

Abstract
The pyruvate dehydrogenase complex (PDC) is the primary metabolic checkpoint connecting glycolysis and mitochondrial oxidative phosphorylation and is important for maintaining cellular and organismal glucose homeostasis. Phosphorylation of the PDC E1 subunit was identified as a key inhibitory modification in bovine tissue ∼50 years ago, and this regulatory process is now known to be conserved throughout evolution. Although Saccharomyces cerevisiae is a pervasive model organism for investigating cellular metabolism and its regulation by signaling processes, the phosphatase(s) responsible for activating the PDC in S. cerevisiae has not been conclusively defined. Here, using comparative mitochondrial phosphoproteomics, analyses of protein–protein interactions by affinity enrichment–mass spectrometry, and in vitro biochemistry, we define Ptc6p as the primary PDC phosphatase in S. cerevisiae. Our analyses further suggest additional substrates for related S. cerevisiae phosphatases and describe the overall phosphoproteomic changes that accompany mitochondrial respiratory dysfunction. In summary, our quantitative proteomics and biochemical analyses have identified Ptc6p as the primary—and likely sole—S. cerevisiae PDC phosphatase, closing a key knowledge gap about the regulation of yeast mitochondrial metabolism. Our findings highlight the power of integrative omics and biochemical analyses for annotating the functions of poorly characterized signaling proteins.
Keywords: mitochondria, phosphoproteomics, phosphorylation, protein phosphatase 2C (PP2C), pyruvate dehydrogenase complex (PDC), Saccharomyces cerevisiae, Gpd1p, Ptc5p, Ptc6p, Ptc7p
Introduction
Cells must to adapt to frequently changing nutritional and environmental conditions to maintain metabolic homeostasis. Post-translational modifications (PTMs)2 are essential cellular “tools” for enabling this metabolic adaptation across short- and long-term time scales. Recent studies have revealed that mitochondrial proteins are replete with PTMs, including phosphorylation (1), acetylation (2), and nitrosylation (3), suggesting that these modifications may play prominent roles in regulating mitochondrial activities. However, it has been challenging to distinguish mitochondrial PTMs that occur in a non-enzymatic fashion, such as certain autophosphorylation and acylation reactions (4, 5), from those that are regulated enzymatically. Moreover, for the latter case, it has proven difficult to pinpoint the specific enzymes (e.g. kinases and phosphatases) responsible for this regulation.
The first example of a regulatory mitochondrial PTM, discovered approximately 50 years ago (6), is the reversible phosphorylation on the E1α subunit of the bovine pyruvate dehydrogenase complex (PDC)—the three-enzyme complex in the mitochondrial matrix that converts pyruvate into acetyl-CoA, thereby linking glycolysis to mitochondrial functions including the TCA cycle, mitochondrial respiration, and fatty acid biosynthesis. The mammalian PDC has three phosphosites (7) that are collectively regulated by four kinase isoenzymes (PDK1–4) (8, 9) and two phosphatase isozymes (PDP1 and PDP2) (10) that differ in their tissue distribution and regulatory characteristics (11). Tracking the altered expression of these enzymes has lent important insight into how and when PDC function is regulated or dysregulated, such as the increased expression of PDK1 during hypoxia (12, 13), the suppression of PDK1 expression accompanied with induction of PDP2 in oncogene-induced senescence (14), and the decreased expression of PDP1 during cardiomyocyte differentiation (15) and in obesity (16).
Saccharomyces cerevisiae is a prominent model organism for diverse areas of biological research. Thanks to its genetic tractability, its short generation time, its conserved genome, and the vast arsenal of available biological research tools, S. cerevisiae has emerged as a dependable model system even for complex human diseases and processes (17), including neurodegeneration and aging (18, 19). S. cerevisiae has also been the focus of major efforts to reconstruct a comprehensive cellular metabolic network (20), and to explore the importance of PTMs in regulating metabolism (21). Despite this, our understanding of PDC regulation in S. cerevisiae surprisingly lags behind that of the mammalian PDC. In particular, despite significant recent progress (22, 23), the identity of the S. cerevisiae PDC phosphatase(s) has not been fully established.
Here, we performed integrative proteomics and biochemical analyses to define Ptc6p as the primary PDC phosphatase. Our data also suggest potential direct targets/physiological roles for Ptc5p and Ptc7p and reveal the dynamic mitochondrial protein phosphorylation changes that accompany respiratory dysfunction in S. cerevisiae.
Results
Primary sequence analyses of PDPs and S. cerevisiae PP2Cs
The mammalian PDC has three phosphosites on its E1α subunit: Ser-264, Ser-271, and Ser-203 (corresponding to the mature human sequence), all of which are inhibitory (7, 24, 25). One of these sites—Ser-264, which has the highest phosphorylation occupancy and is phosphorylated most rapidly (7, 26)—is conserved in S. cerevisiae (Ser-313). The kinases responsible for the phosphorylation of these residues in mammals (PDK1–4) (8, 9) and in S. cerevisiae (Pkp1p,2p) (22, 23) have been defined, as have the phosphatases in the mammalian system (PDP1,2) (10). In contrast, the S. cerevisiae phosphatase(s) is not fully established (Fig. 1).
Figure 1.

Current model of PDC regulation by reversible phosphorylation. Activity of the PDC is regulated by reversible phosphorylation, but the phosphatase that dephosphorylates the S. cerevisiae PDC remains unclear.
