR) and U5 sequence of the 5′-LTR and part of the gag gene. This surprising result prompted us to look for the presence of both TAR and TAR-gag RNAs in exosomes from a few of our infected cell lines, including U1, OM10.1, and J1.1. We utilized the same nanoparticles as before (NT080/082) to trap and concentrate exosomes, followed by RNA isolation and analysis. Total exosomal RNA was subjected to RT with primers specific for the 3′-end, env, pol, gag, U5, and the R region (TAR). Data in Fig. 9 (B–D) indicate an abundance of TAR found in exosomes from all tested infected cells, with varying amounts from different cell types. High copy numbers were observed in OM10.1 (5.7 × 106) and J1.1 (2.0 × 105) exosomes, whereas lower copy numbers were observed in U1 (4.6 × 104) exosomes. The amount of RNA quantified decreased with the U5 reverse primer to 2.8 × 104 from U1, 2.0 × 106 from OM10.1, and 9.8 × 104 from J1.1 exosomes. The novel TAR-gag transcript was found in significant quantities from U1 exosomes (1.5 × 104 copies), from OM10.1 exosomes (1.8 × 106 copies), and from J1.1 exosomes (8.5 × 104). The amounts of RNA quantified from pol, env, and the 3′-end were 5% or less of the total viral transcripts. Collectively, these data indicate that TAR and TAR-gag RNAs are packaged into exosomes from infected cells.
TAR-gag RNA does not translate into pr55 or p24 in recipient cells
Because TAR-gag RNA is a novel RNA transcript, we asked whether this RNA could potentially be translated in the recipient cells into pr55 (Gag precursor) and processed into products such as capsid p24 protein. To do this, we treated Jurkat and U937 cells with either ACH2/U1 supernatant alone or ACH2/U1 exosomes concentrated with NT080/082 beads. The cells were treated for 3 days at 37 °C and were lysed and processed for Western blotting using pr55 and p24 combined antibodies. As controls we used uninfected and infected cell lysates (Fig. 10A, lanes 1, 2, and 7). Additional controls included the use of infected supernatants either alone or concentrated with nanoparticles for the presence of pr55 and p24 (Fig. 10A, lanes 3, 4, 8, and 9). Results from Fig. 10A indicate that pr55 and p24 are present in the ACH2-concentrated supernatants, whereas only p24 was found in the U1-concentrated supernatants. However, when these supernatants were added to Jurkat recipient cells, no new pr55 or p24 was observed (Fig. 10A, lanes 5, 6, 10, and 11). We then repeated the same experiment in recipient U937 cells and found similar results compared with the Jurkat cells (Fig. 10B). It is important to note that, using mass spectrometry, we have previously shown that exosomes from infected cells do contain pr55 protein, which may explain how the new TAR-gag RNA is packaged into exosomes (30), and we and others have also previously shown that RNA from exosomes enter recipient cells (30, 59, 60). Collectively, these data indicate that the novel TAR-gag RNA does not translate into pr55 or p24 and therefore may serve as a long non-coding RNA.
Figure 10.
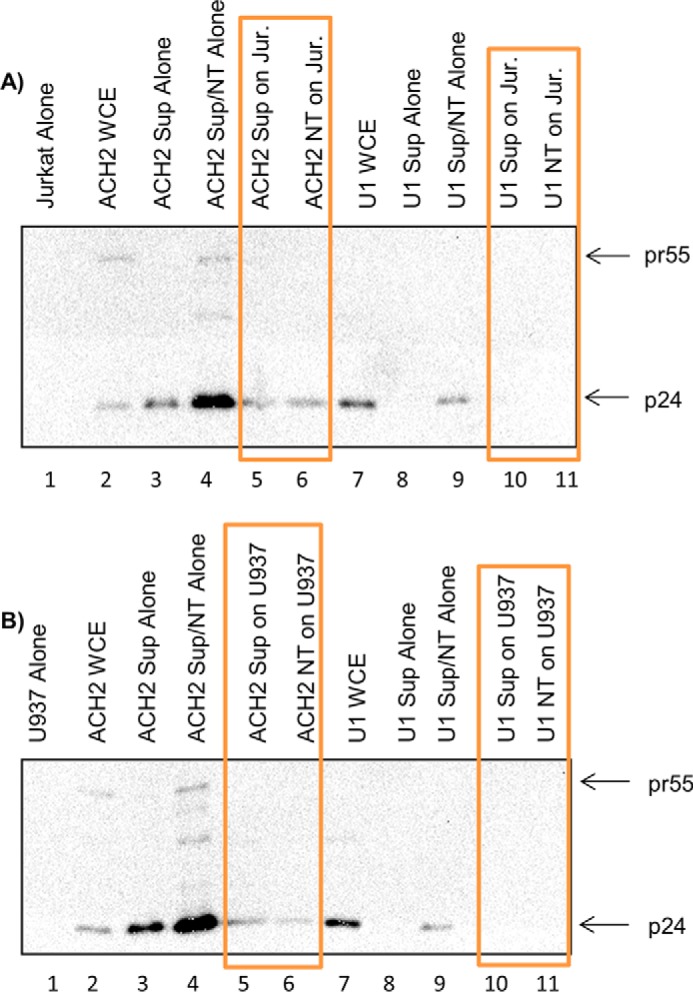
TAR-gag is a long non-coding RNA. ACH2 cells and U1 cells were pretreated for 5 days with a cART mixture. One milliliter of cell supernatant was collected and treated with nanotraps (NT), specifically a 30% NT080/082 slurry, prior to rotating for 16 h at 4 °C to concentrate exosomes. Jurkat and U937 cells (3.5 × 106; 0.75 and 1.0 ml, respectively) were treated with 1 ml of either ACH2 or U1 supernatants or the concentrated exosomes from the infected cells (emphasized by orange boxes). Jurkat (A) and U937 (B) were incubated for 72 h prior to harvest for Western blotting using p24 and pr55 combined antibodies. Whole-cell extract (WCE) was used as a positive control for Western blots.
High-throughput sequencing to define the 3′-end of the HIV-1 RNAs in exosomes
Based on the aforementioned results, we next characterized the 3′-end of all potential TAR-associated RNA sequences. We utilized exosomes from infected cells (OM10.1 and J1.1) and isolated RNA for 3′-end sequencing. Poly(A) tail was added prior to cDNA synthesis, and the final DNA product was used to construct a library followed by Illumina Nextgen sequencing. Sequencing results identified four main clusters of sequences ending at ∼135, 300, 408, and 615 bp from the transcription start site (+1; Fig. 11, A–C). The first two clusters were found to end within the LTR region, whereas the last two clusters were found to end in the Gag (p17) region. The left reads (pink) were anchored at the TAR sequence, and the right reads (blue) indicated the 3′-ends. The added poly(A) tail is depicted as a green box within the right read, and the mismatches at the tail end of the right reads indicate the adaptor sequences. The final 3′-end RNA sequences are shown as clusters of arrows in Fig. 11D. The longest 3′-RNA molecules were present at the beginning of the Nuc-2 region within the Gag (p17) region. Collectively, these data indicate that RNA pol II pausing takes place at multiple regions within the gag gene, resulting in the accumulation of various incomplete viral transcripts.
