

Abstract
The inositol 1,4,5 trisphosphate receptor (IP3R) is an intracellular Ca2+ release channel expressed predominately on the membranes of the endoplasmic reticulum. IP3R1 can be cleaved by caspase or calpain into at least two receptor fragments. However, the functional consequences of receptor fragmentation are poorly understood. Our previous work has demonstrated that IP3R1 channels, formed following either enzymatic fragmentation or expression of the corresponding complementary polypeptide chains, retain tetrameric architecture and are still activated by IP3 binding despite the loss of peptide continuity. In this study, we demonstrate that region-specific receptor fragmentation modifies channel regulation. Specifically, the agonist-evoked temporal Ca2+ release profile and protein kinase A modulation of Ca2+ release are markedly altered. Moreover, we also demonstrate that activation of fragmented IP3R1 can result in a distinct functional outcome. Our work suggests that proteolysis of IP3R1 may represent a novel form of modulation of IP3R1 channel function and increases the repertoire of Ca2+ signals achievable through this channel.
Keywords: calcium; calpain; caspase; inositol 1,4,5 trisphosphate receptor (IP3R); PKA; calcium signaling
Introduction
Calcium ions (Ca2+) are utilized widely as an intracellular second messenger and interact with effectors to induce a diverse array of cellular activities. These events include gene expression, cell migration, muscle contraction, secretion, autophagy, and cell death (1–3). To use Ca2+ to finely control cellular activities with high specificity and accuracy, cells have evolved a “calcium signaling toolbox” consisting of Ca2+ channels, pumps, and binding proteins (4). These components function in concert and encode unique information in the forms of amplitude, frequency, and subcellular location of Ca2+ signals. An important component of the toolbox is the inositol 1,4,5-trisphosphate receptor (IP3R).2 IP3R are intracellular Ca2+ release channels expressed predominately in the membrane of the endoplasmic reticulum (ER) in most of eukaryotic species (5–8). In response to IP3 binding, IP3R are activated, resulting in Ca2+ release from the intracellular store into the cytosol (9).
A series of complex regulatory events allows IP3R to encode Ca2+ signals with distinct temporal and spatial characteristics. First, following binding of IP3, the interaction of Ca2+, nucleotides, and binding proteins with IP3R can regulate channel activity (10–18). Second, posttranslational modifications, including phosphorylation, ubiquitination, and oxidation, can also influence channel activity and shape the IP3R Ca2+ release profile (19–23). As a further level of complexity, our laboratory has recently demonstrated that the particular isoform complement of the IP3R heterotetramer can either contribute to or largely determine the specific characteristics of IP3R-mediated Ca2+ signals (24, 25).
A further potential route of regulation of IP3R activity is through proteolytic fragmentation (26–28). The functional consequence of IP3R fragmentation has been a subject of debate for years (29–35). One of the major controversies centered over the question whether IP3R1 is a preferred substrate of caspase (35). Although several reports have demonstrated that caspase and calpain can cleave IP3R1 into at least two fragments (26, 27), other evidence has suggested that IP3R1 is not generally cleaved under apoptotic conditions and is, in fact, only a late substrate of caspase during intense staurosporine-induced apoptosis (35). The latter evidence argued that fragmentation of IP3R1 does not play a key role in the process of apoptosis.
A further area of contention surrounds the biophysical properties of fragmented IP3R. Proteolytic fragmentation of IP3R1 by caspase and calpain results in at least two receptor fragments: the N-terminal fragment consists of the IP3 binding core and much of the cytosolic peptide chain, whereas the C-terminal fragment contains the Ca2+ permeation pore and the C-terminal cytosolic tail (26, 27, 33). One proposal suggests that receptor fragmentation physically dissociates the N-terminal region from the ER-associated C-terminal channel domain. Such cleavage and dissociation would be predicted to functionally uncouple the regulation of IP3 binding from channel gating, leading to leaky C-terminal fragments retained in the ER, disruption of Ca2+ homeostasis, and apoptosis (31, 36). However, our laboratory proposed and demonstrated an alternative scenario by showing that IP3R1 retains its tetrameric architecture even after proteolytic fragmentation. Moreover, co-expression of complementary IP3R1 peptides, designed based on caspase- and calpain-fragmented IP3R1, can reconstitute the tetrameric channel structure that is functionally gated by IP3 binding (33). These data unambiguously demonstrate that IP3R1 is still tightly regulated by its endogenous ligand even after proteolytic fragmentation.
In this study, we have continued to investigate the consequences of proteolytic fragmentation of IP3R1. Given that fragmented IP3R1 are still functional in response to IP3 binding, we hypothesized that disruption of peptide continuity by proteolytic cleavage may affect the fine regulation of IP3R1 and, subsequently, alter IP3R1-mediated Ca2+ signals. This study demonstrates that proteolytic fragmentation has profound effects on IP3-mediated Ca2+ signals, resulting in alteration of the signature temporal pattern of Ca2+ release through IP3R1. Further, fragmentation can abolish PKA regulation of the receptor in a cleavage region-specific manner. More importantly, we show that the altered Ca2+ signals elicited by fragmented IP3R1 can specifically activate distinct downstream effectors compared with IP3R WT. Our results therefore strongly suggest that proteolytic fragmentation may represent a novel form of regulation of IP3R activity that expands the repertoire of signaling through IP3R1 activation.
Results
Functional fragmented IP3R1 are assembled from complementary IP3R1 fragments
We have reported previously that heterologous expression of complementary polypeptide fragments, designed based on putative caspase and calpain proteolytic cleavage sites, can be assembled into IP3R1 tetramers (33). This strategy provides an experimental platform to investigate the functional consequences of IP3R fragmentation. Our previous work strongly suggested that tetramers assembled from complementary IP3R1 fragments are functional in terms of IP3-gated Ca2+ release. To further characterize the functional consequence of receptor fragmentation, we designed additional complementary IP3R1 fragments according to defined IP3R1 trypsin cleavage sites in the rat IP3R1 (Fig. 1, A and B). Previous studies have shown that exposure of purified IP3R1 protein to a low concentration of trypsin in vitro results in five receptor fragments (37–39). These data have been interpreted to indicate that IP3R1 consists of five compact globular domains connected by four solvent-exposed linker regions. Throughout this work, cleavage sites introduced after the second or third trypsin cleavage sites, respectively, are denoted as IP3R1 I–II+III–V (tryp) and IP3R1 I–III+IV–V (tryp) (Fig. 1B). Caspase and calpain both cleave IP3R1 in the same solvent-exposed region (after tryptic fragment IV) and are denoted as IP3R1 I–IV+V(casp) and IP3R1 I–IV+V (calp) (Fig. 1B). To confirm that all fragmented IP3R1 are functional, DT40-3KO cells (null for all IP3R) (40) expressing various complementary receptor fragments (Fig. 2, A–D) were loaded with the Ca2+ indicator fura-2 and then challenged with the Gαq/IP3-linked, protease-activated receptor 2 (PAR2) agonist trypsin. Consistent with our previous work, all types of complementary receptor fragments were capable of inducing an elevation in [Ca2+]i in response to maximal PAR2 activation (Fig. 2, E–I). Further, DT40-3KO cells stably expressing only the channel fragment (IP3R1 V) showed no Ca2+ response when PAR2 was activated (Fig. 2J). Remarkably, functional channels were assembled by co-transfecting the remaining four complementary receptor fragments into these cells (Fig. 2K). To confirm this finding, we performed the co-transfection experiments in HEK cells. We and others have thoroughly characterized and demonstrated that HEK-3KO cells (a HEK cell line null for all IP3R) are completely devoid of any functional IP3R (41, 42). Consistently, trypsin-stimulated PAR2 activation induced Ca2+ release in HEK-3KO cells only when the complementary five tryptic fragments in a monomer were transiently expressed in cells (Fig. 2L). Together, these data confirm that complementary receptor fragments are assembled into functional channels that are gated by IP3 binding. Next we investigated whether IP3R fragmentation results in functionally equivalent IP3R or, alternatively, whether the biophysical profile or regulation of the fragmented IP3R differs from IP3R formed from conventional peptide monomer chains with linear continuity.