Given the overall conservation of the PDC regulatory system between mammals and S. cerevisiae, we first attempted to identify the most likely candidate(s) for the S. cerevisiae PDC phosphatase(s) via primary sequence analyses. The mammalian PDC phosphatases belong to the metal-dependent serine/threonine protein phosphatase 2C (PP2C) family. S. cerevisiae has seven PP2C family members (Ptc1p–7p), three of which have been localized to mitochondria (Fig. 2A). A phylogenetic analysis of S. cerevisiae PP2C phosphatases (Fig. 2A and supplemental Fig. S1) with PDPs from diverse species did not reveal a clear PDP ortholog. The three S. cerevisiae mitochondrial phosphatases were more closely related to human PDP1 or PDP2 than were the others, with Ptc5p and Ptc6p having the highest homology. However, these phosphatases share only modest sequence identity with each other and with human PDP1 or PDP2 (Fig. 2B). Importantly, both Ptc5p and Ptc6p have been implicated as PDC phosphatases in S. cerevisiae (22, 23); however, whether either or both of these phosphatases act directly on the PDC or instead affect PDC activity indirectly remains unclear. As such, the bona fide PDC phosphatase(s) has not been conclusively defined and cannot be established confidently via primary sequence analyses.
Figure 2.

Primary sequence analyses of PDPs and S. cerevisiae PP2Cs. A, phylogenetic tree of vertebrate PDP1 and PDP2, human PPM1A, and seven S. cerevisiae PP2C phosphatases. The blue region highlights homologs of PDP2, and the green region highlights homologs of PDP1. B, chart showing the percentage similarity and identity between human PPM1A, PDP1, PDP2, and seven S. cerevisiae PP2C phosphatases.
Comparative mitochondrial phosphoproteomic analyses in Δptc yeast strains
To begin identifying potential S. cerevisiae PDC phosphatase(s) experimentally, we performed a quantitative phosphoproteomics analysis of WT and KO strains (Fig. 3A and supplemental Tables S1 and S2). We focused on strains lacking Ptc5p, Ptc6p, and Ptc7p because of their mitochondrial localization (22, 27, 28). Additionally, because deletion of each of these phosphatases causes varying levels of respiratory dysfunction, we analyzed strains lacking Coq8p and Coq9p—proteins essential for mitochondrial coenzyme Q (CoQ) production, and thus respiratory competency—as controls (29, 30). These experiments were designed to identify potential substrates, including Pda1p (the S. cerevisiae PDC E1α subunit), for three mitochondrial phosphatases, and to assess the overall phosphoproteomics changes that accompany general mitochondrial respiratory incompetency.
Figure 3.

Comparative mitochondrial phosphoproteomic analyses of Δptc yeast strains. A, experimental phosphoproteomics workflow. Crude mitochondria from six yeast strains (WT, Δptc5, Δptc6, Δptc7, Δcoq8, and Δcoq9) were used. Peptides were tagged with 6-plex TMT for MS-based quantification. The red and white boxes in the Phospho Enrichment panel represent the reporter and balancing groups of the TMT, respectively. B, hierarchical clusters of Δgene yeast strains and all quantified mitochondrial phosphoisoforms. C–E, fold changes of mitochondrial phosphoisoforms with values larger than 1.5 (log2 (Δgene/WT)) from Δptc5 (C), Δptc6 (D), and Δptc7 (E). The red arrowheads in D denote Pda1p phosphoisoforms.
All yeast strains were grown in rich medium containing 2% glucose (w/v) until 4 h past the diauxic shift. At this point, yeast transition from a fermentative metabolism to mitochondria-dependent respiratory metabolism. A crude mitochondrial enrichment was performed, and 6-plex tandem mass tags (TMTs) were used for proteomic and phosphoproteomic quantification. Phosphoisoform fold changes were normalized to the corresponding protein fold change. Overall, we quantified 3184 proteins, including 823 mitochondrial proteins (annotated using the Yeast MitoMiner database (31)), and 2335 phosphoisoforms (267 mitochondrial) (supplemental Table S1 and S2). All KO strains exhibited the expected abundance decrease of the proteins encoded by the deleted genes (supplemental Fig. S2, A–E). Unsupervised hierarchical clustering of the fold changes for both mitochondrial phosphoisoforms and proteins showed distinct profiles for each phosphatase KO strain (Fig. 3B and supplemental Fig. S2F). Consistent with their shared function, strains lacking Coq8p and Coq9p generated similar proteomic and phosphoproteomic profiles (Fig. 3B and supplemental Fig. S2F). Additionally, the abundance of Coq9p was diminished in the Δcoq8 strain (supplemental Fig. S2E), consistent with previous reports that these proteins exist in a biosynthesis complex (complex Q) (32, 33). Together, these data argue against Ptc5p, Ptc6p, or Ptc7p possessing largely redundant functions, unlike mammalian PDP1 and PDP2.
To identify potential direct substrates for each phosphatase, we analyzed the individual phosphoproteomic changes in each strain, with the hypothesis that deletion of a phosphatase would cause an increased phosphorylation level of its substrate(s). Consistent with prior studies (34), we used a fold change cutoff of 1.5 (50%; log2 fold change of 0.585) to define increased phosphorylation. Based on this criterion and filtering for proteins in the Yeast MitoMiner database, Ptc5p, Ptc6p, and Ptc7p had 25, 5, and 68 hits, respectively (Fig. 3, C–E, and supplemental Table S1). Surprisingly, despite having the fewest overall changes, only the Δptc6 strain exhibited a marked increase in Pda1p Ser-313 phosphorylation (Fig. 3D).
Based on our phosphoproteome data, we propose that Ptc5p possesses in vivo function(s) distinct from direct PDC regulation. For example, phosphorylation of Ser-25 and/or Ser-27 and Ser-24 on Gpd1p are increased in Δptc5 yeast (Fig. 3C). Gpd1p is one of the two glycerol-3-phosphate dehydrogenase isoforms in S. cerevisiae (35), and its activity is regulated by the kinases Ypk1p and Ypk2p (36); however, the corresponding phosphatase(s) remains unknown. Gpd1p is found in the cytosol and peroxisomes (37) but has been localized to mitochondria by high-throughput studies (38). Thus, it is possible that Ptc5p is responsible for dephosphorylating the mitochondrial fraction of Gpd1p.