Figure 11.

Sequence alignment of exosomal RNA using Integrated Genomics Viewer. The paired-end reads are depicted by left reads (pink) and right reads (purple) connected by a thin gray line. The mismatch nucleotides are individually colored for A (green), T (red), G (brown), and C (blue). The figure depicts results from OM10.1 exosomes, which were similar to the J1.1 exosomes. A, cluster of reads ending at ∼135 bp (+1 is the start of transcription) in the LTR region. For this cluster, the left and right reads overlap, and thus the connecting gray line is not shown. B, cluster of reads ending at ∼300 bp in the LTR region. C, cluster of reads ending at ∼408 bp in the beginning and ∼615 bp toward the end of the Gag (p17) region. D, diagram and sequence of HIV-1 LTR and p17 region of gag that maps to multiple clusters of RNA reads using NexGen sequencing.
ESCRT pathway regulates exosome release from HIV-1-infected cells
We have shown that treatment with exosomes increased transcription of short and long RNA transcripts in recipient infected cells. Furthermore, previous studies have shown that these transcripts are packaged into exosomes and cause recipient uninfected cells to become more susceptible to future infection using the Toll-like receptor and IKK pathways (44). To minimize this adverse effect, we sought to decrease the amount of exosomes being released from infected cells. Previous studies have used siRNA to knock down ESCRT proteins involved in viral budding but failed to take into account how exosomal release was also affected (61). Therefore, we transfected monocytes with siRNA specific for five ESCRT proteins, representing each of the four complexes, and CD63 as a positive control. The siRNA-treated cells were tested with PCR for mRNA of targeted ESCRT components to confirm that siRNA can knock down protein translation (seen in supplemental Fig. 4). Exosomes were captured from culture supernatants of siRNA-transfected cells using nanoparticles (58). The exosomes collected by the nanoparticles were then processed for Western blot and RT-qPCR analysis. Results in Fig. 12A indicate that considerably less TAR RNA was detected in exosomes released from monocytes when using siRNAs against components of ESCRT II and III and Vps4 (ESCRT IV) but not against ESCRT I. TSG101 is a component of ESCRT I, which is essential for HIV-1 release (52, 53). The levels of TAR RNA in these exosomes decreased significantly with siEAP20, a component of ESCRT II. A similar pattern of RNA decrease was also observed for TAR-gag RNA when using siRNA against all components (Fig. 12B). A similar trend of less genomic RNA was also observed in siEAP20-treated cells as compared with other components of the ESCRT machinery (Fig. 12C). Therefore, ESCRT II knockdown reduced TAR, TAR-gag, and full-length genomic RNA, whereas ESCRT III and IV knockdown reduced only TAR and TAR-gag RNA packaging into exosomes. Finally, there were no dramatic changes in the exosomal release of cargo proteins, such as Alix or CD63, in these cells (Fig. 12D). Collectively, the data suggest that ESCRT II (i.e. EAP20) potentially regulates packaging of TAR present in short and long HIV-1 transcripts (TAR is present at the 5′-end of all transcripts) into exosomes.
Figure 12.

Exosome release from infected monocytes treated with siRNA against the ESCRT pathway. Infected U1 cells were treated with various siRNAs against TSG101, EAP45, EAP20, CHMP6, Vps4A, and CD63 (at 20 nm final concentration) using Lipofectamine. Supernatants were isolated 72 h post-treatment and utilized for RNA isolation or Western blotting. RT-qPCRs were utilized for TAR RNA (A), TAR-gag (B), and genomic RNA (C). Lipofectamine treatment alone served as a negative control. Error bars, S.D. of three independent measurements. D, Western blotting for components of exosomes, including Alix, CD63, and actin, was performed.
The previous experiments were performed with latent HIV-1–infected monocytes. We then asked whether differentiation of infected monocytes to macrophages could affect the packaging of viral RNA into exosomes via the ESCRT pathway (62). We next asked whether induction of HIV-1 transcription with PMA in monocytes also could increase non-functional RNA release through the exosomal pathway. A similar set of experiments compared with Fig. 12 were performed, where infected cells were first treated with siRNA and then with PMA to activate HIV-1 transcription. Data in Fig. 13 indicate that PMA activation increased the overall copy number of viral RNA transcripts packaged into exosomes by 2 logs (compared with RNA counts in Fig. 12). Data in Fig. 13A indicate that siRNAs against components of ESCRT II and IV, but not against ESCRT I and III, decreased levels of TAR-containing exosomal RNA. Interestingly, the levels of TAR-gag and full-size genomic RNA were not dramatically decreased in any exosomes from the ESCRT knockdown cells (Fig. 13, B and C). We did, however, observe a slight decrease of the genomic RNA in the exosomes from ESCRT II knockdown cells. As expected, positive control siCD63 results showed a decrease in all three transcripts. Surprisingly, siTSG101, which shuts down viral packaging (52, 53), increased the level of all three transcripts into exosomes. We assume that the TSG101 knockdown inhibited virion assembly and release, which, in turn, allowed excess RNA transcripts accumulated in the cell to be packaged instead into exosomes. Finally, the Western blot analysis showed a slight increase in CD63 and actin levels following PMA activation (Fig. 13D). Collectively, these data imply that only short transcripts are reduced in ESCRT II and IV siRNA– and PMA-treated cells, indicating a potential selective packaging of short versus long RNAs into exosomes from activated macrophages.
Figure 13.

Exosome release from infected macrophages treated with siRNA against the ESCRT pathway. Experimental designs were similar to the ones described in the legend to Fig. 12, with the exception of PMA treatment. U1 cells were first treated with various siRNAs; the next day, they were treated with PMA at 10 μm. RT-qPCR samples were performed for TAR (A), TAR-gag (B), and genomic RNA (C). D, Western blot analysis for exosomal proteins. Error bars, S.D.
To gain a better understanding of the effects of siRNA knockdown of ESCRT, we then turned our focus on another myeloid cell line, namely CHME5/HIV. It is important to note that this cell line includes GFP and that the gag gene is partially present but is not translated (63). We performed an experiment similar to the one shown in Fig. 12 using various siRNAs against the ESCRT pathway. We observed overall fewer RNA transcripts (by 1 log) being produced from these cells when looking at TAR RNAs. When using siRNAs, we observed a dramatic decrease of TAR and TAR-gag in ESCRT II and IV knockdowns (Fig. 14, A and B). Interestingly, ESCRT III knockdown showed a slight increase in both transcripts. Positive control siCD63 decreased packaging of both RNAs. Finally, we observed virtually no genomic RNAs being packaged into exosomes from these cells (Fig. 14C). It is important to note that these cells do not produce pr55 and therefore would not be able to package full-length RNA. Taken together, these data suggest that ESCRT II and IV knockdowns decrease the amount of shorter transcripts in exosomes from various HIV-1–infected cells.