Figure 1.
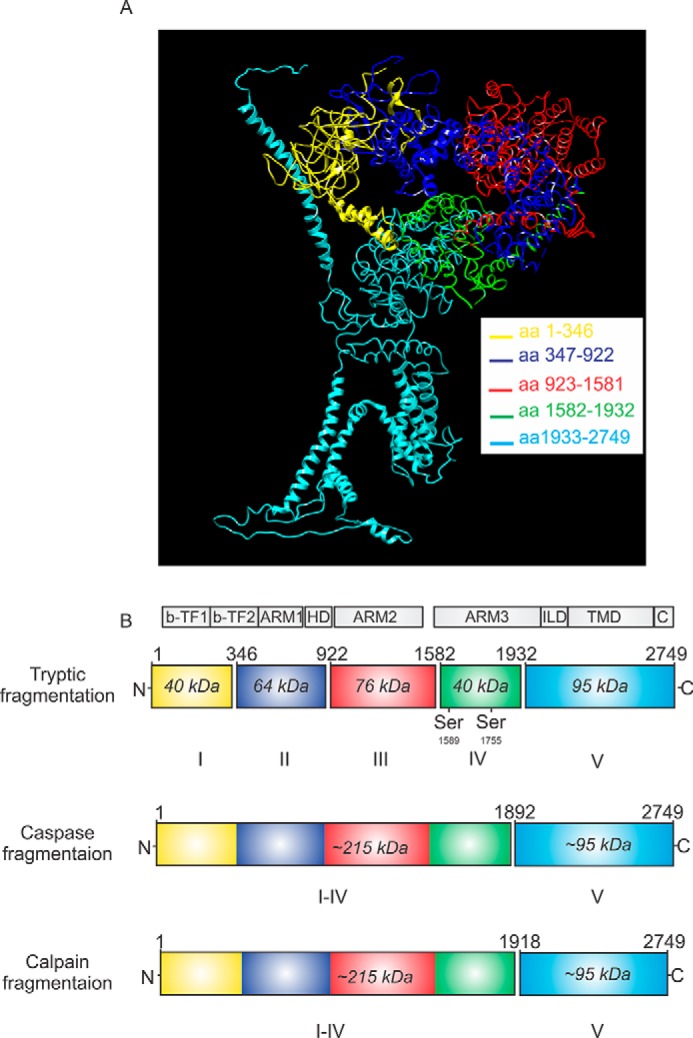
Schematic showing the proteolytic fragmentation sites on IP3R1. A, 3D structure of IP3R1 utilizing the cryo-EM structure published in Ref. 50, emphasizing five receptor fragments derived from limited trypsin exposure. Each fragment is color-coded. aa, amino acid. B, ribbons with corresponding colors represent the linear structure of fragmented IP3R1. Gaps between ribbons indicate the sites of proteolytic cleavages. Numbers above the gap indicate the amino residues at the C-terminal end of cleavage sites. Numbers in the ribbons indicate the relative molecular weight of each receptor fragment. b-TF, β trefoil domain; ARM, armadillo solenoid folds; HD, helical domain; ILD, intervening lateral domain; TMD, transmembrane domain; C, C-terminal domain.
Figure 2.

IP3R1 reconstituted from complementary receptor fragments are functional. A–D, expression of each type of IP3R1 receptor fragments in DT40-3KO cells was confirmed by Western blotting (WB) using IP3R1 antibodies probing the N terminus (A and C) or the C terminus of the receptor (B and D). Each lane was loaded with 18 μg of protein. Arrows indicate the fragments of interest. E–I, DT40-3KO cells stably expressing IP3R1 WT (E), IP3R1 I–II+III–V (tryp) (F), IP3R1 I–III+IV–V (tryp) (G), IP3R1 I–IV+V (calp) (H), or IP3R1 I–IV+V (casp) (I) were loaded with 2 μm Fura-2/AM, followed by stimulation with the PAR2 agonist trypsin (500 nm). Averaged traces of Ca2+ release were measured as a change in the 340/380 fluorescence ratio. J, DT40-3KO cells stably expressing IP3R1 V (tryp) did not respond to trypsin (500 nm) stimulation. K, transient co-expression of IP3R1 I (tryp), IP3R1 II (tryp), IP3R1 III (tryp), and IP3R1 IV (tryp) into DT40-3KO cells stably expressing IP3R1 V (tryp) constituted functional Ca2+ release channels. L, three representative HEK-3KO cells demonstrating that functional IP3R1 can be assembled from 20 IP3R1 polypeptide fragments corresponding to those produced by trypsinization of each monomer into five fragments. Ca2+ imaging assays were repeated five times for each set of complementary receptor fragments.
Proteolytic fragmentation alters the temporal pattern of IP3R1-mediated Ca2+ release in a region-specific manner
IP3R exhibit subtype-specific Ca2+ release profiles when continuously exposed to IP3. This is best exemplified following activation of B cell receptors (BCR) on DT40-3KO cells evoked by cross-linking with α-IgM (43). For example, stimulation of DT40-3KO cells stably expressing IP3R1 WT with α-IgM characteristically evokes only a few transient increases in [Ca2+]i (Fig. 3A and Refs. 41, 43, 44), whereas stimulation of cells expressing mouse IP3R2 WT elicits robust Ca2+ oscillations (13, 41, 43). A higher concentration of α-IgM (2 μg/ml) had no impact on the profile of Ca2+ release evoked in cells expressing IP3R1 WT (Fig. 3G). In addition, this general pattern was observed in various clones of DT40-3KO cells with different IP3R1 WT expression levels (data not shown), indicating that the temporal pattern of [Ca2+]i signal likely reflects an intrinsic property of the particular IP3R. Next we examined the Ca2+ release profile of cells expressing various complementary receptor fragments. When the cleavage site was located closer to the N terminus of the receptor, such as with IP3R1 I–II+III–V (tryp), similar to IP3R1 WT, a low number of Ca2+ transients were evoked upon α-IgM stimulation (Fig. 3, B and C). Remarkably, when cleavage sites were introduced further toward the C terminus of the receptor (for example, IP3R1 I–III+IV–V (tryp), IP3R1 I–IV+V (casp), and R1 I–IV+V (calp), a significant increase in the ability of BCR stimulation to evoke oscillatory activity was observed (Fig. 3, D–G). These data provide evidence that IP3R fragmentation at sites corresponding to cleavage by calpain and caspase can markedly alter the activity of the receptor and subsequently alter the temporal profile of Ca2+ release. We reported previously that a significant increase in the ability of IP3R1 to support Ca2+ oscillations occurred following PKA-mediated phosphorylation of the receptor (44). To investigate whether PKA phosphorylation plays a role in the increased oscillatory activity of particular fragmented IP3R1, BCR-stimulated [Ca2+]i signals were studied in cells expressing PKA non-phosphorylatable (S1589A, S1755A) and phosphomimetic (S1589E, S1755E) IP3R1 I–IV+V (calp). These mutations had no impact on the Ca2+ release profile mediated by IP3R1 I–IV+V (calp) (Fig. 4, A–D), indicating that PKA phosphorylation does not underlie the gain of oscillatory activity observed in the specific fragmented IP3R1. In addition, Ca2+ oscillations were retained for an extended period of time in the absence of extracellular Ca2+ (Fig. 5, A and B), suggesting that Ca2+ influx is not necessary to promote Ca2+ oscillations from these fragmented IP3R1.
Figure 3.

Fragmentation pattern determines the temporal Ca2+ release profile of the complementary receptor fragments. A–F, DT40-3KO cells stably expressing IP3R1 WT (A), IP3R1 I-II+III-V (tryp) (B), IP3R1 I-III+IV-V (tryp) (D), IP3R1 I-IV+V (calp) (E), or IP3R1 I-IV+V (casp) (F) were loaded with 2 μm Fura-2/AM, followed by cross-linking the B cell receptor using α-IgM (500 ng/ml). Two representative Ca2+ traces are shown for each pair of complementary receptor fragments. The numbers of Ca2+ transients in 15 min of experiments were calculated. C and G, scatterplots indicate that a cleavage site in solvent-exposed region II has no effect on the temporal Ca2+ release profile (C, Student's t test), whereas fragmentation sites more toward to the C terminus significantly increase the number of Ca2+ transients mediated by complementary receptor fragments (G, one-way ANOVA followed by Dunnett post-test). *, statistical significance determined by Dunnett post-test; n.s., not significant. Ca2+ imaging assays were repeated seven times, with more than 40 cells in each experimental run for each set of complementary receptor fragments.