Of course, it is likely that some elevated phosphoisoforms across our data set represent downstream compensatory changes instead of being direct targets of these phosphatases, such as Thr-31 on Gph1p in Δptc6 (Fig. 3D). Gph1p is the S. cerevisiae glycogen phosphorylase, which catalyzes the breakdown of glycogen into glucose subunits for glycolysis in the cytoplasm, and is activated by phosphorylation of Thr-31 (39). In Δptc6 yeast, the enhanced phosphorylation of Pda1p could slow entry of pyruvate-derived acetyl-CoA into the TCA cycle, thereby causing a compensatory increase in glycolytic flux for ATP generation by increasing the phosphorylation of Gph1p. These data lend insight into the specific metabolic adaptations of each KO strain and demonstrate that additional experiments are needed to distinguish direct substrates from indirect adaptive changes.
Affinity enrichment–mass spectrometry (AE-MS) analyses directly link Ptc6p to the PDC
To help distinguish direct phosphatase substrates from general adaptive changes, we next performed AE-MS. AE-MS is an approach to identify protein–protein interactions (PPIs) across multiple “bait” proteins that combines quantitative MS with a scoring algorithm (CompPASS (40)) to differentiate between informative interactions and nonspecific background co-enrichment (33).
We began our AE-MS analyses by immunoprecipitating each bait protein (Ptc5p, Ptc6p, Ptc7p, Coq8p, and Coq9p), which had been modified to include a C-terminal FLAG tag (Fig. 4A). The resulting protein eluate was analyzed using nanoflow liquid chromatography coupled to high-resolution MS (Q Exactive Hybrid Quadrupole-Orbitrap). Collectively, analysis of these five baits yielded 118 high-confidence mitochondrial PPIs from among 1750 total PPIs identified. These included interactions between our Cop9p control and seven other proteins involved in CoQ biosynthesis (Coq3p, Coq4p, Coq5p, Coq6p, Cat5p (Coq7p), Coq8p, and Coq11p (Ylr290c)) (supplemental Table S3 and Fig. S3B). These observations are consistent with recent reports that CoQ-related proteins interact to form a biosynthetic complex (complex Q) (32, 33), thereby validating the robustness of our approach.
Figure 4.

AE-MS analyses directly link Ptc6p to the PDC. A, experimental workflow for AE-MS analyses. Six baits were used: empty vector (EV), Ptc5p, Ptc6p, Ptc7p, Coq8p, and Coq9p. Eluates from immunoprecipitation were analyzed by LC-MS/MS, and interactions were scored using the CompPASS (WD) scoring algorithm. B–D, mitochondrial interaction networks of Ptc5p (B), Ptc6p (C), and Ptc7p (D). Only mitochondrial protein interactors are shown here. Line width correlates with the number of PSMs identified for each interactor. The shading in C indicates the Ptc6p interactions with PDC-related proteins. E, overlapping hits of mitochondrial interactors and mitochondrial proteins with phosphoisoforms that increased by ≥1.5-fold.
Our data revealed distinct interaction networks for Ptc5p, Ptc6p, and Ptc7p (Fig. 4, B–D, and supplemental Table S3), further suggesting that these phosphatases perform non-overlapping functions in vivo. Although Ptc5p and Ptc7p each exhibited a fairly large number of mitochondrial PPIs (∼30), perhaps suggesting more pervasive roles in mitochondrial phosphoprotein regulation, Ptc6p exhibited a minimal interaction network of only eight mitochondrial proteins. Strikingly, four of these eight are subunits of the PDC, including Pda1p, Lat1p (dihydrolipoamide acetyltransferase, the PDC E2 subunit), Pdb1p (the E1β subunit), and Pdx1p (an E3-binding protein) (Fig. 4C). No PDC subunits were observed in the larger Ptc5p and Ptc7p networks (Fig. 4, B and D).
To further home in on potential direct substrates for these phosphatases, we integrated our phosphoproteomics data with our AE-MS data (Fig. 4D). Phosphatases might only exhibit low-energy, fleeting interactions with their substrate(s), which may not be readily detectible by our PPI analysis method. Nonetheless, we reasoned that proteins whose phosphorylation levels increased following KO of a phosphatase and that also physically interacted with that phosphatase, would likely be direct substrates. Although Ptc6p had very few hits in either category, Pda1p stood out in both analyses (Fig. 4E). Ptc5p had no overlapping targets, whereas Ptc7p had three, albeit with lower interaction scores than that for Ptc6p-Pda1p (Fig. 4D and supplemental Table S3). Collectively, these analyses implicate each phosphatase in diverse mitochondrial activities and strongly suggest that Pda1p is a direct substrate of Ptc6p.
In vitro biochemical assays verify Ptc6p as a Pda1p phosphatase
We next performed a series of biochemical experiments to validate the results from our large-scale analyses and to further test the hypothesis that Ptc6p is the primary PDC phosphatase. First, using a phospho-specific antibody against Pda1p Ser-313 (supplemental Fig. S4A), we performed immunoblots as an orthogonal approach to assess the relative change in this phosphoisoform between WT and each of our five KO strains. Consistent with our MS analyses, the Δptc6 strain exhibited a marked increase in Pda1p Ser-313 phosphorylation, whereas the Δptc5 and Δptc7 strains showed little difference from WT (Fig. 5A and supplemental Fig. S4, A and B). Notably, our immunoblotting results suggest an even larger increase in Pda1p Ser-313 phosphorylation (∼3-fold change) in Δptc6 yeast than was estimated by our MS analyses (supplemental Fig. S4B), potentially because of dynamic range suppression—a known caveat of MS isobaric tagging methods that can cause an underestimation of fold-change values (41).