Figure 14.

Exosome release in siRNA-treated CHME5/HIV cells. The CHME5/HIV cells were treated with various siRNAs (similar to Figs. 12 and 13), and exosomes were enriched with NT080/082 beads overnight. RNA was isolated for RT-qPCR using TAR (A), TAR-gag (B), and genomic RNA (C) primers. D, Western blot analyses of the treated samples for exosomal markers, including Alix, CD63, and actin, were performed. Error bars, S.D.
Discussion
Exosomes are known to carry cargo, including proteins and RNAs, from donor to recipient cells, which may control signal transduction and cell fate (54, 64). Previous studies have shown that virally infected cells can spread viral proteins and/or RNA via exosomes to uninfected cells, contributing to either disease pathogenesis or increased susceptibility to the next round of infection (43, 44). Our current data also indicate that, similar to exosomes from infected cells, exosomes generated from uninfected cells control transcription and potentially translation of viral proteins in the recipient infected cells.
We have observed that exosomes derived from uninfected cells could increase levels of short transcripts in infected cells containing a reporter gene (Jurkat E4). These cells contain a partial deletion of the gag gene and nef, which is replaced by GFP. Interestingly, we did observe an increase of both short and long transcripts when using wild-type virally infected recipient cells, as well as an increase in p24 protein levels. Furthermore, similar increases in short and long RNA transcripts were observed in infected cells treated with cART once exosomes from normal cells were added to these cells.
We observed a consistently better and more reproducible set of results when treating infected monocytes with uninfected monocyte-derived exosomes. This could potentially be due to greater specificity because they originated from the same cell type. Interestingly, neither Jurkat-derived nor U937-derived exosomes caused any notable increase in TAR RNA transcription in OM10.1 cells. This could be due to lack of factor(s) in exosomes from T cells and monocytes that would be needed to activate HIV-1 in OM10.1 cells. Experiments are in progress using parental HL-60 cells and their exosomes, along with T cell and monocyte-derived exosomes, to better understand this mechanism. Also, it may very well be possible that OM10.1 cells contain unintegrated DNA along with integrated genome, which could potentially explain the higher basal transcription observed in these cells. Experiments conducted using Hirt supernatants indicated that OM10.1 cells did contain unintegrated DNA (using TAR primers) with ∼100-fold more DNA compared with ACH2 and U1 cells (data not shown).
Importantly, we were able to reproduce these results in human PBMCs from three different donors. Our data show that when exosomes from uninfected PBMCs were added back to the latent HIV-1–infected cells under cART conditions, both short and long viral transcript levels increased. In donors 1 and 2, there was a more dramatic increase in genomic RNA than in TAR RNA (>1000- and 51-fold versus 16- and 6-fold, respectively). In donor 3, there was an equal increase in genomic RNA and TAR RNA, each increasing by 4-fold upon the addition of exosomes. The less dramatic increase in viral RNA observed in donor 3 may be explained by high background levels of genomic RNA already present prior to the addition of exosomes. Therefore, the addition of exosomes to an already saturated system would not show an increased effect on transcription.
A second aspect of this study was to find a mechanism to explain the increases in TAR RNA and genomic RNA levels observed within infected recipient cells. Preliminary data indicated that upon the addition of U937-derived exosomes to U1 cells, there was an increase in the amount of RNA polymerase II binding to the proviral DNA but not p65, a component of the NF-κB transcription factor (data not shown). A subsequent ChIP assay suggested that T-cell exosome addition to infected T-cells and monocytes promoted increased Cdk9 binding to the transcriptional complex (Fig. 7, A and B), implying that uninfected exosomes can enhance elongation by RNA polymerase II. This could be explained by the presence of one or more kinases from the donor cell exosomes that, upon entry into the recipient infected cell, contribute to RNA polymerase II activity. Candidates for these kinases are AKT1, MAP2K, mTOR, DNAPK, and SRC, which have all been previously shown to be packaged into exosomes (26). We are currently utilizing inhibitors of these kinases to better understand which kinase(s) contribute to pol II phosphorylation or general transcription.
A third aspect of this study examines what happens to the viral transcripts from infected cells once treated with uninfected exosomes. We isolated exosomes containing these transcripts from the infected cells and used various reverse primers in an RT reaction to quantify the amounts of six different HIV-1 transcripts that could potentially be packaged into these exosomes. We expected to observe even levels of all of the transcripts in the exosomes; however, we instead observed a very dramatic decrease in slightly longer RNA transcripts. Using these primers, we observed that the amount of RNAs that went beyond the gag gene was significantly lower when compared with TAR alone. We named the new transcript “TAR-gag” because it dropped off in the gag gene (within the p17 region). We speculate that this new transcript is caused from a paused polymerase (potentially due to the presence of nucleosomes) or that the full-length transcript was processed differentially to give rise to a more stable, hence detectable, RNA species. The presence of paused pol II at downstream LTR sites has previously been observed, which may explain the presence of TAR-gag (65). We next asked whether this RNA can be translated in the recipient cells. The gag gene is normally expressed from the full-length transcript and is made into a polyprotein, which subsequently is cleaved and processed to make Gag proteins, including p24. Western blot analysis from the recipient uninfected cells showed no new synthesis of HIV-1 p24 protein, indicating that TAR-gag may be a long non-coding RNA. It is also important to note that recently, the presence of TAR-gag and other variants has been observed as cell-associated unspliced HIV-RNA in patient samples under cART (termed zombie viruses); however, they were deemed to translate into proteins (56). Future experiments will better define whether TAR-gag is translated in primary cells or other cancer cell types.
Finally, because exosomes containing viral RNA can cause negative effects in recipient cells, we investigated whether we could potentially regulate the amount of exosomes released from infected cells. Previous studies have used siRNA to knock down ESCRT proteins to decrease the amount of virus released from infected cells (52, 53). They have further shown that TSG101, part of ESCRT I, is involved in the viral release pathway, in addition to ESCRT III (52, 53, 61, 66, 67). Because exosome formation and viral release are both dependent upon members of the ESCRT pathway, we utilized knockdown of ESCRT proteins to examine an increase and decrease in viral transcripts within exosomes. Our data indicate that ESCRT II and ESCRT IV knockdown led to a decrease of viral TAR, TAR-gag, and genomic RNA found within exosomes secreted from infected cells. Interestingly, knockdown of ESCRT I and ESCRT III in PMA-activated cells led to an increase in all three RNA transcripts. We assume that this occurred because ESCRT I and III are involved in viral release and, as the cells were activated, they were producing virus. The inhibition of viral release probably led to a buildup in viral RNA levels within the cell that was instead packaged into exosomes to be removed from the cell. Overall, our data indicate that knockdown of ESCRT II and IV may restrict HIV-1 pathogenesis because less viral RNA is delivered to recipient cells via exosomes.