Figure 4.

Altered temporal Ca2+ release profile mediated by IP3R1 I–IV+V (calp) is not due to PKA-mediated receptor phosphorylation. A–C, DT40-3KO cells stably expressing IP3R1 I–IV+V (calp) (A), IP3R1 I–IV+V (calp) (S1589A, S1755A) (B), or IP3R1 I–IV+V (calp) (S1589E, S1755E) (C) were loaded with 2 μm Fura-2/AM, followed by cross-linking the B cell receptor using α-IgM (500 ng/ml). Two representative Ca2+ traces are shown for each set of complementary receptor fragments. D, scatterplot indicating that there is no significant difference among three types of receptor fragments with respect to the number of Ca2+ transients in 30 min of recording (one-way ANOVA). Ca2+ imaging assays were repeated three times, with more than 40 cells in each experimental run for each complementary receptor fragments. n.s., not significant.
Figure 5.
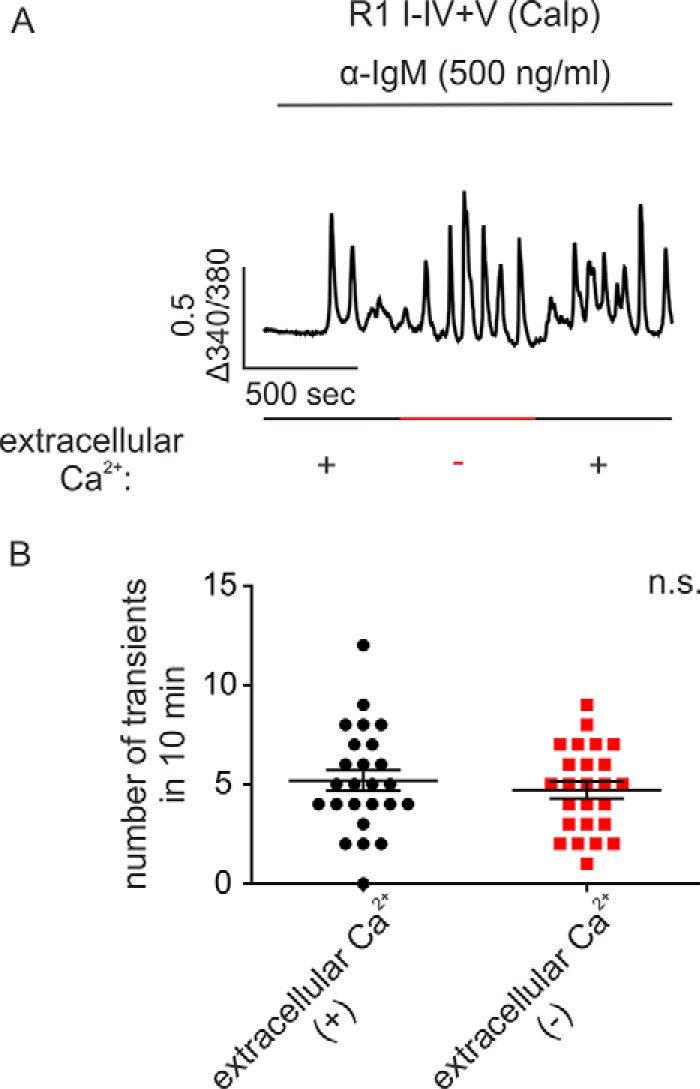
Ca2+ influx is not necessary for complementary receptor fragments to induce robust Ca2+ oscillation. A, DT40-3KO cells stably expressing IP3R1 I-IV+V (calp) were loaded with 2 μm Fura-2/AM, followed by cross-linking the cell surface B cell receptor using α-IgM (500 ng/ml). Perfusion buffers with or without extracellular Ca2+ were alternated every 10 min during recording as indicated. B, column statistics suggest that there is no significant difference between conditions with or without extracellular Ca2+ with respect to the number of Ca2+ transients (Student's t test). Experiments were repeated five times, with more than 40 cells in each experimental run. n.s., not significant.
We next investigated the profile of Ca2+ signals mediated by fragmented IP3R1 more directly by photorelease of a cell-permeable, poorly degradable, caged IP3 (45, 46). DT40-3KO cells expressing IP3R1 WT showed a sustained monophasic [Ca2+]i signal in response to photorelease of caged IP3 (Fig. 6A). In contrast, cells expressing IP3R2 WT exhibited robust oscillatory Ca2+ signals (Fig. 6B). Notably, IP3R1 I–IV+V (calp) showed both sustained monophasic and oscillatory responses (Fig. 6, C and D). A distribution of the frequency of Ca2+ oscillations shows that cells expressing IP3R1 WT mainly evoked a limited number of Ca2+ transients in response to IP3 exposure (Fig. 6E), whereas cells expressing IP3R1 I–IV+V (calp) displayed an increase in the population of cells that exhibit robust oscillatory Ca2+ release (Fig. 6F). This observation was consistent with the statistics showing that cells expressing IP3R1 I–IV+V (calp) induced significantly more Ca2+ transients compared with IP3R1 WT in response to photorelease of caged IP3 (Fig. 6G).
Figure 6.

Photorelease of caged IP3 induces distinct Ca2+ signals mediated by complementary receptor fragments. A and B, DT40-3KO cells stably expressing IP3R1 WT, IP3R2 WT, and IP3R1 I–IV+V (calp) were loaded with 1 μm Fluo-8/AM and 2 μm caged 6-O-[(4,5-dimethoxy-2-nitrophenyl)methyl]-2,3-O-(1-methylethylidene)-d-myo-inositol 1,4,5-tris[bis[(1-oxopropoxy)methyl]phosphate] (ci-IP3) for 30 min. A UV flash (200 ms) was introduced at the indicated time to photolyse caged IP3, and Ca2+ signals were recorded for 15 min. In response to ci-IP3, cells stably expressing IP3R1 WT mainly evoked a sustained single Ca2+ release event (A), whereas cells stably expressing IP3R2 WT evoked robust Ca2+ oscillations (B). C and D, both types of Ca2+ signals in A and B were observed in cells stably expressing IP3R1 I–IV+V (calp). E and F, distribution plots indicate that the majority of cells expressing IP3R1 WT induced either a sustained single Ca2+ release or few Ca2+ transients (E) whereas cells expressing IP3R1 I–IV+V (calp) gave rise to an increased level of Ca2+ transients during the 15-min recording. (F). G, box plot with whiskers showing the 10–90 percentile suggests a significant increase in the ability of IP3R1 I–IV+V (calp) to induce Ca2+ oscillations compared with IP3R1 WT (Student's t test). Experiments were repeated four times for each IP3R or complementary pairs of receptor fragments.
Proteolytic fragmentation alters the single-channel open probability of IP3R1
One caveat of the single-cell imaging assay above is that fragmented IP3R1 is not generated from the proteolytic cleavage of full-length IP3R1 WT but from the assembly of complementary receptor fragments. Therefore, we next performed patch-clamp recording in the “on nucleus” configuration to fragment IP3R1 WT in situ and directly investigate the biophysical consequences of IP3R1 fragmentation at the single-channel level (18). A submaximal concentration of IP3 (1 μm) resulted in an increase in the steady-state open probability (Po) of IP3R1 to ∼20% (Fig. 7, A and G). Addition of active caspase-3 (3 ng/ml) in the patch pipette in the presence of IP3 (1 μm) significantly augmented the channel Po to ∼70% (Fig. 7, B and G). Higher concentrations of active caspase-3 (10 ng/ml or 30 ng/ml) either diminished the conductance of the channel or completely inactivated the receptor, likely because of nonspecific digestion followed by destruction of the receptor (Fig. 7, C, D, and G). Addition of the caspase inhibitor Z-VAD (20 μm) completely blocked the effect of active caspase on channel Po (Fig. 7, E and G). In addition, in cells expressing constructs where the caspase cleavage site (DEVD at amino acids 1888–1891) was mutated to be non-cleavable (IEVA) (Fig. 7, F and G) (30), no increase in Po was observed in the presence of active caspase-3. These data strongly suggest that specific receptor fragmentation by caspase-3 at Asp-1891 enhances IP3R1 channel activity (Fig. 7G), and this likely reflects the single-channel correlate of the increase in oscillatory activity observed in intact cells expressing fragmented IP3R1 following stimulation with IP3.