Figure 5.
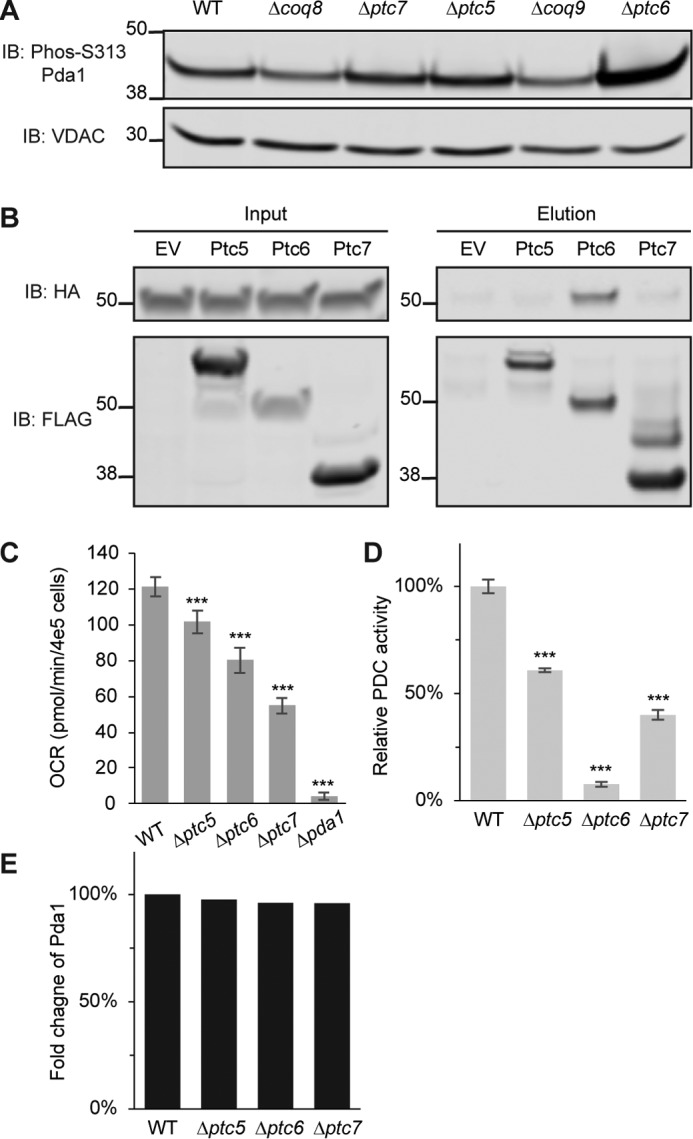
In vitro biochemical assays verify Ptc6p as a Pda1p phosphatase. A, immunoblot (IB) of phosphorylated Pda1p on Ser-313 or voltage-dependent anion channel (VDAC) (loading control). The samples are mitochondrial lysates from WT, Δcoq8, Δptc7, Δptc5, Δcoq9, and Δptc6 yeast. B, immunoblot of HA (Pda1p was endogenously tagged with HA) or FLAG (bait proteins were C-terminally FLAG-tagged). Samples in the left panels are input lysates for the anti-FLAG immunoprecipitations from WT yeast strains overexpressing empty vector (EV), Ptc5p, Ptc6p, and Ptc7p. Samples in the right panels are the corresponding eluates from the same immunoprecipitations. C, oxygen consumption rates (OCR) of WT, Δptc5, Δptc6, Δptc7, and Δpda1 in synthetic complete medium containing 2% pyruvate (mean ± S.D., n = 6). D, relative PDC activity of mitochondrial lysates from WT, Δptc5, Δptc6, and Δptc7 (mean ± S.D., n = 3). E, fold change of Pda1p protein quantified by MS from same samples in D (normalized to WT). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Next, to further test the interaction between Ptc6p and Pda1p identified by AE-MS, we immunoprecipitated each FLAG-tagged phosphatase and blotted directly for Pda1p. Despite comparable immunoprecipitation efficiencies, Pda1p again only stood out as an interacting protein of Ptc6p (Fig. 5B). These data confirm the in vivo physical interaction between Ptc6p and the PDC, likely via its direct interaction with Pda1p (Fig. 5B).
All three phosphatase deletion strains exhibited lower oxygen consumption rates than WT when grown on pyruvate-based medium, with Δptc6 less able to utilize pyruvate for oxygen consumption than Δptc5 (Fig. 5C). However, this does not directly reveal the relative contribution of PDC inhibition to the growth deficiency of each strain. Phosphorylation of Pda1p is an inhibitory modification (42); as such, the relative changes in Pda1p phosphorylation generally should be anti-correlated with PDC activity. To test whether this indeed holds true across our strains, we measured PDC activity in mitochondrial lysates from WT, Δptc5, Δptc6, and Δptc7 yeast. The Δptc6 strain possessed very low PDC activity (less than 10% compared with WT; Fig. 5D), suggesting that PDC inhibition plays a major role in the compromised oxygen consumption of this strain on pyruvate-based medium. The Δptc5 and Δptc7 strains also showed a more modest decrease in PDC activity (∼50% of WT). This decrease is not surprising both because the Δptc5 and Δptc7 strains exhibited a slightly increased phosphorylation level of Pda1p Ser-313 by immunoblot or MS analyses (supplemental Table S1 and Fig. S4B) and because the loss of these phosphatases is known to cause general mitochondrial dysfunction, which can lead to PDC inactivation by other means. Importantly, the protein abundance of Pda1p did not change appreciably across these four strains based on our proteomics analysis (Fig. 5E).