Because we observed a difference in packaging of transcripts before and after PMA activation, we hypothesized that the cells may be utilizing different mechanisms of packaging RNA (short versus long). We have previously shown that exosomes derived from HIV-1–infected cells contain unprocessed Gag polyprotein, pr55 (30), which can potentially bind to genomic RNA and bring it into an exosome. The p7 region of pr55 binds to genomic RNA between nucleotides 240 and 400 (68), which is also included within the TAR-gag RNA sequence. Other studies have also shown that the HIV-1 matrix protein (p17, which is processed from pr55) can also bind to RNA (69), around nucleotides 1433–1446, to assist in viral packaging of genomic RNA (70). This may explain why the genomic RNA is not packaged into the exosomes from CHME5/HIV cells that do not make pr55 polyprotein. The cellular protein heterogeneous nuclear ribonucleoprotein (hnRNP) is known to bind to cellular mRNAs and miRNAs (71). A class of these proteins, hnRNPA2B1, can bring RNAs into exosomes by binding to a specific sequence motif: GGAG (72). This sequence is present within TAR and TAR-gag transcripts; therefore, hnRNPA2B1 may be responsible for packaging of TAR and TAR-gag into exosomes. This may explain how TAR-gag is found in exosomes from the CHME5/HIV cells. Finally, it is important to note that hnRNPA2B1 can bind to methylated RNA and convert pri-miRNA to pre-miRNA with recruitment of Pasha (73). Taken together, we hypothesize that TAR alone (either methylated or unmethylated) may be packaged into exosomes by hnRNPA2B1, whereas TAR-gag could be packaged by either hnRNPA2B1 and/or pr55 (supplemental Fig. 5); however, genomic RNA is packaged into exosomes mostly by pr55.
Overall, we have shown that exosomes from uninfected donor cells cause increased transcription of both short and long RNA transcripts within infected recipient cells. A potential mechanism for this increase could be phosphorylation of RNA polymerase II on the HIV-1 promoter. It is possible that three sites on the CTD 52 repeats are hyperphosphorylated with the recruitment of p-TEFb responsible for phosphorylation at Ser-2/5 and one of the dominant kinases from exosomes responsible for assisting in phosphorylation at Ser-2/5 and/or Ser-7. Furthermore, these cells produce a novel RNA transcript, TAR-gag, which can be packaged into exosomes and released from infected cells, where siRNA specific for ESCRT II caused the most dramatic decrease in exosomal release (Fig. 15). Collectively, this leads us to the conclusion that true “transcriptional” latency of HIV-1 may not occur in vivo because cells are constantly surrounded by various types of exosomes (i.e. 108–1011 in various fluids (74)), which are taken up by the cells, leading to a constant production of low levels of viral RNAs. Although clinical latency, which is defined as a lack of disease symptoms (1), may still occur, we speculate that transcriptional latency (at least for short transcripts) is highly unlikely in vivo. There are many blocks, including lack of components of p-TEFb (i.e. low cyclin T1 levels in resting T cells), chromatin block, etc., that may be playing various inhibitory roles; however, a lack of true transcriptional inhibitors in cART mixture may be one reason for accumulation of HIV-1 coding and non-coding transcripts in cells, which eventually find their ways into exosomes (this work) (57, 75). It may also alternatively be possible that one or more components of the cART (i.e. NRTIs) could control the exit of exosomes (i.e. biogenesis), resulting in a lower level of productive viral RNA transcripts being packaged and released. Future experiments will better define the mechanism of this block in latent cells treated with cART.
Figure 15.

A proposed model for exosomal activation of latent HIV-1 genome. Exosomes from uninfected cells increase HIV-1 transcription in infected cells under cART. Short viral RNA transcripts (i.e. TAR) are increased more than genomic RNA transcripts. Exosomes released from these cells contain an increased amount of non-coding viral RNA transcripts, including TAR and TAR-gag RNA.
Materials and methods
Cells
CEM cells (uninfected T cells), Jurkat cells (uninfected T cells), Jurkat E4 cells (HIV-1–infected T cells; generous gift of J. Karn), ACH2 cells (HIV-1–infected T cells), U937 cells (uninfected promonocytic cells), U1 cells (HIV-1–infected monocytes), and OM10.1 cells (HIV-1–infected myeloid-derived cells) were grown in RPMI complete medium at 37 °C and 5.0% CO2. CHME5/HIV cells (infected microglia) were grown in DMEM complete medium at 37 °C and 5.0% CO2. Bovine exosomes were excluded from culture media by ultracentrifugation of FBS at 100,000 × g for 70 min prior to addition to media for growing cells.
Reagents and antibodies
Complete culture media consisted of RPMI 1640 or DMEM supplemented with 10% FBS, 1% l-glutamine, and 1% streptomycin/penicillin. Antibodies used for ChIP assay included IgG (sc2027), RNA polymerase II (sc899), p65 (ab7970), RNA polymerase II (Ser-2/5) (catalog no. 4735), and Cdk9 (C12F7). Antibodies used for Western blots were p24 (NIH AIDS Reagent, catalog no. 4121), TSG101 (H-270) (sc22774), Vps4 (H-165) (sc32922), Eap45 (T-20) (sc79931), VPS25 (B-4) (sc271648), CHMP6 (FL-201) (sc67231), Alix (sc49268), CD63 (ab134045), and actin (ab49900).
Exosome isolation
CEM, Jurkat, and U937 cells were grown in appropriate media supplemented with 10% exosome-free FBS. Exosome preparation was made with 100 ml of cell culture supernatants (produced from a culture of one million cells/ml for 5 days). Cells were pelleted by centrifugation at 300 × g for 10 min. An additional centrifugation at 2000 × g for 10 min was used to pellet dead cells. The supernatant was then filtered through a 0.22-μm filter and ultracentrifuged at 10,000 × g for 30 min to remove any cell debris. This was followed by two ultracentrifugations at 100,000 × g for 70 min to pellet the exosomes. The resulting pellet was resuspended in 100 μl of PBS. All spins were performed at 4 °C. Protein levels of the exosomes were determined using a BCA assay and an AchE enzymatic assay.
Exosome titration of cells from cell lines
Exosomes were added in a dose-dependent manner to HIV-1–infected cells that were both pretreated and not treated with cART. The exosomes were added once a day for 3 days. Then the cells were allowed to incubate for 48 h at 37 °C, after which they were harvested, spun down into pellets, and washed in preparation for RNA isolation and RT-qPCR.