Figure 7.

Fragmentation of IP3R1 by caspase-3 increases the channel open probability. A–D, patch clamp recording in the “on nucleus” configuration demonstrated that a low concentration of active caspase-3 (3 ng/ml) significantly increased the IP3R1 single-channel open probability in the presence of 1 μm IP3 (A and B), whereas high concentrations of active caspase-3 (10 and 30 ng/ml) abolished channel activity (C and D). E and F, the increase in channel open probability mediated by low concentrations of active caspase (3 ng/ml) can be blocked by addition of the caspase-3 inhibitor Z-VAD-fmk (E) or mutating the putative caspase cleavage motif DEVD to IEVA to make the IP3R1 non-cleavable (F). G, pooled data (heteroscedastic t test). Each condition was repeated six times. *, P < 0.01.
PKA regulation is abolished in calpain-fragmented IP3R1
Although fragmented IP3R is still gated by IP3 binding, how important individual modes of regulation of IP3R1 are impacted by receptor fragmentation remains unclear. Phosphorylation of IP3R1 at Ser-1589 and Ser-1755 by PKA significantly increases channel Po at the single-channel level (21, 22). This biophysical alteration is manifested as an increase in Ca2+ release at the single-cell level determined in Ca2+ imaging assays. Interestingly, the calpain fragmentation site is located between the PKA phosphorylation sites and the receptor channel domain. As a result, proteolytic receptor fragmentation by calpain separates PKA phosphorylation sites and the Ca2+ permeation pore into two different peptide fragments. Given the location of the calpain fragmentation site in IP3R1, we next investigated the effects of fragmentation on PKA regulation of IP3R1. Activation of PKA resulted in IP3R1 phosphorylation and, subsequently, a significant increase in Ca2+ release in cells expressing IP3R1 compared with DMSO-treated controls in response to all PAR2 agonist concentrations (Fig. 8, A, B, and F). The increase in phosphorylation and regulation of Ca2+ signals was completely abolished in cells expressing non-phosphorylatable IP3R1 mutants at both PKA phosphorylation sites (Fig. 8, C and F). Notably, when a disruption of peptide continuity was introduced at the third trypsin cleavage site, IP3R1 I–III+IV–V (tryp), PKA phosphoregulation was maintained (Fig. 8, D and F). In contrast, although forskolin pretreatment increased the level of phosphorylated IP3R1 I–IV+V (calp), no significant difference in terms of the amplitude of Ca2+ response was observed. (Fig. 8, E and G). These data demonstrate that PKA regulation of IP3R activity requires the phosphorylated residues to be proximal or, possibly, in the same fragment as the channel domain and further suggest that other particular modes of regulation of IP3R1 activity may be altered depending on the specific site of fragmentation.
Figure 8.

The increase in Ca2+ release in IP3R1 by PKA phosphorylation is regulated by receptor fragmentation in a cleavage region-specific manner. A, cells were loaded with 1 μm Fluo-2/AM for 1 h, followed by a FlexStation assay to monitor the change in [Ca2+]i. Preincubation of cells stably expressing IP3R1 WT with forskolin significantly increased the amplitude of Ca2+ release in response to PAR2 activation. B and D, this increase was observed at all agonist concentrations tested for IP3R1 WT (B) and at high agonist concentrations tested for IP3R1 I–III+IV–V (tryp) (D). C and E, forskolin had no effect on the amplitude of Ca2+ release in cells expressing IP3R1 (S1589A, S1755A) (C) or IP3R1 I-IV+V (calp) (E). F and G, pretreatment of forskolin results in IP3R1 phosphorylation at residue Ser-1755 in both full-length and fragmented receptors. Statistics were performed using one-way ANOVA followed by Dunnett post-test. Experiments were repeated three times for each set of IP3R1 or complementary receptor fragments. WB, Western blot.
Fragmented IP3R1 can activate distinct downstream effectors
Based on the observation that specific fragmented IP3R1 can induce different Ca2+ signals, we next investigated whether fragmented IP3R1 can specifically activate distinct downstream effectors. Oscillatory Ca2+ signals, but not single Ca2+ transients, have been shown to specifically activate the transcription factor nuclear factor of activated T cells (NFAT). Ca2+ oscillations are thought to deliver signals with the appropriate spatial and temporal properties necessary to activate the phosphatase calcineurin, which dephosphorylates NFAT and facilitates its translocation to the nucleus (45, 47, 48). We hypothesized that caspase- or calpain-fragmented IP3R1, but not IP3R1 WT, might provide the necessary Ca2+ signal to activate NFAT translocation. GFP-tagged NFAT (NFAT-GFP) was transfected into cells expressing either IP3R1WT, IP3R1 I–IV+V (casp), or IP3R1 I–IV+V (calp). In the quiescent state, NFAT-GFP was mainly located in the cytosol (Fig. 9, B, E, and H). Upon BCR stimulation, NFAT-GFP efficiently translocated from the cytosol into the nucleus in cells expressing either IP3R1 I–IV+V (casp) or IP3R1 I–IV+V (calp) (Fig. 9, A–F, J, and K). Translocation of NFAT-GFP was not observed in cells expressing IP3R1 WT (Fig. 9, G–K). These data strikingly illustrate that the distinct patterns of Ca2+ signal evoked through particular fragmented IP3R have the ability to activate distinct downstream events compared with the intact IP3R1.
Figure 9.

Ca2+ signals induced by IP3R1 I–IV+V can activate distinct downstream effectors. B, E, and H, DT40-3KO cells stably expressing IP3R1 I–IV+V (casp), IP3R1 I–IV+V (calp), and IP3R1 WT were transiently transfected with NFAT-GFP, followed by 12-h recovery in a 39 °C, 5% CO2 incubator. Cells were then mounted in a perfusion chamber, the location of NFAT-GFP was monitored following excitation with 488 nm, and emitted fluorescence recorded above 510 nm. Prior to stimulation, NFAT-GFP was mainly located in the cytosol (B, E, and H). A, D, and G, 3D heat plots, with the x and y axes indicating spatial coordinates the and z axis indicating the amplitude of the GFP signal. A, C, D, F, G, I, and J, addition of α-IgM (500 ng/ml) activated and translocated NFAT-GFP from the cytosol into the nucleus in cells expressing IP3R1 I–IV+V (casp) (A, C, and J) and IP3R1 I–IV+V (calp) (D, F, and J). This translocation was not observed in cells expressing IP3R1 WT (G, I, and J). C, F, and I, nucleus NFAT-GFP signals are shown in red and cytosolic NFAT-GFP signals in black and were used to calculate the nucleus/cytosol ratio (N/C, blue). J, the percentage of cells where translocation could be observed in individual trials in which more than 20 cells were imaged per trial. K, the magnitude of the N/C ratio in individual cells from each trial that showed translocation. These data demonstrate a significant increase in nucleus/cytosol ratio for fragmented IP3R1 compared with IP3R1 WT in response to α-IgM stimulation. Experiments were repeated four times for IP3R1 and three times for each type of complementary receptor fragments. *, P < 0.01.