Finally, to test the ability of Ptcp5, Ptc6p, and Ptc7p to dephosphorylate Pda1p more directly, we purified recombinant versions of each phosphatase from Escherichia coli, along with Hfd1p (a fatty aldehyde dehydrogenase involved in CoQ biosynthesis that served as a negative control) (43) (supplemental Fig. S4C). We then incubated equal amounts of each protein with mitochondrial lysate from Δptc6 (where Pda1p phosphorylation is highest) and analyzed Pda1p Ser-313 phosphorylation at different time points. Only Ptc6p was able to dephosphorylate Pda1p in vitro (Fig. 6A), despite the fact that all three purified phosphatases were able to dephosphorylate para-nitrophenyl phosphate (pNPP), albeit with differing rates (Fig. 6B). Last, we measured the PDC activity in these lysates at the 30-min incubation time point and found, consistently, that the Ptc6p-treated lysates clearly had the highest PDC activity (Fig. 6C).
Figure 6.
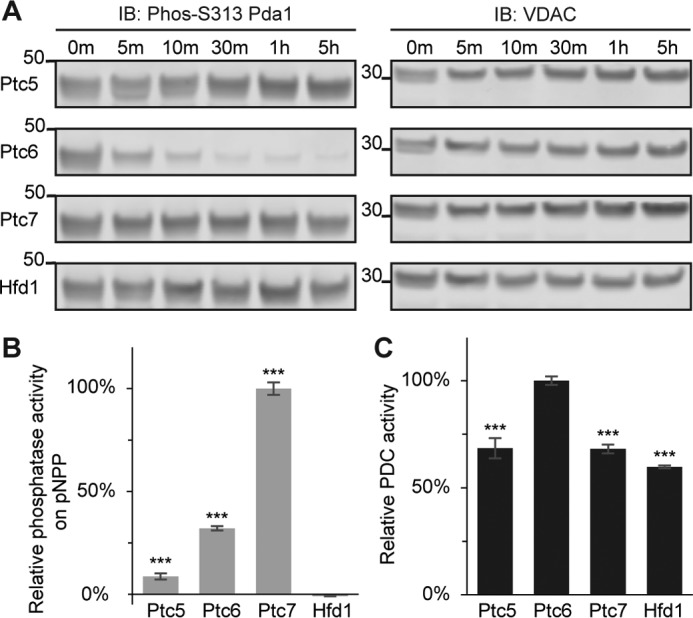
Only Ptc6p dephosphorylates and activates Pda1p in vitro. A, immunoblots (IB) of phosphorylated Pda1p on Ser-313 (S313, left panels) or voltage-dependent anion channel (VDAC) (loading control, right panels). Mitochondrial lysate from Δptc6 was treated with recombinant Ptc5p, Ptc6p, Ptc7p, or Hfd1p. Samples from six different time points were taken and analyzed by immunoblotting. B, relative in vitro phosphatase activity against pNPP for recombinant Ptc5p, Ptc6p, Ptc7p, or Hfd1p (mean ± S.D., n = 3). The activity is normalized to total protein amount. C, relative PDC activity of mitochondrial lysates from Δptc6 yeast after 30 min of treatment with recombinant Ptc5p, Ptc6p, Ptc7p, or Hfd1p (mean ± S.D., n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Collectively, our integration of quantitative MS-based comparative phosphoproteomics with AE-MS assessment of PPIs and in vitro biochemical analyses reveals that Ptc6p is the primary—and likely sole—S. cerevisiae PDC phosphatase. Our data point to distinct, non-overlapping functions for the closely related mitochondrial phosphatases Ptc5p and Ptc7p. Finally, or work provides a prioritized list of direct substrates for each phosphatase and describes the overall phosphoproteomic changes that accompany mitochondrial respiratory dysfunction.
Discussion
Metabolic processes are under exquisite control by signaling pathways that use PTMs, such as phosphorylation, to alter the function of target proteins. However, defining how a given signaling pathway imparts such post-transcriptional control of a metabolic event is difficult and demands that at least three parameters be established: 1) the identification of the PTMs themselves, 2) the determination of the functional consequence(s) of the modification(s), and 3) the identification of the regulatory enzymes (e.g. kinases and phosphatases) that control the PTM abundance. Recent advances in MS-based analyses have rapidly accelerated the first of these challenges (44), but the latter two remain difficult and laborious.
The mammalian mitochondrial PDC is a classic metabolic target of reversible phosphorylation. In this case, the phosphosites (Ser-264, Ser-271, and Ser-203) (7, 24), the regulatory effect (inhibition) (25), and the regulatory machinery (PDK1-4; PDP1,2) (8–10) have all been established. Control of PDC activity is essential for regulating the entry of glycolysis-derived pyruvate into the TCA cycle and the overall maintenance of cellular and organismal glucose homeostasis. Dysfunction of the core PDC or the phosphatases that regulate it leads to pyruvate dehydrogenase deficiency (45, 46). Additionally, dichloroacetate, a small molecule inhibitor of PDK, has been explored as a potential therapy for cancer (47). More broadly, understanding when PDC kinases and phosphatases are induced or repressed lends important insight into the metabolic state of a cellular system.
S. cerevisiae has emerged as an important model organism for studying basic metabolism and its regulation by signaling processes, including the PDC system. Despite this, surprisingly, the identity of the S. cerevisiae PDC phosphatase has not been clearly established. Recent work by Krause-Buchholz and co-workers (22, 23) linked two of the S. cerevisiae PP2C-type phosphatases, Ptc5p and Ptc6p, to PDC function. In these studies, the authors demonstrated that loss of either enzyme resulted in decreased PDC activity concomitant with elevated PDC phosphorylation and that each phosphatase potentially associated with Pda1p. However, direct evidence of their ability to dephosphorylate PDC and their physical interaction with Pda1p were lacking, and the sequence divergence between these phosphatases suggests that they are unlikely to perform redundant functions. As such, we decided to revisit whether one of these phosphatases—or a distinct, but related phosphatase—serves as the primary PDC phosphatase.