Treatment of PBMCs
PBMCs were activated with phytohemagglutinin and IL-2 treatment (44). One week later, exosomes were isolated using the ultracentrifugation method. The PBMCs enriched in T cells were infected with HIV-1 (89.6; multiplicity of infection = 1.0) and, 3 days postinfection, treated with cART and IL-7 to enable cells to enter latency. Ten days postinfection, the PBMC exosomes previously isolated from healthy culture were added back in a dose-dependent manner to the infected cells and incubated for 72 h, after which the cells were harvested, spun down into pellets, washed, and processed for RNA isolation and RT-qPCR using HIV-1 primers.
cART treatment
ACH2, U1, and OM10.1 cells were pretreated with cART for 3 days prior to exosome addition. An equal-parts mixture of indinavir (protease inhibitor), lamivudine (NRTI), tenofovir disoproxil fumarate (NtRTI), and emtricitabine (NRTI) were added to cells (10 μm each). The antiretrovirals were obtained from the AIDS Research and Reference Reagent Program. This treatment was repeated every 3 days over the course of 8 days. Finally, cells were processed for RNA isolation, and RT-qPCR was performed.
GFP assay
Jurkat E4 (105) cells were plated and treated with varying concentrations (0.24, 0.78, and 2.4 units/ml) of CEM-, Jurkat-, or U937-derived exosomes. After incubation for 5 days, fluorescence was measured using the Promega GloMax multidetection system. A 425-nm wavelength was used for GFP measurement.
AchE assay
The Amplex® acetylcholine/acetylcholine esterase activity assay kit (Thermo A12217) was used to quantify exosomes following the manufacturer's instructions. Briefly, a negative control of 1× running buffer (consisting of 20 ml of H2O and 5 ml of 5× reaction buffer, which included 250 mm Tris-HCl, pH 8.0) and two positive controls, one consisting of acetylcholine esterase and one consisting of hydrogen peroxide, were made and plated on a 96-well plate. Exosomes were treated as per the manufacturer's instructions and plated on the 96-well plate. Acetylcholine esterase activity was measured every 15 min for 1 h to obtain optimal enzymatic activity.
Nanoparticle capture of exosomes
One milliliter of cell supernatants was processed and centrifuged at 25,000 × g for 5 min to remove cells. The supernatant was then filtered through a 0.22-μm filter into a new tube. Thirty microliters of a 30% slurry of NT080/082 beads were added to the filtrate and placed on a rotator at 4 °C overnight. Next, the filtrate was centrifuged at 25,000 × g for 10 min, and the supernatant was removed. The NT pellet was washed once with 1 ml of PBS and resuspended in 50 μl of PBS prior to RNA isolation and RT-qPCR analysis.
RNA isolation and RT-qPCR
RNA was isolated from cells and exosomes using TRI Reagent-LS (MRC) according to the manufacturer's protocol. A cDNA library was then created with the GoScript reverse transcription system from Promega using 7 μl of isolated RNA, 1 μl of reverse primer (100 μm) (see supplemental Fig. 2), 4.25 μl of nuclease free water, 1 μl of PCR nucleotide mix (10 mm), 4 μl of 5× GoScript buffer, 2 μl of MgCl2 (25 mm), 0.25 μl of recombinant RNasin ribonuclease inhibitor (40 units/μl), and 0.5 μl of GoScript Reverse Transcriptase per sample. Real-time qPCR was then performed with 2 μl of cDNA, 10 μl of iQ Supermix (Bio-Rad), 7.84 μl of nuclease-free water, 0.06 μl each of forward and reverse primers (100 μm), and 0.04 μl of probe (supplemental Fig. 2) per sample.
ChIP assay
Cells were harvested, and their DNA and protein were cross-linked and processed using the Imprint chromatin immunoprecipitation kit (Sigma). Samples were then sonicated, and mono-disomes were used for immunoprecipitation. Antibodies were added, and the samples were allowed to rotate overnight at 4 °C. A 50% (v/v) protein A-Sepharose/protein G-Sepharose mix was added and allowed to rotate for 2 h at 4 °C. The samples were washed twice with IP Wash Buffer (Sigma) prior to the addition of proteinase K (800 units/ml). After a 15-min incubation at 65 °C, reversing solution (Sigma) was added, and the samples were incubated at 65 °C for 90 min. DNA was purified, and real-time qPCR was performed using NF-κB site 1 forward primer and TAR +59R reverse primer (supplemental Fig. 1).
cDNA synthesis for RNA sequencing
The 3′-end of the extracted exosomal RNA was processed using a miRNA cDNA synthesis kit (Applied Biological Materials catalog no. G902). Briefly, poly(A) tail was added using Poly(A) polymerase, and the first strand of cDNA was synthesized using oligo(dT) adaptor. The synthesized cDNA was subject to amplification using TAR-specific primer (ggtctctctggttagac) and the 3′-adaptor–specific primer. The amplified DNA was purified using AMpure XP beads (Beckman Coulter) prior to library construction using the Illumina TruSeq library preparation kit (Illumina). Quality control of the sequencing library was performed using an Agilent 2100 Bioanalyzer (Agilent) to confirm library fragment sizes and qPCR to quantify the amount of the library. Sequencing was performed on the Illumina NextSeq system with 2 × 75-bp paired-end sequencing at Applied Biological Materials Inc.
Bioinformatics
Sequencing reads were aligned to the HIV-1 genome, and cluster analysis was performed using in-house PERL scripts. Only read pairs with R1 anchored at the TAR sequence were considered. Because the amplified cDNAs were expected to have a poly(A) tail, the confidence of the 3′-boundary was considered until the last non-A sequence.
siRNA transfection
The Lipofectamine RNAiMAX reagent protocol (Life Technologies, Inc.) was used to transfect cells. Briefly, ∼2 × 105 adherent cells were seeded in a 24-well plate. The next day, Lipofectamine RNAiMAX reagent was diluted in Opti-MEM medium by adding 25 μl of Opti-MEM medium to 1.5 μl of Lipofectamine RNAiMAX reagent. siRNA was then diluted in Opti-MEM medium with 25 μl of Opti-MEM medium, and 1.5 μl of siRNA (10 μm) was used. Twenty-five microliters of the diluted siRNA were then added to 25 μl of the diluted Lipofectamine RNAiMAX reagent and allowed to incubate at room temperature for 5 min. Cell medium was then removed, and 50 μl of siRNA–lipid complex and ∼250 μl of fresh medium were added to the cells (the final concentration of siRNA used was 20 nm) and allowed to incubate overnight. The siRNA–lipid complex was then removed, 1 ml of complete medium was added, and cells were incubated at 37 °C. When U1 cells were used, PMA (10 μm) was also added to the fresh medium. The cells were incubated for 72 h to differentiate prior to harvest and processed for downstream assays.