Discussion
The data presented here expand on our earlier findings exploring the functional consequences of fragmentation of IP3R1 by intracellular proteases. We now demonstrate that expression of complementary polypeptides that correspond to any pair of fragments representing the five globular domains generated in vitro by limited tryptic exposure similarly result in functional channels (37). Indeed, co-expression of individual cDNA encoding the five domains separately, remarkably, leads to the assembly of a functional channel gated by IP3. Although these data firmly establish that peptide continuity is not required for channel opening per se, the major finding of this study is that fragmentation of IP3R1 markedly alters allosteric regulation of the channel by key modulators to alter Ca2+ release activity. Specifically, as an example, we show that a prominent mode of regulation of IP3R1, augmented Ca2+ release following PKA phosphorylation, is lost when fragments are expressed that mimic calpain cleavage to generate fragmented IP3R1 I–IV+V. However, regulation by PKA is clearly evident when peptide continuity is lost more proximal to the N terminus to yield IP3R1 I–III+IV–V. Notably, in this case, the key phosphorylation sites at Ser-1589 and Ser-1755 are present in the same fragment as the channel pore in fragment V, suggesting that peptide continuity is required to communicate the conformation change imparted by phosphorylation to modulation of gating of the pore. Further, these data are consistent with the observation that PKA phosphorylation does not alter IP3 binding to the binding core in the N terminus, which is present in the complementary fragment (49). These data clearly demonstrate that alterations in allosteric modulation of IP3R activity are dependent on the site of cleavage.
A further demonstration that the activity of IP3R1 is dramatically altered by cleavage is the observation that the temporal profile of Ca2+ release following agonist stimulation is temporally transformed when the channel is assembled from particular complementary polypeptide chains. We and others have consistently reported that sustained stimulation of cells expressing individual IP3R subtypes in isolation supports specific patterns of Ca2+ release that can be considered a “signature” for that subtype (13, 41, 43, 44). As the signal is largely independent of extracellular Ca2+, these signatures are the result of the integrated regulatory input received by the particular IP3R expressed. Notably, in this study, we show that expression of pairs of complementary fragments, in particular those corresponding to products derived from caspase or calpain proteolytic fragmentation, results in a transformation of the Ca2+ signal from a largely transient increase into an oscillatory profile consisting of numerous organized transients present throughout stimulation. In addition, single-channel recordings indicate that in situ cleavage of IP3R1 by caspase significantly increased the channel open probability. This provides a potential underlying mechanism for altering IP3R1 activity through proteolytic fragmentation. Consistent with the widely held view that the spatial and temporal properties of Ca2+ signals are important for the activation of downstream effectors, these oscillatory Ca2+ signals were capable of driving the nuclear localization of NFAT-GFP whereas signals through the IP3R1 WT could not. Taken together, these data provide support for the hypothesis that cleavage by proteases can potentially have significant consequences for IP3R1 activity and might be considered a novel mode of regulation influencing multiple modulatory inputs.
The cryo-EM structure of IP3R1 was recently solved to near atomic resolution and provides structural details consistent with our findings (50). The suppressor domain (TFβ1) and the IP3 binding core (TFβ2 and the N-terminal region of ARM1) at the N terminus of the receptor physically interact with the C-terminal domain of the adjacent subunit. This interaction provides a physical basis for a direct communication between IP3 binding at the N terminus and the distant channel opening at the C terminus without requiring signaling propagation along the whole peptide sequence. These data are consistent with our conclusion that peptide continuity in the receptor coupling domain, which comprises the helical domain, armadillo solenoid folds (ARM), and the intervening later domain, is not required for channel opening following IP3 binding. However, the coupling domain is crucial for integrating intracellular input and, accordingly, imposing regulations on the channel. This concept was again strongly supported by the recent structure that shows that flexible structures of the ARM domains were amenable to generating interfaces for recognition and binding of various regulators. Noticeably, both caspase and calpain cleavage sites as well as the fragmentation site for IP3R1 I–III+IV–V are located in ARM regions. Given the critical role of the coupling domain in terms of receptor regulation, it is conceivable that disruption of peptide continuity in the ARM regions (IP3R1 I–III+IV–V and IP3R1 I–IV+V) are likely to either interfere with the intrinsic regulation of ARM on the channel domain or affect other mediators that regulate channel activity through binding to or modifying ARM domains and thus alter IP3R1-induced Ca2+ signals.
What is the mechanism underlying the ability of particular fragmented IP3R1 to support robust Ca2+ oscillations? A canonical model for class I Ca2+ oscillations provides a possible explanation for this remarkable alteration (51). This model suggests that cells can control the Ca2+ oscillation period by modulating the rate of Ca2+ activation of IP3R in response to the change in [Ca2+]i. Of note, although there was no EF-hand Ca2+ binding motif found in IP3R1, the ARM3 region in the coupling domain contains a putative Ca2+ binding region with a highly conservative Glu-2100 critical for Ca2+ binding and regulation of IP3R1 (52, 53). Based on the model and the location of the Ca2+ sensor in the ARM regions, we speculate that fragmentation of IP3R1 at particular ARM regions impacts the Ca2+ modulation of channel gating and, consequently, alters the Ca2+ activation rate for IP3R1. This hypothesis is supported by mathematical simulations showing that, by solely changing the rate of IP3R1 activation by Ca2+, the normal monotonic Ca2+ release pattern through wild-type IP3R1 Ca2+ can be converted into robust oscillatory Ca2+ signals observed with IP3R1 I–III+IV–V and IP3R1 I–IV+V (51).
Although IP3R1 has been reported to be a substrate of caspase and calpain for more than a decade, any cellular role of cleavage has yet to be firmly established. We and other laboratories report that, even when caspase and calpain are massively activated during apoptosis, only a small fraction of IP3R1 is actually fragmented. Therefore, we envision that, when caspase or calpain are mildly activated under non-apoptotic conditions, a small proportion of a particular fragmented IP3R1 is generated in cells that might then locally activate specific downstream effectors and fulfill unique roles. When might lower levels of protease activity occur? Recent reports have suggested that caspase and calpain activity is also essential for processes other than initiating cell death. These include protein maturation, cell proliferation and differentiation, and myoblast fusion (54–61). More importantly, there is burgeoning evidence suggesting that, beyond negating protein activity, caspase can activate its substrates by proteolysis (54, 56, 60, 62). Consistent with this idea, this study provides evidence that enzymatic cleavage per se might uniquely regulate IP3R1 activity and thus might potentially contribute to the regulation of different cellular activities rather than cell death.
In summary, our work provides the first evidence showing that proteolytic fragmentation may serve as a novel regulatory event for IP3R1 by altering its Ca2+ release profile. Future work is necessary to specifically investigate the pathophysiological conditions under which IP3R1 is cleaved and the corresponding significance of receptor fragmentation for those processes. We envision that spatially confined protease activity may cleave the IP3R1 to obtain a transient alteration of receptor-mediated Ca2+ signals, which can rapidly switch on alternative signaling pathways for unique cellular activities. In addition, there is evidence showing that both IP3R2 and IP3R3 can also be fragmented by proteases (63). We are currently investigating the functional consequence of proteolytic fragmentation for IP3R2 and IP3R3. Our study will answer the important question of whether proteolytic fragmentation may be a general regulatory event for all isoforms of IP3R.
Materials and methods
Reagents
All restriction enzymes and T4-DNA ligase were from New England Biolabs. RPMI 1640 medium, penicillin/streptomycin, G418 sulfate, β-mercaptoethanol, and chicken serum were purchased from Invitrogen. Fetal bovine serum was from Gemini Bioproducts. Iso-Ins(1,4,5)P3/PM (caged), Z-VAD-fmk, and active caspase-3 were from Enzo Life Science. Fura-2 was from Teflabs. All reagents for SDS-PAGE were from Bio-Rad. The N-terminal antibody for IP3R1 and phospho-IP3R1 were from Cell Signaling Technology. DyLightTM 800CW secondary antibody was from Thermo Scientific. Forskolin was from Sigma-Aldrich. Mouse α-chicken IgM was from Southern Biotech. The antibody against the C-terminal 19 amino acids of IP3R1 was generated by Pocono Rabbit Farms and Laboratories.