Through a combination of comparative phosphoproteomics, AE-MS, and in vitro biochemistry, we demonstrated that Ptc6p is the primary PDC phosphatase in S. cerevisiae and suggest orthogonal functions for the related PP2C phosphatases Ptc5p and Ptc7p. Ptc5p exhibited a distinct set of potential substrates, including a possible role in regulating mitochondrial Gpd1p. Unique substrates likewise emerged for Ptc7p, including Yml6p, which was a top substrate candidate in our recent analysis (48). Notably, some different potential substrates for Ptc7p were identified between this study and our previous study (48), in which the growth conditions of the yeast were different. This suggests that it is still possible for Ptc5p to work directly on the PDC under a different metabolic state. However, given our data, we believe that a more auxiliary role for Ptc5p in PDC function—such as in complex assembly, as previously suggested (23)—is more likely.
Overall, our quantitative proteomics and biochemical analyses have built upon recent work (22) to help establish Ptc6p as the S. cerevisiae PDC phosphatase, thereby filling a key gap in knowledge regarding the regulation of yeast mitochondrial metabolism. More generally, our work reveals the power of integrative omics and biochemical analyses for annotating the functions of poorly characterized signaling proteins.
Experimental procedures
Yeast strain used in this study
WT, Δptc5, Δptc6, Δptc7, Δcoq8, and Δcoq9 BY4741 yeast (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) were purchased from Open Biosystems. The identity of each KO strain was verified by PCR. The endogenously three tandem hemagglutinin (3×HA)-tagged pda1 strain was made as previously described (49).
Quantitative proteomics and phosphoproteomics analyses
Briefly, crude mitochondria were lysed, digested, TMT-labeled, and fractionated by strong cation-exchange chromatography (1). Phosphopeptide enrichment was done with immobilized metal-affinity chromatography. All samples were analyzed by reverse phase liquid chromatography on an EASY-nLC system (Thermo) coupled to a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometry (Thermo). A customized software Coon OMSSA Proteomics Software Suite (COMPASS) was used to analyze the MS raw data.
Protein interaction study
C-terminal FLAG-tagged protein constructs in p416 GPD plasmid were transformed into WT BY4741 yeast. Yeast cells were harvested and lysed. The clarified supernatant was incubated with anti-FLAG M2 magnetic beads (Sigma) followed with five washes and eluted with FLAG peptide. The samples were subjected to MS analysis on a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo). CompPASS (WD) scoring was used to filter interaction partners (33, 40). For immunoblot verification of the Ptc6p and Pda1p interaction, an endogenously 3×HA-tagged pda1 strain was transformed with an empty vector or C-terminal FLAG-tagged protein construct (ptc5, ptc6, ptc7, coq8, and coq9) in p416 GPD plasmid. Clarified cell lysate (input) and final elution were collected and used for immunoblotting.
PDC activity assay
Purified mitochondria were lysed, clarified, and quantified via bicinchoninic acid assay. The PDC activity assay was done in a 96-well plate containing 50 μl of reaction buffer (150 mm MOPS, 3 mm MgCl2, 0.03 mm CaCl2, pH 8.0), 20 μl of 3 mm thiamine pyrophosphate, 20 μl of 25 mm NAD+, 20 μl of 38 mm cysteine, pH 7.4, 20 μl of 50 mm sodium pyruvate, and 50 μl of 0.5g/liter mitochondrial lysate (50). 20 μl of 1.5 mm coenzyme A sodium salt was added just before starting the assay. A340 nm was monitored to calculate the NADH production rate.
In vitro phosphatase assays
In vitro pNPP phosphatase assays were done in a 96-well plate. 30 μl of 50 ng/μl protein was added to 150 μl of reaction buffer (66.7 mm Tris, 13.3 mm MnCl2, pH 8.0). The reaction was initiated by the addition of 20 μl of 500 mm pNPP, and A405 nm was recorded over time. The initial slope was calculated to represent the reaction rate. The relative phosphatase activity is normalized to the total protein amount. For in vitro phosphatase assays on Pda1p, purified mitochondria from Δptc6 yeast were lysed. 200 μl of 100 mm MnCl2 and 0.2 μl of 100 mm CaCl2 were added into each aliquot of clarified supernatant before the addition of 60 μl of 50 ng/μl recombinant protein. The mixture was incubated at room temperature. 20 μl of sample was taken out at different time points, mixed with 4× sample buffer, and boiled at 95 °C for 5 min before Western blotting. A separate aliquot of 10 μl of sample after 30 min of treatment was used for the PDC activity assay.
Protein alignment and generation of phylogenetic tree
Protein sequences were downloaded from Uniprot (http://www.uniprot.org/) or NCBI (http://www.ncbi.nlm.nih.gov/). Protein alignment and phylogeny were done using MacVector. ClustalW algorithm with default settings was chosen for protein alignment.
Statistics
The p values were calculated by a two-tailed Student's t test.
Author contributions
X. G. and D. J. P. conceived of the project and its design, analyzed data, and wrote the manuscript. X. G. and N. M. N. performed all experiments. J. J. C. provided key resources and experimental infrastructure.
Supplementary Material
Acknowledgments
We thank the members of the Pagliarini and Coon laboratories for helpful discussions and technical support, including Brendan Floyd, Jon Stefely, Adam Jochem, Greg Potts, Arne Ulbrich, Catie Minogue, and Emily Wilkerson.
This work was supported by National Institutes of Health Grants R01DK098672 (to D. J. P.) and R35GM118110 (to J. J. C.) and a United Mitochondrial Disease Foundation (UMDF) grant (to N. M. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental text, Tables S1–S3, and Figs. S1–S4.