SDS-PAGE and Western blot analysis
Cell extracts were resolved by SDS-PAGE on 4–20% Tris-glycine gels (Novex). For Western blot analysis, proteins were transferred to Immobilon membranes (Millipore) at 50 mA overnight for ∼16 h. Membranes were blocked with Dulbecco's PBS + 0.1% Tween 20 + 5% dry milk for 30 min at 4 °C. Primary antibodies (at 1:200 to 1:50,000) against specified proteins were incubated with the membranes overnight at 4 °C. Membranes were washed twice with PBS + 0.1% Tween 20 and incubated with HRP-conjugated secondary antibody for 2 h at 4 °C. Membranes were washed two times with PBS + 0.1% Tween 20 and once with PBS prior to imaging. HRP luminescence was elicited with Super Signal West Dura Extended Duration Substrate (Pierce) and visualized by a Molecular Imager ChemiDoc XRS system (Bio-Rad).
Statistical analysis
S.D. was found from three independent measurements. Student's t tests were also performed between the control and treated samples to test for the significance: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
R. A. B., A. S., C. D., and Y. A. carried out most of the experiments and contributed to cell culture work, RNA isolation, RT-qPCR, Western blotting, ChIP assays, and data analysis. B. L. provided nanotrap particles NT080/082. S. K. provided participant samples. S. I. contributed to experimental design, and F. K. contributed to the overall direction and coordination of the study as well as to experimental design and data interpretation.
Supplementary Material
Acknowledgments
We thank the members of the Kashanchi laboratory for experimental designs and assistance with the manuscript. Dr. Gavin Sampey also assisted with the data in Fig. 1. We are also grateful to the AIDS Research and Reference Reagent Program for the contribution of various reagents. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health: U1, J1.1, and ACH-2 cells from Dr. Thomas Folks; HIV-1 89.6 dual-tropic viral strain from Dr. Ronald Collman; and anti-HIV-1 p24 mouse monoclonal antibody from Dr. Michael Malim. Four clinical samples utilized in this study were provided by the Washington, D. C., Metropolitan Site of the Women's Interagency HIV Study Collaborative Study Group (Principal Investigator, S. K.). The Women's Interagency HIV Study is supported by NIAID, National Institutes of Health, Grant U01-AI-34994. The RNA sequencing data from exosomes were generated by Dr. Jeff Chu from Applied Biological Materials Inc. (Richmond, Canada).
This work was supported by National Institutes of Health (NIH) Grants AI078859, AI074410, AI127351-01, AI043894, and NS099029 (to F. K.) and in part by NIAID/NIH, NCI/NIH, and the National Institute on Drug Abuse. The Washington, D. C., Metropolitan Site of the Women's Interagency HIV Study (WIHS) (principal investigator S. K.) is supported by NIAID/NIH, Grant U01AI034994 and co-funded by NCI/NIH, the National Institute on Drug Abuse, and the Eunice Kennedy Shriver NICHD/NIH. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- cART
- combination antiretroviral therapy
- LRA
- latency-reversing agent
- ESCRT
- endosomal sorting complex required for transport
- TAR
- trans-activation response
- qPCR
- quantitative PCR
- HTLV-1
- human T-lymphotropic virus-1
- AchE
- acetylcholinesterase
- IDV
- indinavir
- 3TC
- lamivudine
- TDF
- tenofovir disoproxil fumarate
- FTC
- emtricitabine
- pol
- polymerase
- PMA
- phorbol 12-myristate 13-acetate
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- PBMC
- peripheral blood mononuclear cell
- NRTI
- nucleoside reverse transcriptase inhibitor.
References
- 1. Kumar A., Abbas W., and Herbein G. (2014) HIV-1 latency in monocytes/macrophages. Viruses 6, 1837–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mbonye U., and Karn J. (2014) Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 454, 328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Archin N. M., Sung J. M., Garrido C., Soriano-Sarabia N., and Margolis D. M. (2014) Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 12, 750–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shan L., Yang H.-C., Rabi S. A., Bravo H. C., Shroff N. S., Irizarry R. A., Zhang H., Margolick J. B., Siliciano J. D., and Siliciano R. F. (2011) Influence of host gene transcription level and orientation on HIV-1 latency in a primary-cell model. J. Virol. 85, 5384–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verdin E., Paras P. Jr., and Van Lint C. (1993) Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 12, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman J., Cho W.-K., Chu C. K., Keedy K. S., Archin N. M., Margolis D. M., and Karn J. (2011) Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 85, 9078–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. du Chéné I., Basyuk E., Lin Y.-L., Triboulet R., Knezevich A., Chable-Bessia C., Mettling C., Baillat V., Reynes J., Corbeau P., Bertrand E., Marcello A., Emiliani S., Kiernan R., and Benkirane M. (2007) Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 26, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wei P., Garber M. E., Fang S. M., Fischer W. H., and Jones K. A. (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 [DOI] [PubMed] [Google Scholar]
- 9. Van Lint C., Bouchat S., and Marcello A. (2013) HIV-1 transcription and latency: an update. Retrovirology 10, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saayman S., Ackley A., Turner A.-M. W., Famiglietti M., Bosque A., Clemson M., Planelles V., and Morris K. V. (2014) An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol. Ther. 22, 1164–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Acheson N. H. (2011) Fundamentals of Molecular Virology, 2nd Ed., Wiley Global Education, Hoboken, NJ [Google Scholar]
- 12. Rasmussen T. A., and Lewin S. R. (2016) Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS 11, 394–401 [DOI] [PubMed] [Google Scholar]
- 13. Sgarbanti M., and Battistini A. (2013) Therapeutics for HIV-1 reactivation from latency. Curr. Opin. Virol. 3, 394–401 [DOI] [PubMed] [Google Scholar]
- 14. Fujiwara T., Ohira K., Urushibara K., Ito A., Yoshida M., Kanai M., Tanatani A., Kagechika H., and Hirano T. (2016) Steric structure-activity relationship of cyproheptadine derivatives as inhibitors of histone methyltransferase Set7/9. Bioorg. Med. Chem. 24, 4318–4323 [DOI] [PubMed] [Google Scholar]
- 15. Mehla R., Bivalkar-Mehla S., Zhang R., Handy I., Albrecht H., Giri S., Nagarkatti P., Nagarkatti M., and Chauhan A. (2010) Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One 5, e11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choudhary S. K., Archin N. M., and Margolis D. M. (2008) Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J. Infect. Dis. 197, 1162–1170 [DOI] [PubMed] [Google Scholar]