Constructs
The method for generation of IP3R1 I–IV+V (casp) and IP3R1 I–IV+V (calp) was first described elsewhere (33). In brief, to create IP3R1 I–IV+V (casp), cDNA encoding rat IP3R1 flanked by the NheI and NotI sites at the 5′ and 3′ ends in pcDNA3.1 was used as the template. All PCR modifications described here were conducted using Pfu Ultra II Hotstart 2X Master Mix (Agilent), and only forward primers are shown here. To generate the construct coding for N-terminal and C-terminal fragments predicted to result from caspase cleavage of IP3R1 at the DEVD1891 consensus site, IP3R1 cDNA was modified by PCR (forward, 5′-GAAAGATGATGAAGTGGACTAGAATTCGCGGCCGCGCTAGCATGCGGGATGCCCCATCCCGAA-3′). This modification introduced a stop codon after residue Asp-1891 and also a Kozak sequence and an initiation methionine in-frame with the sequence coding for the membrane fragment, designed to ensure efficient expression. To obtain a two-promoter vector (TPV) encoding both N- and C-terminal fragments, two-step ligation was performed (33). First, NheI-NotI IP3R1 I–IV (casp) was inserted into the TPV digested with NheI and PspOMI (NotI and PspOMI have compatible cohesive ends). Second, the NotI-NotI fragment coding IP3R1 V (casp) was inserted into the TPV that was digested with NotI. The TPV encoding IP3R1 I–II+III–V (tryp), IP3R1 I–III+IV–V (tryp), and IP3R1 I–V+V (calp) were constructed in an identical manner using corresponding primers: forward 5′-GGCAGCAACGTGATGAGATAGGCGGCCGCGCTAGCATGTCTATCCATGGAGTTGG-3′) for IP3R1 I–II+III–V (tryp), forward 5′-CTGGCGGTTATCAGCCCGCTAGGCGGCCGCGCTAGCATGAACGCTGCTCGTAGAG-3′) for IP3R1 I–III+IV–V (tryp), and forward 5′-CCGGGATCAGCTCTTGGAATAGAATTCGCGGCCGCGCTAGCATGGCATCTGCTGCCACCAGAAAAGCC-3′ for IP3R1 I–V+V (calp). To generate caspase non-cleavable rIP3R1, forward primer 5′-GGGAAACAAAAAGAAAGATATCGAAGTGGCCAGGGATGCCCC-3′ was used to mutate the sequence encoding amino acid DEVD to be IEVA. Two-step PCR was performed sequentially using forward primers 5′-TCAGGAAGAAGAGAGGCTCTTACCAGCTTTGGCA-3′ and 5′-GCTGCTCGTAGAGACGCTGTCCTGGCAGCTTCC-3′ to generate rIP3R1 (S1589A, S1755A) and forward primers 5′-TCAGGAAGAAGAGAGGAGCTTACCAGCTTTGGCA-3′ and 5′-GCTGCTCGTAGAGACGAGGTCCTGGCAGCTTCC-3′ to generate rIP3R1 (S1589E, S1755E).
Western blot analysis
Cells were harvested by centrifugation, washed once with ice-cold PBS, and solubilized in cell lysis buffer containing 10 mm Tris-HCl, 10 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mm NaF, 20 mm Na4P2O7, 2 mm Na3VO4, 1% Triton X-100 (v/v), and 10% glycerol with a mixture of protease inhibitors. After 30 min on ice, cell lysates were precleared by centrifugation at 16,000 × g for 10 min at 4 °C. Cleared lysates were transferred into fresh tubes, and protein concentrations were measured using a Dc protein assay kit (Bio-Rad). Protein were resolved on 5–7.5% SDS-PAGE gels, transferred to nitrocellulose, and processed for immunoblotting with the indicated primary antibodies and corresponding secondary antibodies. Proteins were detected using an Odyssey infrared imaging system (LI-COR Biosciences).
Fluorescence imaging assay
DT40 cells expressing defined IP3R constructs were loaded with 2 μm Fura-2/AM on a glass coverslip mounted onto a Warner chamber at room temperature for 20–30 min. Loaded cells were perfused with HEPES imaging buffer (137 mm NaCl, 4.7 mm KCl, 1.26 mm CaCl2, 1 mm Na2HPO4, 0.56 mm MgCl2,10 mm HEPES, and 5.5 mm glucose (pH 7.4)) and stimulated with the indicated agonist. Ca2+ imaging was performed using an inverted epifluorescence Nikon microscope with a ×40 oil immersion objective (NA = 1.3). Cells were alternately excited at 340 and 380 nm, and emission was monitored at 505 nm. Images were captured every second with an exposure of 10 ms and 4 × 4 binning using a digital camera (Cooke Sensicam QE) driven by TILL Photonics software.
Cell culture and plasmid transfection
DT40-3KO cells, a chicken B lymphocyte line with targeted deletion of the three endogenous IP3R isoforms, were grown in RPMI 1640 medium supplemented with 1% chicken serum, 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 39 °C with 5% CO2. DT40-3KO cell transfection and generation of stable cell lines was performed as described previously using the Amaxa nucleofector (Lonza Laboratories). HEK-3KO cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C with 5% CO2. HEK-3KO cells were transfected using Lipofectamine 2000 (Invitrogen) following the protocol of the manufacturer.
FlexStation assay
DT40-3KO cells (5 × 105 cells/well) expressing the receptor of interest were washed with PBS once, followed by incubation in imaging buffer containing 1% BSA and 5 μm Fluo-2/AM for 1 h. Cells were then spun down, washed with imaging buffer containing 1% BSA, and seeded onto a 96-well plate that was precoated with 0.1% poly-l-lysine. The plate was spun at 500 × g for 2 min and rested at room temperature for 15 min. During FlexStation recording, cells were treated with 20 μm forskolin (final concentration) or the same volume of DMSO for 3 min, followed by stimulation with different concentrations of PAR2 agonist. For each type of IP3R or receptor fragments, a control response was defined as the amplitude of Ca2+ response stimulated by 500 μm PAR2 agonist following DMSO preincubation. The amplitudes of Ca2+ responses for all other conditions were calculated and displayed as the percentage of the control response.
NFAT-GFP translocation assay
DT40-3KO cells expressing the desired receptor were transiently transfected with NFAT-GFP. 12 h after recovery, cells were transferred onto coverslips mounted in a Warner chamber. Cells were perfused with HEPES imaging buffer (137 mm NaCl, 4.7 mm KCl, 1.26 mm CaCl2,1 mm Na2HPO4, 0.56 mm MgCl2,10 mm HEPES, and 5.5 mm glucose (pH 7.4)) and stimulated with mouse α-chicken IgM. GFP imaging was recorded using an inverted epifluorescence Nikon microscope with a ×40 oil immersion objective. Cells were excited at 488 nm, and emission was monitored at 509 nm. Images were captured every second with an exposure of 10 ms and 2 × 2 binning using a digital camera driven by TILL Photonics software.
Preparation of DT40 cell nuclei
Isolated DT40 nuclei were prepared by homogenization as described previously (64). The homogenization buffer contained 250 mm sucrose, 150 mm KCl, and 10 mm Tris (pH 7.5). Cells were washed and resuspended in homogenization buffer prior to nuclear isolation using a RZR 2021 homogenizer (Heidolph Instruments) with 25 strokes at 1200 rpm. A 3-μl aliquot of nuclear suspension was placed in 3 ml of bath solution that contained 140 mm KCl, 10 mm HEPES, 500 μm BAPTA and 246 nm free Ca2+, pH 7.1. Nuclei were allowed to adhere to a plastic culture dish for 10min prior to patching.
On-nuclei patch clamp experiments
Single InsP3R channel currents using Potassium ions as the charge carrier (ik) were measured in the on nucleus patch clamp configuration using pCLAMP 9 and an Axopatch 200B amplifier (Molecular Devices, Sunnydale, CA, USA) as previously described (64). Pipette solution contained 140 mm KCl, 10 mm HEPES, 1 μm InsP3, 5 mm ATP, and 200 nm free Ca2+ (pH 7.1). Free [Ca2+] was calculated using Max Chelator freeware and verified fluorometrically. Active caspase-3 and/or Z-VAD-fmk were included in the pipette solution for the corresponding experiments. Traces were consecutive 3-s sweeps recorded at −100 mV, sampled at 20 kHz, and filtered at 5 kHz. A minimum of 15 s of recordings was considered for data analyses. Pipette resistances were typically 20 megohms, and seal resistances were >5 gigaohms.