- PTM
- post-translational modification
- PDC
- pyruvate dehydrogenase complex
- PPI
- protein–protein interaction
- PP2C
- protein phosphatase 2C
- CoQ
- coenzyme Q
- TMT
- tandem mass tags
- AE-MS
- affinity-enrichment mass spectrometry
- pNPP
- para-nitrophenyl phosphate.
References
- 1. Grimsrud P. A., Carson J. J., Hebert A. S., Hubler S. L., Niemi N. M., Bailey D. J., Jochem A., Stapleton D. S., Keller M. P., Westphall M. S., Yandell B. S., Attie A. D., Coon J. J., and Pagliarini D. J. (2012) A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 16, 672–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He W., Newman J. C., Wang M. Z., Ho L., and Verdin E. (2012) Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 23, 467–476 [DOI] [PubMed] [Google Scholar]
- 3. Doulias P.-T., Tenopoulou M., Greene J. L., Raju K., and Ischiropoulos H. (2013) Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 6, rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Phillips D., Aponte A. M., Covian R., and Balaban R. S. (2011) Intrinsic protein kinase activity in mitochondrial oxidative phosphorylation complexes. Biochemistry 50, 2515–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wagner G. R., and Payne R. M. (2013) Widespread and enzyme-independent Nϵ-acetylation and Nϵ-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Linn T. C., Pettit F. H., and Reed L. J. (1969) α-Keto acid dehydrogenase complexes: X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. U.S.A. 62, 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yeaman S. J., Hutcheson E. T., Roche T. E., Pettit F. H., Brown J. R., Reed L. J., Watson D. C., and Dixon G. H. (1978) Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 17, 2364–2370 [DOI] [PubMed] [Google Scholar]
- 8. Gudi R., Bowker-Kinley M. M., Kedishvili N. Y., Zhao Y., and Popov K. M. (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J. Biol. Chem. 270, 28989–28994 [DOI] [PubMed] [Google Scholar]
- 9. Rowles J., Scherer S. W., Xi T., Majer M., Nickle D. C., Rommens J. M., Popov K. M., Harris R. A., Riebow N. L., Xia J., Tsui L.-C., Bogardus C., and Prochazka M. (1996) Cloning and characterization of PDK4 on 7q21. 3 encoding a fourth pyruvate dehydrogenase kinase isoenzyme in human. J. Biol. Chem. 271, 22376–22382 [DOI] [PubMed] [Google Scholar]
- 10. Huang B., Gudi R., Wu P., Harris R. A., Hamilton J., and Popov K. M. (1998) Isoenzymes of pyruvate dehydrogenase phosphatase DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 273, 17680–17688 [DOI] [PubMed] [Google Scholar]
- 11. Holness M. J., and Sugden M. C. (2003) Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 31, 1143–1151 [DOI] [PubMed] [Google Scholar]
- 12. Kim J.-W., Tchernyshyov I., Semenza G. L., and Dang C. V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 [DOI] [PubMed] [Google Scholar]
- 13. Papandreou I., Cairns R. A., Fontana L., Lim A. L., and Denko N. C. (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 [DOI] [PubMed] [Google Scholar]
- 14. Kaplon J., Zheng L., Meissl K., Chaneton B., Selivanov V. A., Mackay G., van der Burg S. H., Verdegaal E. M., Cascante M., Shlomi T., Gottlieb E., and Peeper D. S. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112 [DOI] [PubMed] [Google Scholar]
- 15. Heo H. J., Kim H. K., Youm J. B., Cho S. W., Song I.-S., Lee S. Y., Ko T. H., Kim N., Ko K. S., Rhee B. D., and Han J. (2016) Mitochondrial pyruvate dehydrogenase phosphatase 1 regulates the early differentiation of cardiomyocytes from mouse embryonic stem cells. Exp. Mol. Med. 48, e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piccinini M., Mostert M., Alberto G., Ramondetti C., Novi R. F., Dalmasso P., and Rinaudo M. T. (2005) Down-regulation of pyruvate dehydrogenase phosphatase in obese subjects is a defect that signals insulin resistance. Obes. Res. 13, 678–686 [DOI] [PubMed] [Google Scholar]
- 17. Smith M. G., and Snyder M. (2006) Yeast as a model for human disease. Curr. Protoc. Hum. Genet. Chapter 15, Unit 15.16 [DOI] [PubMed] [Google Scholar]
- 18. Khurana V., and Lindquist S. (2010) Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat. Rev. Neurosci. 11, 436–449 [DOI] [PubMed] [Google Scholar]
- 19. Longo V. D., Shadel G. S., Kaeberlein M., and Kennedy B. (2012) Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 16, 18–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Förster J., Famili I., Fu P., Palsson B. Ø., Nielsen J. (2003) Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 13, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oliveira A. P., and Sauer U. (2012) The importance of post-translational modifications in regulating Saccharomyces cerevisiae metabolism. FEMS Yeast Res. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 22. Gey U., Czupalla C., Hoflack B., Rödel G., and Krause-Buchholz U. (2008) Yeast pyruvate dehydrogenase complex is regulated by a concerted activity of two kinases and two phosphatases. J. Biol. Chem. 283, 9759–9767 [DOI] [PubMed] [Google Scholar]
- 23. Krause-Buchholz U., Gey U., Wünschmann J., Becker S., and Rödel G. (2006) YIL042c and YOR090c encode the kinase and phosphatase of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. FEBS Lett. 580, 2553–2560 [DOI] [PubMed] [Google Scholar]
- 24. Dahl H. H., Hunt S. M., Hutchison W. M., and Brown G. K. (1987) The human pyruvate dehydrogenase complex. Isolation of cDNA clones for the E1 alpha subunit, sequence analysis, and characterization of the mRNA. J. Biol. Chem. 262, 7398–7403 [PubMed] [Google Scholar]
- 25. Korotchkina L. G., and Patel M. S. (1995) Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase: site-specific regulation. J. Biol. Chem. 270, 14297–14304 [DOI] [PubMed] [Google Scholar]