- 17. Banerjee C., Archin N., Michaels D., Belkina A. C., Denis G. V., Bradner J., Sebastiani P., Margolis D. M., and Montano M. (2012) BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 92, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sherrill-Mix S., Lewinski M. K., Famiglietti M., Bosque A., Malani N., Ocwieja K. E., Berry C. C., Looney D., Shan L., Agosto L. M., Pace M. J., Siliciano R. F., O'Doherty U., Guatelli J., Planelles V., and Bushman F. D. (2013) HIV latency and integration site placement in five cell-based models. Retrovirology 10, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iordanskiy S., and Kashanchi F. (2016) Potential of radiation-induced cellular stress for reactivation of latent HIV-1 and killing of infected cells. AIDS Res. Hum. Retroviruses 32, 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer S., Maldarelli F., Wiegand A., Bernstein B., Hanna G. J., Brun S. C., Kempf D. J., Mellors J. W., Coffin J. M., and King M. S. (2008) Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 105, 3879–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dornadula G., Zhang H., VanUitert B., Stern J., Livornese L. Jr., Ingerman M. J., Witek J., Kedanis R. J., Natkin J., DeSimone J., and Pomerantz R. J. (1999) Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 282, 1627–1632 [DOI] [PubMed] [Google Scholar]
- 22. Jones R. B., Mueller S., O'Connor R., Rimpel K., Sloan D. D., Karel D., Wong H. C., Jeng E. K., Thomas A. S., Whitney J. B., Lim S.-Y., Kovacs C., Benko E., Karandish S., Huang S.-H., et al. (2016) A subset of latency-reversing agents expose HIV-infected resting CD4+ T-cells to recognition by cytotoxic T-lymphocytes. PLoS Pathog. 12, e1005545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mikovits J. A., Lohrey N. C., Schulof R., Courtless J., and Ruscetti F. W. (1992) Activation of infectious virus from latent human immunodeficiency virus infection of monocytes in vivo. J. Clin. Invest. 90, 1486–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schrier R. D., McCutchan J. A., Venable J. C., Nelson J. A., and Wiley C. A. (1990) T-cell-induced expression of human immunodeficiency virus in macrophages. J. Virol. 64, 3280–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muller L., Mitsuhashi M., Simms P., Gooding W. E., and Whiteside T. L. (2016) Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 6, 20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ung T. H., Madsen H. J., Hellwinkel J. E., Lencioni A. M., and Graner M. W. (2014) Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 105, 1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akers J. C., Gonda D., Kim R., Carter B. S., and Chen C. C. (2013) Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 113, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaworski E., Narayanan A., Van Duyne R., Shabbeer-Meyering S., Iordanskiy S., Saifuddin M., Das R., Afonso P. V., Sampey G. C., Chung M., Popratiloff A., Shrestha B., Sehgal M., Jain P., Vertes A., Mahieux R., and Kashanchi F. (2014) Human T-lymphotropic virus type 1-infected cells secrete exosomes that contain Tax protein. J. Biol. Chem. 289, 22284–22305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schorey J. S., and Bhatnagar S. (2008) Exosome function: from tumor immunology to pathogen biology. Traffic 9, 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayanan A., Iordanskiy S., Das R., Van Duyne R., Santos S., Jaworski E., Guendel I., Sampey G., Dalby E., Iglesias-Ussel M., Popratiloff A., Hakami R., Kehn-Hall K., Young M., Subra C., Gilbert C., Bailey C., Romerio F., and Kashanchi F. (2013) Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 288, 20014–20033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teow S.-Y., Nordin A. C., Ali S. A., and Khoo A. S.-B. (2016) Exosomes in human immunodeficiency virus type I pathogenesis: threat or opportunity? Adv. Virol. 2016, 9852494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahsan N. A., Sampey G. C., Lepene B., Akpamagbo Y., Barclay R. A., Iordanskiy S., Hakami R. M., and Kashanchi F. (2016) Presence of viral RNA and proteins in exosomes from cellular clones resistant to Rift Valley fever virus infection. Front. Microbiol. 7, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nour A. M., and Modis Y. (2014) Endosomal vesicles as vehicles for viral genomes. Trends Cell Biol. 24, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valiathan R. R., and Resh M. D. (2008) Differential control of CXCR4 and CD4 downregulation by HIV-1 Gag. Virol. J. 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W., Hu Y., Jiang T., Han Y., Han G., Chen J., and Li X. (2014) Rab27A regulates exosome secretion from lung adenocarcinoma cells A549: involvement of EPI64. APMIS 122, 1080–1087 [DOI] [PubMed] [Google Scholar]
- 36. Bobrie A., Krumeich S., Reyal F., Recchi C., Moita L. F., Seabra M. C., Ostrowski M., and Théry C. (2012) Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 72, 4920–4930 [DOI] [PubMed] [Google Scholar]
- 37. Ostrowski M., Carmo N. B., Krumeich S., Fanget I., Raposo G., Savina A., Moita C. F., Schauer K., Hume A. N., Freitas R. P., Goud B., Benaroch P., Hacohen N., Fukuda M., Desnos C., et al. (2010) Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 12, 19–30 [DOI] [PubMed] [Google Scholar]
- 38. Hsu C., Morohashi Y., Yoshimura S., Manrique-Hoyos N., Jung S., Lauterbach M. A., Bakhti M., Grønborg M., Möbius W., Rhee J., Barr F. A., and Simons M. (2010) Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J. Cell Biol. 189, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., and Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 40. Ghossoub R., Lembo F., Rubio A., Gaillard C. B., Bouchet J., Vitale N., Slavík J., Machala M., and Zimmermann P. (2014) Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 5, 3477. [DOI] [PubMed] [Google Scholar]
- 41. Columba Cabezas S., and Federico M. (2013) Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell Microbiol. 15, 412–429 [DOI] [PubMed] [Google Scholar]
- 42. Laganà A., Russo F., Veneziano D., Bella S. D., Giugno R., Pulvirenti A., Croce C. M., and Ferro A. (2013) Extracellular circulating viral microRNAs: current knowledge and perspectives. Front. Genet. 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fleming A., Sampey G., Chung M.-C., Bailey C., van Hoek M. L., Kashanchi F., and Hakami R. M. (2014) The carrying pigeons of the cell: exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 71, 109–120 [DOI] [PubMed] [Google Scholar]
- 44. Sampey G. C., Saifuddin M., Schwab A., Barclay R., Punya S., Chung M.-C., Hakami R. M., Zadeh M. A., Lepene B., Klase Z. A., El-Hage N., Young M., Iordanskiy S., and Kashanchi F. (2016) Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA. J. Biol. Chem. 291, 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bukong T. N., Momen-Heravi F., Kodys K., Bala S., and Szabo G. (2014) Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 10, e1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Masciopinto F., Giovani C., Campagnoli S., Galli-Stampino L., Colombatto P., Brunetto M., Yen T. S. B., Houghton M., Pileri P., and Abrignani S. (2004) Association of hepatitis C virus envelope proteins with exosomes. Eur. J. Immunol. 34, 2834–2842 [DOI] [PubMed] [Google Scholar]