Data analysis
Single-channel openings were detected by half-threshold crossing criteria using the event detection protocol in Clampfit 9. We assumed that the number of channels in any particular nuclear patch is represented by the maximum number of discrete stacked events observed during the experiment. Even at low Po, stacking events were evident (data not shown). Only patches with one apparent channel were considered for analyses. Po, unitary current (ik), and open and closed times were calculated using Clampfit 9 and Origin 6 software (Origin Lab, Northampton, MA). All-points current amplitude histograms were generated from the current records and fitted with a normal Gaussian probability distribution function. The coefficient of determination (R2) for every fit was >0.95. The Po was calculated using the multimodal distribution for the open and closed current levels. Channel dwell time constants for the open and closed states were determined from exponential fits of all-points histograms of open and closed times. The threshold for an open event was set at 50% of the maximum open current, and events shorter than 0.1 ms were ignored.
Author contributions
This work was performed in the Department of Pharmacology and Physiology at the University of Rochester. L. W. designed and stably expressed some of the constructs, collected and analyzed the data, drafted the manuscript, and prepared the figures. L. E. W. collected and analyzed data obtained through single-channel electrophysiology (Fig. 7). K. J. A. designed and expressed some of the constructs and performed the experiments shown in Fig. 1J. D. I. Y. was responsible for the conception and design of all experiments as well as data analysis, generation of figures, and editing of the manuscript. All authors approved the final version.
Acknowledgments
We thank the members of the Yule laboratory for helpful discussions throughout this study.
This work was supported by National Institutes of Health Grants RO1 DE014756 and DE019245. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IP3R
- inositol 1,4,5 trisphosphate receptor
- ER
- endoplasmic reticulum
- BCR
- B cell receptor
- Po
- open probability
- Z-VAD-fmk
- benzyloxycarbonyl-VAD-fluoromethyl ketone
- ARM
- armadillo
- TPV
- two-promoter vector
- ANOVA
- analysis of variance.
References
- 1. Berridge M. J., Lipp P., and Bootman M. D. (2000) The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21 [DOI] [PubMed] [Google Scholar]
- 2. Carafoli E., Santella L., Branca D., and Brini M. (2001) Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 36, 107–260 [DOI] [PubMed] [Google Scholar]
- 3. Luyten T., Welkenhuyzen K., Roest G., Kania E., Wang L., Bittremieux M., Yule D. I., Parys J. B., and Bultynck G. (2017) Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2. Biochim Biophys Acta 1864, 947–956 [DOI] [PubMed] [Google Scholar]
- 4. Berridge M. J., Bootman M. D., and Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 5. Berridge M. J. (1993) Inositol trisphosphate and calcium signalling. Nature 361, 315–325 [DOI] [PubMed] [Google Scholar]
- 6. Mikoshiba K. (2007) IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem. 102, 1426–1446 [DOI] [PubMed] [Google Scholar]
- 7. Foskett J. K., White C., Cheung K. H., and Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alzayady K. J., Sebé-Pedrós A., Chandrasekhar R., Wang L., Ruiz-Trillo I., and Yule D. I. (2015) Tracing the evolutionary history of inositol 1,4,5-trisphosphate receptor: insights from analyses of Capsaspora owczarzaki Ca2+ release channel orthologs. Mol. Biol. Evol. 32, 2236–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alzayady K. J., Wang L., Chandrasekhar R., Wagner L. E. 2nd, Van Petegem F., and Yule D. I. (2016) Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal 9, ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruce J. I., Straub S. V., and Yule D. I. (2003) Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium 34, 431–444 [DOI] [PubMed] [Google Scholar]
- 11. Bezprozvanny I., Watras J., and Ehrlich B. E. (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754 [DOI] [PubMed] [Google Scholar]
- 12. Park H. S., Betzenhauser M. J., Zhang Y., and Yule D. I. (2012) Regulation of Ca2+ release through inositol 1,4,5-trisphosphate receptors by adenine nucleotides in parotid acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G97–G104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Betzenhauser M. J., Wagner L. E. 2nd, Iwai M., Michikawa T., Mikoshiba K., and Yule D. I. (2008) ATP modulation of Ca2+ release by type-2 and type-3 inositol (1,4,5)-triphosphate receptors: differing ATP sensitivities and molecular determinants of action. J. Biol. Chem. 283, 21579–21587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagner L. E. 2nd, Betzenhauser M. J., and Yule D. I. (2006) ATP binding to a unique site in the type-1 S2-inositol 1,4,5-trisphosphate receptor defines susceptibility to phosphorylation by protein kinase A. J. Biol. Chem. 281, 17410–17419 [DOI] [PubMed] [Google Scholar]
- 15. Yule D. I., Betzenhauser M. J., and Joseph S. K. (2010) Linking structure to function: Recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 47, 469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schulman J. J., Wright F. A., Han X., Zluhan E. J., Szczesniak L. M., and Wojcikiewicz R. J. (2016) The stability and expression level of Bok are governed by binding to inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 291, 11820–11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivanova H., Ritaine A., Wagner L., Luyten T., Shapovalov G., Welkenhuyzen K., Seitaj B., Monaco G., De Smedt H., Prevarskaya N., Yule D. I., Parys J. B., and Bultynck G. (2016) The trans-membrane domain of Bcl-2α, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget 7, 55704–55720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wagner L. E. 2nd, and Yule D. I. (2012) Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J. Physiol. 590, 3245–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Betzenhauser M. J., and Yule D. I. (2010) Regulation of inositol 1,4,5-trisphosphate receptors by phosphorylation and adenine nucleotides. Curr. Top. Membr. 66, 273–298 [DOI] [PubMed] [Google Scholar]
- 20. Betzenhauser M. J., Fike J. L., Wagner L. E. 2nd, and Yule D. I. (2009) Protein kinase A increases type-2 inositol 1,4,5-trisphosphate receptor activity by phosphorylation of serine 937. J. Biol. Chem. 284, 25116–25125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagner L. E. 2nd, Joseph S. K., and Yule D. I. (2008) Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J. Physiol. 586, 3577–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wagner L. E. 2nd, Li W. H., and Yule D. I. (2003) Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2- splice variants. J. Biol. Chem. 278, 45811–45817 [DOI] [PubMed] [Google Scholar]
- 23. Bánsághi S., Golenár T., Madesh M., Csordás G., Ramachandrarao S., Sharma K., Yule D. I., Joseph S. K., and Hajnóczky G. (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 289, 8170–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chandrasekhar R., Alzayady K. J., Wagner L. E. 2nd, and Yule D. I. (2016) Unique regulatory properties of heterotetrameric inositol 1,4,5-trisphosphate receptors revealed by studying concatenated receptor constructs. J. Biol. Chem. 291, 4846–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chandrasekhar R., Alzayady K. J., and Yule D. I. (2015) Using concatenated subunits to investigate the functional consequences of heterotetrameric inositol 1,4,5-trisphosphate receptors. Biochem. Soc. Trans. 43, 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirota J., Furuichi T., and Mikoshiba K. (1999) Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J. Biol. Chem. 274, 34433–34437 [DOI] [PubMed] [Google Scholar]