- 26. Sale G. J., and Randle P. J. (1982) Occupancy of phosphorylation sites in pyruvate dehydrogenase phosphate complex in rat heart in vivo: relation to proportion of inactive complex and rate of re-activation by phosphatase. Biochem. J. 206, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juneau K., Nislow C., and Davis R. W. (2009) Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics 183, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vögtle F.-N., Burkhart J. M., Rao S., Gerbeth C., Hinrichs J., Martinou J.-C., Chacinska A., Sickmann A., Zahedi R. P., and Meisinger C. (2012) Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteomics 11, 1840–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Do T. Q., Hsu A. Y., Jonassen T., Lee P. T., and Clarke C. F. (2001) A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiae abc1 mutants. J. Biol. Chem. 276, 18161–18168 [DOI] [PubMed] [Google Scholar]
- 30. Johnson A., Gin P., Marbois B. N., Hsieh E. J., Wu M., Barros M. H., Clarke C. F., and Tzagoloff A. (2005) COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J. Biol. Chem. 280, 31397–31404 [DOI] [PubMed] [Google Scholar]
- 31. Smith A. C., and Robinson A. J. (2009) MitoMiner, an integrated database for the storage and analysis of mitochondrial proteomics data. Mol. Cell. Proteomics 8, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He C. H., Xie L. X., Allan C. M., Tran U. C., and Clarke C. F. (2014) Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim. Biophys. Acta 1841, 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Floyd B. J., Wilkerson E. M., Veling M. T., Minogue C. E., Xia C., Beebe E. T., Wrobel R. L., Cho H., Kremer L. S., Alston C. L., Gromek K. A., Dolan B. K., Ulbrich A., Stefely J. A., Bohl S. L., et al. (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. cell 63, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rose C. M., Venkateshwaran M., Volkening J. D., Grimsrud P. A., Maeda J., Bailey D. J., Park K., Howes-Podoll M., den Os D., Yeun L. H., Westphall M. S., Sussman M. R., Ané J.-M., and Coon J. J. (2012) Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Mol. Cell. Proteomics 11, 724–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H.-T., Rahaim P., Robbins P., and Yocum R. R. (1994) Cloning, sequence, and disruption of the Saccharomyces diastaticus DAR1 gene encoding a glycerol-3-phosphate dehydrogenase. J. Bacteriol. 176, 7091–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee Y. J., Jeschke G. R., Roelants F. M., Thorner J., and Turk B. E. (2012) Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol. Cell. Biol. 32, 4705–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valadi A., Granath K., Gustafsson L., and Adler L. (2004) Distinct intracellular localization of Gpd1p and Gpd2p, the two yeast isoforms of NAD+-dependent glycerol-3-phosphate dehydrogenase, explains their different contributions to redox-driven glycerol production. J. Biol. Chem. 279, 39677–39685 [DOI] [PubMed] [Google Scholar]
- 38. Renvoisé M., Bonhomme L., Davanture M., Valot B., Zivy M., and Lemaire C. (2014) Quantitative variations of the mitochondrial proteome and phosphoproteome during fermentative and respiratory growth in Saccharomyces cerevisiae. J. Proteomics 106, 140–150 [DOI] [PubMed] [Google Scholar]
- 39. Lin K., Hwang P. K., and Fletterick R. J. (1995) Mechanism of regulation in yeast glycogen phosphorylase. J. Biol. Chem. 270, 26833–26839 [DOI] [PubMed] [Google Scholar]
- 40. Behrends C., Sowa M. E., Gygi S. P., and Harper J. W. (2010) Network organization of the human autophagy system. Nature 466, 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., and Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly.” J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 42. Uhlinger D. J., Yang C. Y., and Reed L. J. (1986) Phosphorylation-dephosphorylation of pyruvate dehydrogenase from bakers' yeast. Biochemistry 25, 5673–5677 [DOI] [PubMed] [Google Scholar]
- 43. Stefely J. A., Kwiecien N. W., Freiberger E. C., Richards A. L., Jochem A., Rush M. J., Ulbrich A., Robinson K. P., Hutchins P. D., Veling M. T., Guo X., Kemmerer Z. A., Connors K. J., Trujillo E. A., Sokol J., et al. (2016) Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol. 34, 1191–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Humphrey S. J., James D. E., and Mann M. (2015) Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 26, 676–687 [DOI] [PubMed] [Google Scholar]
- 45. Brown G. K., Otero L. J., LeGris M., and Brown R. M. (1994) Pyruvate dehydrogenase deficiency. J. Med. Genet. 31, 875–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maj M. C., MacKay N., Levandovskiy V., Addis J., Baumgartner E. R., Baumgartner M. R., Robinson B. H., and Cameron J. M. (2005) Pyruvate dehydrogenase phosphatase deficiency: identification of the first mutation in two brothers and restoration of activity by protein complementation. J. Clin. Endocrinol. Metab. 90, 4101–4107 [DOI] [PubMed] [Google Scholar]
- 47. Michelakis E. D., Webster L., and Mackey J. R. (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 99, 989–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo X., Niemi N. M., Hutchins P. D., Condon S. G., Jochem A., Ulbrich A., Higbee A. J., Russell J. D., Senes A., Coon J. J., and Pagliarini D. J. (2017) Ptc7p dephosphorylates select mitochondrial proteins to enhance metabolic function. Cell Reports 18, 307–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 50. Brown J. P., and Perham R. N. (1976) Selective inactivation of the transacylase components of the 2-oxo acid dehydrogenase multienzyme complexes of Escherichia coli. Biochem. J. 155, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.