- 47. Ramakrishnaiah V., Thumann C., Fofana I., Habersetzer F., Pan Q., de Ruiter P. E., Willemsen R., Demmers J. A. A., Stalin Raj V., Jenster G., Kwekkeboom J., Tilanus H. W., Haagmans B. L., Baumert T. F., and van der Laan L. J. W. (2013) Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. U.S.A. 110, 13109–13113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Z., Zhang X., Yu Q., and He J. J. (2014) Exosome-associated hepatitis C virus in cell cultures and patient plasma. Biochem. Biophys. Res. Commun. 455, 218–222 [DOI] [PubMed] [Google Scholar]
- 49. Longatti A., Boyd B., and Chisari F. V. (2015) Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J. Virol. 89, 2956–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mori Y., Koike M., Moriishi E., Kawabata A., Tang H., Oyaizu H., Uchiyama Y., and Yamanishi K. (2008) Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 9, 1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bleck M., Itano M. S., Johnson D. S., Thomas V. K., North A. J., Bieniasz P. D., and Simon S. M. (2014) Temporal and spatial organization of ESCRT protein recruitment during HIV-1 budding. Proc. Natl. Acad. Sci. U.S.A. 111, 12211–12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langelier C., von Schwedler U. K., Fisher R. D., De Domenico I., White P. L., Hill C. P., Kaplan J., Ward D., and Sundquist W. I. (2006) Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 80, 9465–9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meng B., Ip N. C. Y., Prestwood L. J., Abbink T. E. M., and Lever A. M. L. (2015) Evidence that the endosomal sorting complex required for transport-II (ESCRT-II) is required for efficient human immunodeficiency virus-1 (HIV-1) production. Retrovirology 12, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwab A., Meyering S. S., Lepene B., Iordanskiy S., van Hoek M. L., Hakami R. M., and Kashanchi F. (2015) Extracellular vesicles from infected cells: potential for direct pathogenesis. Front. Microbiol. 6, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barillari G., Iovane A., Bacigalupo I., Labbaye C., Chiozzini C., Sernicola L., Quaranta M. T., Falchi M., Sgadari C., and Ensoli B. (2014) The HIV protease inhibitor indinavir down-regulates the expression of the pro-angiogenic MT1-MMP by human endothelial cells. Angiogenesis 17, 831–838 [DOI] [PubMed] [Google Scholar]
- 56. Imamichi H., Dewar R. L., Adelsberger J. W., Rehm C. A., O'Doherty U., Paxinos E. E., Fauci A. S., and Lane H. C. (2016) Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 113, 8783–8788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ishizaka A., Sato H., Nakamura H., Koga M., Kikuchi T., Hosoya N., Koibuchi T., Nomoto A., Kawana-Tachikawa A., and Mizutani T. (2016) Short intracellular HIV-1 transcripts as biomarkers of residual immune activation in patients on antiretroviral therapy. J. Virol. 90, 5665–5676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jaworski E., Saifuddin M., Sampey G., Shafagati N., Van Duyne R., Iordanskiy S., Kehn-Hall K., Liotta L., Petricoin E. 3rd, Young M., Lepene B., and Kashanchi F. (2014) The use of Nanotrap particles technology in capturing HIV-1 virions and viral proteins from infected cells. PloS One 9, e96778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Record M., Subra C., Silvente-Poirot S., and Poirot M. (2011) Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 81, 1171–1182 [DOI] [PubMed] [Google Scholar]
- 60. Ludwig A.-K., and Giebel B. (2012) Exosomes: small vesicles participating in intercellular communication. Int. J. Biochem. Cell Biol. 44, 11–15 [DOI] [PubMed] [Google Scholar]
- 61. Goff A., Ehrlich L. S., Cohen S. N., and Carter C. A. (2003) Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J. Virol. 77, 9173–9182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sen S., Kaminiski R., Deshmane S., Langford D., Khalili K., Amini S., and Datta P. K. (2015) Role of hexokinase-1 in the survival of HIV-1-infected macrophages. Cell Cycle 14, 980–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., and Naldini L. (1998) A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vlassov A. V., Magdaleno S., Setterquist R., and Conrad R. (2012) Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948 [DOI] [PubMed] [Google Scholar]
- 65. Jadlowsky J. K., Wong J. Y., Graham A. C., Dobrowolski C., Devor R. L., Adams M. D., Fujinaga K., and Karn J. (2014) Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell Biol. 34, 1911–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prescher J., Baumgärtel V., Ivanchenko S., Torrano A. A., Bräuchle C., Müller B., and Lamb D. C. (2015) Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog. 11, e1004677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Votteler J., and Sundquist W. I. (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14, 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abd El-Wahab E. W., Smyth R. P., Mailler E., Bernacchi S., Vivet-Boudou V., Hijnen M., Jossinet F., Mak J., Paillart J.-C., and Marquet R. (2014) Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 5, 4304. [DOI] [PubMed] [Google Scholar]
- 69. Alfadhli A., McNett H., Tsagli S., Bächinger H. P., Peyton D. H., and Barklis E. (2011) HIV-1 matrix protein binding to RNA. J. Mol. Biol. 410, 653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Purohit P., Dupont S., Stevenson M., and Green M. R. (2001) Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7, 576–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jean-Philippe J., Paz S., and Caputi M. (2013) hnRNP A1: the Swiss army knife of gene expression. Int. J. Mol. Sci. 14, 18999–19024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Villarroya-Beltri C., Gutiérrez-Vázquez C., Sánchez-Cabo F., Pérez-Hernández D., Vázquez J., Martin-Cofreces N., Martinez-Herrera D. J., Pascual-Montano A., Mittelbrunn M., and Sánchez-Madrid F. (2013) Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 4, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cao G., Li H.-B., Yin Z., and Flavell R. A. (2016) Recent advances in dynamic m6A RNA modification. Open Biol. 6, 160003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zeringer E., Li M., Barta T., Schageman J., Pedersen K. W., Neurauter A., Magdaleno S., Setterquist R., and Vlassov A. V. (2013) Methods for the extraction and RNA profiling of exosomes. World J. Methodol. 3, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hatano H., Jain V., Hunt P. W., Lee T.-H., Sinclair E., Do T. D., Hoh R., Martin J. N., McCune J. M., Hecht F., Busch M. P., and Deeks S. G. (2013) Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 208, 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bacon M. C., von Wyl V., Alden C., Sharp G., Robison E., Hessol N., Gange S., Barranday Y., Holman S., Weber K., and Young M. A. (2005) The Women's Interagency HIV Study: an observational cohort brings clinical sciences to the bench. Clin. Diagn. Lab. Immunol. 12, 1013–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Barkan S. E., Melnick S. L., Preston-Martin S., Weber K., Kalish L. A., Miotti P., Young M., Greenblatt R., Sacks H., and Feldman J. (1998) The Women's Interagency HIV Study: WIHS Collaborative Study Group. Epidemiology 9, 117–125 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.