- 27. Kopil C. M., Vais H., Cheung K. H., Siebert A. P., Mak D. O., Foskett J. K., and Neumar R. W. (2011) Calpain-cleaved type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1) has InsP3-independent gating and disrupts intracellular Ca2+ homeostasis. J. Biol. Chem. 286, 35998–36010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diaz F., and Bourguignon L. Y. (2000) Selective down-regulation of IP3 receptor subtypes by caspases and calpain during TNFα-induced apoptosis of human T-lymphoma cells. Cell Calcium 27, 315–328 [DOI] [PubMed] [Google Scholar]
- 29. Verbert L., Lee B., Kocks S. L., Assefa Z., Parys J. B., Missiaen L., Callewaert G., Fissore R. A., De Smedt H., and Bultynck G. (2008) Caspase-3-truncated type 1 inositol 1,4,5-trisphosphate receptor enhances intracellular Ca2+ leak and disturbs Ca2+ signalling. Biol. Cell 100, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Assefa Z., Bultynck G., Szlufcik K., Nadif Kasri N., Vermassen E., Goris J., Missiaen L., Callewaert G., Parys J. B., and De Smedt H. (2004) Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J. Biol. Chem. 279, 43227–43236 [DOI] [PubMed] [Google Scholar]
- 31. Nakayama T., Hattori M., Uchida K., Nakamura T., Tateishi Y., Bannai H., Iwai M., Michikawa T., Inoue T., and Mikoshiba K. (2004) The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem. J. 377, 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akimzhanov A. M., Barral J. M., and Boehning D. (2013) Caspase 3 cleavage of the inositol 1,4,5-trisphosphate receptor does not contribute to apoptotic calcium release. Cell Calcium 53, 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alzayady K. J., Chandrasekhar R., and Yule D. I. (2013) Fragmented inositol 1,4,5-trisphosphate receptors retain tetrameric architecture and form functional Ca2+ release channels. J. Biol. Chem. 288, 11122–11134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khan M. T., Bhanumathy C. D., Schug Z. T., and Joseph S. K. (2007) Role of inositol 1,4,5-trisphosphate receptors in apoptosis in DT40 lymphocytes. J. Biol. Chem. 282, 32983–32990 [DOI] [PubMed] [Google Scholar]
- 35. Elkoreh G., Blais V., Béliveau E., Guillemette G., and Denault J. B. (2012) Type 1 inositol-1,4,5-trisphosphate receptor is a late substrate of caspases during apoptosis. J. Cell Biochem. 113, 2775–2784 [DOI] [PubMed] [Google Scholar]
- 36. Szlufcik K., Missiaen L., Parys J. B., Callewaert G., and De Smedt H. (2006) Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol. Cell 98, 1–14 [DOI] [PubMed] [Google Scholar]
- 37. Yoshikawa F., Iwasaki H., Michikawa T., Furuichi T., and Mikoshiba K. (1999) Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor: structural and functional coupling of cleaved ligand binding and channel domains. J. Biol. Chem. 274, 316–327 [DOI] [PubMed] [Google Scholar]
- 38. Bezprozvanny I. (2005) The inositol 1,4,5-trisphosphate receptors. Cell Calcium 38, 261–272 [DOI] [PubMed] [Google Scholar]
- 39. Joseph S. K., Pierson S., and Samanta S. (1995) Trypsin digestion of the inositol trisphosphate receptor: implications for the conformation and domain organization of the protein. Biochem. J. 307, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sugawara H., Kurosaki M., Takata M., and Kurosaki T. (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alzayady K. J., Wagner L. E. 2nd, Chandrasekhar R., Monteagudo A., Godiska R., Tall G. G., Joseph S. K., and Yule D. I. (2013) Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. J. Biol. Chem. 288, 29772–29784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bittremieux M., Gerasimenko J. V., Schuermans M., Luyten T., Stapleton E., Alzayady K. J., De Smedt H., Yule D. I., Mikoshiba K., Vangheluwe P., Gerasimenko O. V., Parys J. B., and Bultynck G. (2017) DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium 62, 60–70 [DOI] [PubMed] [Google Scholar]
- 43. Miyakawa T., Maeda A., Yamazawa T., Hirose K., Kurosaki T., and Iino M. (1999) Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wagner L. E. 2nd, Li W. H., Joseph S. K., and Yule D. I. (2004) Functional consequences of phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 279, 46242–46252 [DOI] [PubMed] [Google Scholar]
- 45. Li W., Llopis J., Whitney M., Zlokarnik G., and Tsien R. Y. (1998) Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392, 936–941 [DOI] [PubMed] [Google Scholar]
- 46. Won J. H., Cottrell W. J., Foster T. H., and Yule D. I. (2007) Ca2+ release dynamics in parotid and pancreatic exocrine acinar cells evoked by spatially limited flash photolysis. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G1166–G1177 [DOI] [PubMed] [Google Scholar]
- 47. Dolmetsch R. E., Xu K., and Lewis R. S. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 [DOI] [PubMed] [Google Scholar]
- 48. Smedler E., and Uhlén P. (2014) Frequency decoding of calcium oscillations. Biochim. Biophys. Acta 1840, 964–969 [DOI] [PubMed] [Google Scholar]
- 49. Joseph S. K., and Ryan S. V. (1993) Phosphorylation of the inositol trisphosphate receptor in isolated rat hepatocytes. J. Biol. Chem. 268, 23059–23065 [PubMed] [Google Scholar]
- 50. Fan G., Baker M. L., Wang Z., Baker M. R., Sinyagovskiy P. A., Chiu W., Ludtke S. J., and Serysheva I. I. (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sneyd J., Han J. M., Wang L., Chen J., Yang X., Tanimura A., Sanderson M. J., Kirk V., and Yule D. I. (2017) On the dynamical structure of calcium oscillations. Proc. Natl. Acad. Sci. U.S.A. 114, 1456–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tu H., Nosyreva E., Miyakawa T., Wang Z., Mizushima A., Iino M., and Bezprozvanny I. (2003) Functional and biochemical analysis of the type 1 inositol (1,4,5)-trisphosphate receptor calcium sensor. Biophys. J. 85, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miyakawa T., Mizushima A., Hirose K., Yamazawa T., Bezprozvanny I., Kurosaki T., and Iino M. (2001) Ca2+-sensor region of IP3 receptor controls intracellular Ca2+ signaling. EMBO J. 20, 1674–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M., Wong W., Kamen R., Tracey D., and Allen H. (1997) Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature 386, 619–623 [DOI] [PubMed] [Google Scholar]
- 55. Mariathasan S., Weiss D. S., Dixit V. M., and Monack D. M. (2005) Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lamkanfi M., Festjens N., Declercq W., Vanden Berghe T., and Vandenabeele P. (2007) Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 14, 44–55 [DOI] [PubMed] [Google Scholar]
- 57. Balcerzak D., Poussard S., Brustis J. J., Elamrani N., Soriano M., Cottin P., and Ducastaing A. (1995) An antisense oligodeoxyribonucleotide to m-calpain mRNA inhibits myoblast fusion. J. Cell Sci. 108, 2077–2082 [DOI] [PubMed] [Google Scholar]
- 58. Fujita J., Crane A. M., Souza M. K., Dejosez M., Kyba M., Flavell R. A., Thomson J. A., and Zwaka T. P. (2008) Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miossec C., Dutilleul V., Fassy F., and Diu-Hercend A. (1997) Evidence for CPP32 activation in the absence of apoptosis during T lymphocyte stimulation. J. Biol. Chem. 272, 13459–13462 [DOI] [PubMed] [Google Scholar]
- 60. Larsen B. D., Rampalli S., Burns L. E., Brunette S., Dilworth F. J., and Megeney L. A. (2010) Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. U.S.A. 107, 4230–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fernando P., Brunette S., and Megeney L. A. (2005) Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 19, 1671–1673 [DOI] [PubMed] [Google Scholar]
- 62. Cathelin S., Rébé C., Haddaoui L., Simioni N., Verdier F., Fontenay M., Launay S., Mayeux P., and Solary E. (2006) Identification of proteins cleaved downstream of caspase activation in monocytes undergoing macrophage differentiation. J. Biol. Chem. 281, 17779–17788 [DOI] [PubMed] [Google Scholar]
- 63. Wojcikiewicz R. J., Ernst S. A., and Yule D. I. (1999) Secretagogues cause ubiquitination and down-regulation of inositol 1,4,5-trisphosphate receptors in rat pancreatic acinar cells. Gastroenterology 116, 1194–1201 [DOI] [PubMed] [Google Scholar]
- 64. Betzenhauser M. J., Wagner L. E. 2nd, Park H. S., and Yule D. I. (2009) ATP regulation of type-1 inositol 1,4,5-trisphosphate receptor activity does not require walker A-type ATP-binding motifs. J. Biol. Chem. 284, 16156–16163 [DOI] [PMC free article] [PubMed] [Google Scholar]